

Introduction
During evolution, multicellular organisms have developed mechanisms to counteract life-threatening events, such as infections and tissue injury, as well as to restore tissue homeostasis. These mechanisms are called ‘the inflammatory response‘. To initiate an appropriate inflammatory response, organisms have developed ways to recognize potentially life-threatening events. Danger signals—the molecules that alert the innate immune system and trigger defensive immune responses—have been classically defined as exogenous, pathogen-associated molecular pattern (PAMP) molecules. PAMPs—for example, lipopolysacharide (LPS), viral RNA and bacterial petidoglycans—interact with dedicated receptors on immune cells, the so-called pattern recognition receptors (PRRs). On ligation, PRRs transduce activation signals that lead to the production of proinflammatory molecules such as tumour necrosis factor (TNF). A well-known family of PRRs is the toll-like receptor (TLR) family in which each member recognizes a specific set of PAMPs.

The EMBO Workshop on Innate Danger Signals and HMGB1 took place between 8 and 11 February 2006 in Milan, Italy, and was organized by M. Bianchi, K. Tracey and U. Andersson.
However, several endogenous molecules also initiate inflammatory responses by interacting with signalling receptors; such innate danger signals are known as endokines and/or alarmins. The term endokine reflects the potential of these molecules with intranuclear and/or intracellular functions also to act extracellularly, in this case to be immunostimulatory on their release from necrotic cells. The endokine family includes high-mobility-group box (HMGB) proteins, interleukins such as IL-1α, cytosolic calcium-binding proteins of the S100 family, heat-shock proteins (HSPs) and nucleosomes. The term alarmin, coined by J. Oppenheim (Frederick, MD, USA) and co-workers, denotes an array of structurally diverse multifunctional host proteins that are rapidly released during infection or tissue damage, and that have mobilizing and activating effects on receptor-expressing cells engaged in host defence and tissue repair. Innate-immune mediators that have alarmin function include defensins, eosinophil-derived neurotoxin, cathelicidins and HMGB1 (Oppenheim & Yang, 2005).
During this EMBO workshop in Milan, 120 participants debated innate danger signal biology. A specific focus of the meeting was HMGB1, a nuclear protein that has been known both as an endokine and as an alarmin. Extracellularly released HMGB1 induces inflammation, proliferation and the migration of cells. All these aspects of HMGB1 biology were discussed during the meeting and compared with other known endokines and alarmins such as IL-1α, S100 proteins, HSPs, uric acid, anti-bacterial peptides and hepatoma-derived growth factor (HDGF). An important issue that emerged at the meeting is the likelihood that the activating effect of several alarmins, such as HMGB1 and HSPs, might be due to their propensity to form complexes with molecules derived from microbial organisms or necrotic cells. This might reflect the pathophysiological ways by which these molecules act on TLRs as well as on other receptors, and might explain the regulation of the various functions that have been ascribed to these molecules. The workshop ended with a lively debate about nomenclature, and as Oppenheim's term alarmin was voted to replace endokine, the former will be used throughout the rest of this report. The talks that we consider to be the highlights of the meeting are summarized below.
Alarmin functions and secretory routes
HMGB1 is a highly mobile nuclear protein that influences transcription and other nuclear transactions (for a thorough review, see Lotze & Tracey, 2005) and also has an important extracellular role as a signal of tissue damage. HMGB1 is either secreted actively by inflammatory cells or released passively as a soluble molecule from necrotic cells to signal tissue injury and initiate the inflammatory response and/or repair. In contrast to necrotic cells, apoptotic cells retain HMGB1 in their nuclei and so do not activate inflammation (Fig 1). This suggests that apoptotic cells modify their chromatin so that HMGB1 binds irreversibly to it.
Figure 1.
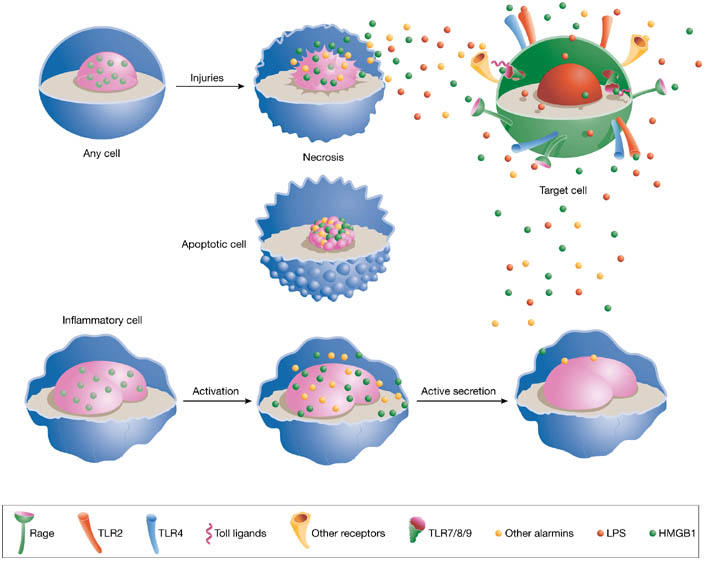
Release of alarmins into extracellular space. High-mobility-group box 1 (HMGB1), and possibly other alarmins or danger-associated molecular patterns (DAMPs), is actively secreted by inflammatory cells and passively released by necrotic cells. By contrast, HMGB1 is irreversibly bound to the chromatin of apoptotic cells. Once in the extracellular environment, HMGB1 activates target cells by signalling through the receptor for advanced glycation end products (RAGE), toll-like receptor 2 (TLR2) or TLR4. HMGB1 might also cooperate with TLR ligands, such as bacterial DNA or lipopolysaccharide, for TLR activation. RAGE might be involved in these phenomena.
In this respect, M. Bianchi (Milan, Italy) presented data for the role of histone H2B serine 14 phosphorylation in the immobilization of HMGB1 on chromatin during apoptosis. Originating from the observation that treatment with the histone deacetylase inhibitor trichostatin A (TSA) inhibits HMGB1 binding to apoptotic chromatin and makes apoptotic cells inflammatory, Bianchi's hypothesis is that histone hypoacetylation is not sufficient to immobilize HMGB1 onto chromatin. Indeed, histone H2B is actually phosphorylated on Ser 14 during apoptosis, and TSA treatment of apoptotic cells is able to inhibit such phosphorylation.
Y. Yamamoto (Osaka, Japan) reported data on a new candidate alarmin, HDGF, which has biochemical features similar to that of HMGB1 (Zhou et al, 2004). HDGF has a bipartite nuclear localization signal, lacks a signal peptide sequence and shares 32% amino-acid sequence homology with HMGB1. Originally isolated from the conditioned medium of a human hepatoma-derived cell line, extracellular HDGF stimulates the growth of several cell types and acts as a neurotrophic factor for neurons. In neurons, HDGF is mainly localized in the nucleus but can be actively secreted. Necrotic cells passively release HDGF, whereas it is tightly retained in the nucleus of apoptotic cells.
R. Apte (Beer-Sheva, Israel) presented data on the ‘intracrine' function of cytosolic IL-1α (Werman et al, 2004). After stimulation by LPS, the precursor of IL-1α translocates to the nucleus of macrophages, where it binds to DNA and functions as a transcription factor. Expression of the precursor of IL-1α in cells in which the surface expression of the receptor for IL-1 is blocked either induces the immediate secretion of cytokines or sensitizes cells to sub-threshold levels of inflammatory inducers for subsequent cytokine secretion.
Many alarmins, including HMGB1, are leaderless proteins, which are not routed through the endoplasmic reticulum and the Golgi apparatus but follow alterative secretory pathways (Nickel, 2003). After the activation of inflammatory cells, HMGB1 translocates from the nucleus to the cytoplasm owing to the acetylation of specific lysines in its nuclear localization signals. Once in the cytoplasm, HMGB1 is loaded into secretory lysosomes. Bianchi reported that the accumulation of HMGB1 in secretory lysosomes and its eventual exocytosis is abolished by inhibitors of multidrug resistance proteins (MRPs), a subgroup of ABC transporters. In support of this, he showed that peritoneal macrophages from Mrp1−/− mice are inefficient in HMGB1 secretion. In addition, HMGB1 secretion is associated with glutathionylation, a modification required for the transport of several drugs by MRP1.
D. Beer Stolz (Pittsburgh, PA, USA) presented data on an additional secretory pathway for HMGB1 in human colonic adenocarcinoma Caco-2 cells. In response to immunostimulators, these cells actively secrete HMGB1 from their apical membrane in exosomes—vesicles that form when multivesicular endosomes fuse with the plasma membrane.
J. Ritzerfeld (Heidelberg, Germany) discussed a possible secretory mechanism for another leaderless alarmin, galectin 1 (Gal1). Gal1 is a ligand of β-galactoside-containing cell-surface counter-receptors, and its secretion involves direct translocation from the cytoplasm across the plasma membrane. Ritzerfeld's data indicate that β-galactoside-containing surface molecules are export receptors for intracellular Gal1.
Leaderless cytokines, for example IL-1β, IL-18 and macrophage migration inhibitory factor (MIF), require free cysteines for their correct folding and/or bioactivity. A. Rubartelli (Genova, Italy) suggested an interesting, potential link between the bioactivity/stability of leaderless cytokines and the microenvironment. She reported that several human tumour cell lines, as well as human solid tumours and their inflammatory infiltrates, overexpress cytokines or danger factors such as HMGB1, MIF and thioredoxin (TRX), all of which are involved in tumour progression. Large amounts of free thiols in the culture media of tumour cells and the production of free thiols by neoplastic tissues in vivo were also observed. As extracellular HMGB1, MIF and TRX are oxidation-sensitive and the presence of extracellular thiols might maintain the reducing environment required for their function, resulting in their excessive and pathological bioactivity. In other types of necrosis involving non-neoplastic tissues—such as infarction—the small amounts of extracellular thiols could lead to the oxidation of leaderless inflammatory mediators and thereby to reduced activity, resulting in a milder inflammatory response.
HMGB1: an immune enhancer?
Four consecutive meeting sessions were devoted to the role of alarmins in immunity, inflammatory responses, cancer, development, tissue regeneration and viral infections.
P. Matzinger (Bethesda, MD, USA) stimulated an interesting discussion about danger signals and how the body decides whether to respond, not to respond or to heal. Matzinger suggested that the type of response induced by danger is governed by general endogenous alarmins such as HMGB1 as well as by tissue-specific sets of molecules. Bianchi noted that the tissues that Matzinger stated as being ‘immunoprivileged' (that is, non-responsive), for example testis and brain, contain low levels of HMGB1. Matzinger also introduced the term ‘Hyppo', which stands for hydrophobic protein parts. Hyppos are common motifs in both PAMPs and endogenous molecules and are recognized by TLRs through specific binding sites. Therefore, Hyppos might be a common denominator for PAMPs and alarmins and both sets of molecules could be called danger-associated molecular patterns (DAMPs; Seong & Matzinger, 2004).
P. Rovere-Querini (Milan, Italy) reported that HMGB1 is important for dendritic cell maturation. LPS-induced maturation of monocyte-derived dendritic cells is inhibited by treatment with either HMGB1-blocking antibodies or the antagonistic HMGB1 box A peptide. Both the expression of co-stimulatory molecules and the production of IL-10, IL-12 and TNF were affected. Immature dendritic cells also had higher expression of the receptor for advanced glycation end products (RAGE)—a proposed receptor for HMGB1 (see section on HMGB1 receptors below)—than did mature dendritic cells. The proliferation of naive CD4+ T cells cultured with mature dendritic cells could be inhibited by blocking HMGB1. Thus, HMGB1 can influence the initiation of both innate and adaptive immune responses (Dumitriu et al, 2005).
Several talks highlighted a new aspect of HMGB1 biology: as a cofactor for several TLR ligands to activate TLR-dependent functions (Fig 1). In particular, A. Coyle (Gaithersburg, MD, USA) showed that recombinant HMGB1 (rHMGB1) generated in Escherichia coli induces inflammatory cytokine production whereas thymus-derived HMGB1 does not, and that the ability of rHMGB1 to induce cytokines is significantly reduced in the presence of benzonase (an RNA/DNA endonuclease). Coyle and collaborators revealed that the ability of rHMGB1 to induce cytokines is partly due to its bacterial nucleic acid content. Thymus-derived HMGB1 synergizes with TLR7, TLR9, TLR2 and TLR4 ligands in the induction of cytokines in murine bone marrow cells, and the synergy occurs through a RAGE-dependent mechanism. These data suggest that HMGB1 lacks a direct pro-inflammatory property but primes cells to augment the response through distinct PAMPs.
G. LaRosa (Lexington, MA, USA) also presented data supporting synergy between HMGB1 and TLR ligands. In particular, rHMGB1 treated with Triton X-114 (Tx-HMGB1), to remove hydrophobic/lipophilic contaminants, loses its ability to stimulate cytokine release. Conversely, cells treated with suboptimal concentrations of LPS along with Tx-HMGB1 greatly increase cytokine and chemokine release, more than that induced by either component alone. The release is blocked by anti-TLR4 antibodies and does not occur in TLR4-defective cells. Thus, rHMGB1 acts as a cofactor for TLR4 ligands. Interestingly, Tx-HMGB1 alone is able to induce an increase in pro-inflammatory mRNAs, suggesting that it acts as an ‘immune enhancer'.
Although the above topics stimulated lively debates, other subjects unfortunately received less attention than they deserved. Most of the knowledge that we have about the extracellular activity of HMGB1 has been gained using bacterially expressed rHMGB1. The above data indicate an important regulatory mechanism by which HMGB1 might have different biological effects depending on whether it is part of a complex with bacterial products. It is tempting to speculate that HMGB1 released in the presence of TLR ligands acts to promote inflammation whereas HMGB1 released in an environment devoid of TLR ligands promotes tissue repair. To evaluate properly the different reported functions of HMGB1, it is of the utmost importance to define the source of the protein, the amount (even low grade) of endotoxins and other TLR ligands in the preparations used, and possibly also the degree of acetylation and the redox status of the protein. LaRosa has worked to purify active, native secreted HMGB1 from primary human monocytes or a monocytic cell line. At least part of the HMGB1 from these sources was acetylated, but differentially active with respect to stimulation of cytokine release. A further issue is the detection of HMGB1 in biological fluids using different methods. Although an enzyme-linked immunosorbent assay (ELISA; V. Urbonaviciute, Erlangen-Nuremberg, Germany) and an enzyme-linked immunosorbent spot (ELISPOT) method (H. Wähämaa, Stockholm, Sweden) were presented, there was no discussion about the conformity of these assays with the multitude of inhouse western blot methods used. This subject warrants its own session during the next HMGB1 meeting.
HMGB1 and tissue regeneration
On the role of HMGB1 as a mediator of tissue regeneration, H. Rauvala (Helsinki, Finland) discussed HMGB1 as an inducer of neuronal growth and migration. He showed that HMGB1 is necessary for the transfilter migration of glioma cells and that migration is inhibited by antibodies against the RAGE-binding domain of HMGB1. By studying nerve crush injuries, Rauvala determined that HMGB1 messenger RNA is present in cell bodies before injury but is only translated into protein after injury. Thus, Rauvala postulated that HMGB1 is a danger signal that is locally translated in injured axons to enhance regeneration of the nerve processes.
M. Capogrossi (Rome, Italy) reported that HMGB1 can induce myocardial regeneration after infarction. Injection of HMGB1 into mouse hearts after ischaemic damage resulted in the formation of new myocytes. Indeed, HMGB1 induced c-kit+ cardiac stem-cell proliferation and differentiation. Interestingly, differentiation towards the myocardial lineage was not observed in non-infarcted HMGB1-treated hearts, suggesting that signals released from damaged tissue are required for differentiation (Limana et al, 2005).
Given its potent extracellular functions, it is likely that HMGB1 is counteracted by inhibiting molecules. I. Maruyama (Kagoshima, Japan), showed that endothelial thrombomodulin binds HMGB1 through its lectin-binding domain and thereby dampens inflammation. Thrombomodulin inhibited HMGB1-induced NF-κB translocation in vitro and decreased ultraviolet-induced skin inflammation in vivo (Abeyama et al, 2005).
HMGB1 receptors
RAGE was the first receptor identified for extracellular HMGB1, but other receptors probably exist. Indeed, TLR2 and TLR4 have been shown to contribute to HMGB1-mediated signalling (Park et al, 2003). Rauvala identified receptor-type tyrosine phosphatase (RPTP) β/ζ—a transmembrane receptor able to mediate invasive migration—as a new putative HMGB1 receptor. HMGB1 binds to phosphacan, the soluble splice variant of RPTP β/ζ, with a high affinity similar to that of RAGE. As HMGB1 can induce neurite extension in RAGE-knockout embryonic neurons, Rauvala suggested that RPTP β/ζ functions as an HMGB1 receptor mediating the migration of neuronal growth cones.
RAGE is a multi-ligand receptor that binds advanced glycation end products (AGEs), S100/calgranulins, amyloid peptide and HMGB1 (Bierhaus et al, 2005). All these ligands contribute to several chronic diseases such as diabetes, amyloidosis, inflammatory conditions and tumours, by promoting cellular dysfunction through binding to RAGE. Engagement of RAGE leads to sustained cellular dysfunction and tissue destruction. In trying to define the role of RAGE in adaptive immunity, A. Bierhaus (Heidelberg, Germany) identified activated leukocyte cell adhesion molecule (ALCAM/CD166) as a putative substitute/homologue of RAGE, with a similar quaternary structure. Indeed, ALCAM-neutralizing antibodies and soluble ALCAM reduced signs of inflammation during delayed-type hypersensitivity, and to a much higher extent in Rage−/− mice compared with wild type.
Bierhaus also presented data indicating that RAGE, as a “molecule with a sense for danger”, might modulate behaviour and anxiety. In the elevated plus maze test, Rage−/− mice have a decreased level of anxiety compared with wild-type mice. These observations couple the concept of danger at a cellular level to that of danger as we perceive it. Such a coupling, of course, would be favoured by evolution.
HMGB1 in disease and its clinical applications
HMGB1 has been detected outside the nucleus of cells in several pathogenic conditions. During this meeting, reports were presented of HMGB1 as a mediator of sepsis (K. Tracey and H. Wang, Manhasset, NY, USA), arthritis (U. Andersson, Stockholm, Sweden), systemic lupus erythematosus (SLE; Urbonaviciute), and as a tumour-derived alarmin that activates eosinophils (M. Lotze, Pittsburgh, PA, USA). The cumulative knowledge of the inflammatory properties of HMGB1 suggests that it could be a target for anti-inflammatory therapy, as well as a therapeutic agent able to promote healing. One session during the meeting and many abstracts concerned strategies for blocking HMGB1 secretion and for inhibiting its extracellular functions, and the effects of such therapies in experimental inflammatory conditions.
Both LaRosa and S. Mao (Gaithersburg, MD, USA) reported the anti-inflammatory effects of treatment with humanized anti-HMGB1 monoclonal antibodies. LaRosa has investigated an experimental Pseudomonas infection and reported a 60% survival rate in treated mice compared with 10% survival in the control group. Mao has studied the effects of treatment in rat adjuvant arthritis and reported a 28% inhibition of disease, which was similar to results obtained with a soluble TNF receptor–immunoglobulin fusion protein. Treatment suppressed HMGB1, IL-6 and IL-1 levels in joint extracts and also reduced bone destruction. Microarray analysis of gene activation/suppression performed on hindpaw extracts from anti-HMGB1-treated and anti-TNF-treated rats revealed that only two genes overlapped between the treatment groups. This indicates that anti-HMGB1 therapy might be effective in anti-TNF-resistant patients.
Using models of sepsis, K. Suda (Tokyo, Japan) reported that the neutrophil elastase inhibitor Sivelestat suppressed HMGB1 expression in the lung and improved survival. Exploring the affinity of HMGB1 for distorted DNA, T. Östberg (Stockholm, Sweden) showed that oxaliplatin, which causes platinum adducts in DNA, blocked the translocation of HMGB1 in activated macrophages and suppressed the development of experimental arthritis. Using in vitro assays, S. Fumero (Turin, Italy) showed that synthetic, bent oligonucleotides had an affinity for HMGB1 and that treatment with such oligonucleotides suppressed HMGB1-induced proliferation and migration of smooth muscle cells.
Concluding remarks
In summary, this EMBO workshop was a successful meeting during which investigators presented and discussed important new data about a structurally diverse set of endogenous molecules that share several important properties in innate and adaptive immunity as well as in tissue repair. The meeting also heralded unification of the nomenclature for this class of molecules, now termed alarmins. HMGB1, HDGF, the proteins of the S100 family and IL-1α can be considered as the prototypes of this new class of proteins. The following criteria to identify and classify alarmins were also proposed: first, they are released from necrotic cells, but not apoptotic cells; second, they induce innate and adaptive immunity, tissue repair and cancer; third, they activate inflammation-related pathways such as NF-κB; and fourth, they have an unconventional secretory mechanism.
Finally, it is clear that the field is now entering a phase in which the previous promising results about the therapeutic potential of HMGB1 blockade and of HMGB1 as a therapeutic agent will be brought closer to clinical reality. To us, this sounds promising rather than alarmin(g).

Helena Erlandsson Harris

Angela Raucci
Acknowledgments
We thank our colleagues for sharing information and allowing their work to be described. We apologize to the participants whose work could not be cited owing to space limitations.
References
- Abeyama K et al. (2005) The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest 115: 1267–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP (2005) Understanding RAGE, the receptor for advanced glycation end products. J Mol Med 83: 876–886 [DOI] [PubMed] [Google Scholar]
- Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P (2005) HMGB1: guiding immunity from within. Trends Immunol 26: 381–387 [DOI] [PubMed] [Google Scholar]
- Limana F et al. (2005) Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res 97: 73–83 [DOI] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5: 331–342 [DOI] [PubMed] [Google Scholar]
- Nickel W (2003) The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes. Eur J Biochem 270: 2109–2119 [DOI] [PubMed] [Google Scholar]
- Oppenheim JJ, Yang D (2005) Alarmins: chemotactic activators of immune responses. Curr Opin Immunol 17: 359–365 [DOI] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E (2003) Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 279: 7370–7377 [DOI] [PubMed] [Google Scholar]
- Seong SY, Matzinger P (2004) Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 4: 469–478 [DOI] [PubMed] [Google Scholar]
- Werman A, Werman-Venkert R, White R, Lee JK, Werman B, Krelin Y, Voronov E, Dinarello CA, Apte RN (2004) The precursor form of IL-1α is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci USA 101: 2434–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Yamamoto Y, Sugai F, Yoshida K, Kishima Y, Sumi H, Nakamura H, Sakoda S (2004) Hepatoma-derived growth factor is a neurotrophic factor harbored in the nucleus. J Biol Chem 279: 27320–27326 [DOI] [PubMed] [Google Scholar]