

Abstract
Background
Pseudomonas aeruginosa is a genetically complex bacterium which can adopt and switch between a free-living or biofilm lifestyle, a versatility that enables it to thrive in many different environments and contributes to its success as a human pathogen.
Results
Transcriptomes derived from growth states relevant to the lifestyle of P. aeruginosa were clustered using three different methods (K-means, K-means spectral and hierarchical clustering). The culture conditions used for this study were; biofilms incubated for 8, 14, 24 and 48 hrs, and planktonic culture (logarithmic and stationary phase). This cluster analysis revealed the existence and provided a clear illustration of distinct expression profiles present in the dataset. Moreover, it gave an insight into which genes are up-regulated in planktonic, developing biofilm and confluent biofilm states. In addition, this analysis confirmed the contribution of quorum sensing (QS) and RpoS regulated genes to the biofilm mode of growth, and enabled the identification of a 60.69 Kbp region of the genome associated with stationary phase growth (stationary phase planktonic culture and confluent biofilms).
Conclusion
This is the first study to use clustering to separate a large P. aeruginosa microarray dataset consisting of transcriptomes obtained from diverse conditions relevant to its growth, into different expression profiles. These distinct expression profiles not only reveal novel aspects of P. aeruginosa gene expression but also provide a growth specific transcriptomic reference dataset for the research community.
Background
Pseudomonas aeruginosa is an opportunistic pathogen which causes a variety of infections including bacteremia in burns patients, urinary tract catheter infections and chronic colonisation of the lungs of Cystic Fibrosis patients [1]. This bacterium is a common nosocomial contaminant and its persistence as a major cause of human disease is linked to its intrinsic resistance to many antibiotics. In addition to this natural resistance, in many infections P. aeruginosa survives as a biofilm – adhered communities which themselves are more resistant to antibiotic therapy than free-living or planktonic organisms [2,3]. Given the decreased therapeutic options for this organism, there is a clear need for novel approaches to treat or prevent P. aeruginosa infection, and the key to this is to understand more about its biology. A greater knowledge of the genetics of growth of P. aeruginosa will help unravel the molecular basis of infection and in particular the mechanics of survival in the biofilm mode of growth.
The success of P. aeruginosa as a human pathogen, and its ability to thrive in diverse environmental habitats, is thought to be due to its large and complex genome (6.3 million base-pairs, 5570 open reading frames) which includes a variety of virulence factors and also its ability to alternate between a free-living and biofilm state [4]. In order to understand this flexibility we have used whole-genome microarrays to obtain transcriptomes from six conditions relevant to the growth of this organism. These conditions were two planktonic phases (logarithmic and stationary) and multiple biofilm time points (8, 14, 24 and 48 hrs). Using confocal microscopy we have previously characterised the static biofilm model used for this study, and found that confluent biofilms develop in a sequential manner from microcolonies [5]. In a preliminary analysis of this transcriptomic dataset, we found major differences in gene expression when comparing actively growing growth states with stationary phase growth states; 19.4% of the PAO1 genome was differentially expressed between log phase and stationary phase planktonic cultures and >15.5% of the PAO1 genome was differentially expressed when developing biofilms (8 hrs) was compared to confluent biofilm time-points (14, 24 and 48 hrs) [5]. In contrast we determined that developing and confluent biofilm transcriptomes were related to those of logarithmic and stationary phase planktonic cultures, respectively and that gene expression was conserved between confluent biofilms [5]. We also identified a unique set of 20 and 26 novel genes that were ≥ 2.5 fold up-regulated (P < 0.05) in developing biofilms and confluent biofilms respectively, when compared to all other conditions.
Although our previous analysis gave information regarding the relatedness of these different P. aeruginosa growth states, in this study we provide a more comprehensive analysis of this data using clustering. We performed such analysis using three different clustering methods: K-means, K-means spectral and hierarchical clustering. This analysis provides an overview of P. aeruginosa gene expression in conditions relevant to its lifestyle and allowed us to reveal the existence of distinct expression profiles in the transcriptomic data from these six different conditions. We were then able to observe gene expression that is unique to single conditions, or common to two or more culture conditions.
Results and discussion
Growth conditions used for microarray analysis
The biofilm microarray data used in this study was derived from static biofilms grown on nitrocellulose filters placed on 20% LB agar [5]. Reduced strength LB (20%) was used in order to reflect a low nutrient environment which would be encountered in vivo. Visual monitoring using strain PAO1 tagged with the green fluorescent protein (GFP) and confocal scanning laser microscope (CSLM) imaging demonstrated that after filters were inoculated with single P. aeruginosa cells, large microcolonies had formed after 8 hours which then further developed into confluent biofilms after 14 hrs [5]. No further change in biofilm architecture was observed after 24 and 48 hrs incubation. Microarray analysis using P. aeruginosa Affymetrix GeneChip arrays was carried out using RNA derived from four biofilm time points; 8, 14, 24, and 48 hours [5].
The planktonic culture microarray data used in this study was derived from logarithmic and stationary phase planktonic cultures grown in 250 ml Erlenmeyer flasks. Briefly, 100 ml LB broth (20%) was inoculated with 1 ml PAO1 overnight LB culture (diluted 10 fold), and the resulting cultures were grown at 37°C with agitation (200 rpm). Microarray analysis was carried out on RNA extracted after 4 hours (OD600 0.1) (Logarithmic phase – LP) and 24 hours growth (OD600 0.3) (Stationary phase – SP).
Transcriptome analysis: clustering of microarray data reveals different expression profiles
On analysis of the microarray data, 1429 genes (26% of genome) [see Additional file 2 – Supplementary Table S1] were found to show low expression (under 50 signal units for all three replicates) for all six different conditions. The most abundant functional classes in this list were: 'transport of small molecules', 'hypothetical, unclassified, unknown', 'putative enzymes' and 'transcriptional regulators'. This list contained two characterised multidrug efflux resistance systems (MexC-MexD-OprJ and MexE-MexF-OprN) and a putative multidrug efflux resistance system (PA3521-PA3523). Also, in this group was the gene encoding the β-lactamase precursor (ampC) and katB, one of the four genes encoding possible catalases in the P. aeruginosa genome. Both, ampC and katB have been shown previously to be induced in biofilms by the presence of sub-inhibitory doses of imipenem [6,7] and by hydrogen peroxide [7], respectively.
We began the transcriptome analysis by filtering out these 1429 genes showing very low expression for all six different conditions. A 1-way ANOVA (assuming unequal variances) was then performed which removed a further 805 genes that did not vary significantly between different conditions [see Additional file 2 – Supplementary Table S2]. As we are interested in the behaviour of genes between the six conditions and not within the 6 conditions, for the remaining 3315 genes we averaged the values of the three replicates for each of the six conditions, and then normalized these averaged profiles to zero mean and unit variance. We used the correlation coefficient as distance measure between gene profiles, and we clustered them using K-means clustering [8], K-means spectral clustering [9,10] and hierarchical clustering [8]. The first two methods were applied using a value of K (i.e. number of clusters) of 4, 7, 8, 9, 10, 12, 13, 15, 16 and 19. For the hierarchical clustering we tried the complete, centroid and ward linkage metrics for building the hierarchical tree and different values of the threshold for cutting it. An important finding is that for equivalent settings of the parameters of these different algorithms we obtained broadly the same groupings of the genes [see Additional file 1 – Supplementary Figure S1]. This revealed the existence of distinct expression profiles in the transcriptomic data from these six conditions. In the following we shall discuss the results obtained using K-means clustering with K = 10 (Figure 1a and 1b). These expression profiles are defined in Table 1.
Figure 1.
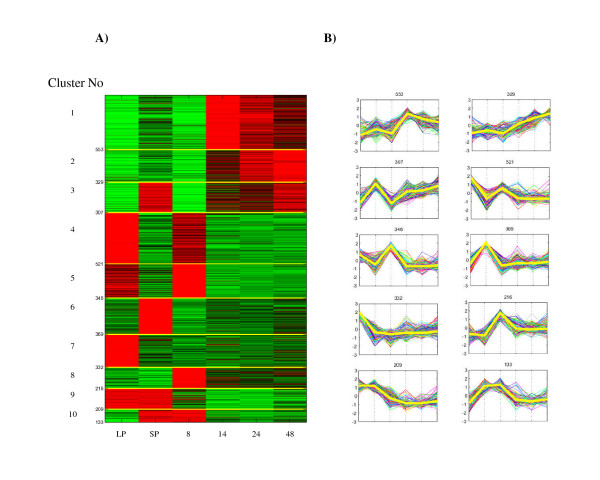
Results of K-means clustering of the expression data (K = 10). The three replicates for each of the six conditions were averaged, and then normalized to zero mean and unit variance. A) The Eisen diagram [56] of the expression profiles grouped according to the results of the clustering. Red denotes that the value observed is above the mean of the observation across the dataset. The y-axis shows the number of genes in each cluster. The labels on the X-axis denote the different expression conditions: LP, LP planktonic culture; SP, SP planktonic culture; 8, 14, 24 and 48, biofilm time points. B) The plot of the profiles in each cluster. The X-axis shows the conditions in the same order as A). The thick yellow lines represent the learned cluster centres.
Table 1.
Description of expression profiles obtained by K-means clustering (K = 10) of microarray data
| Cluster No | Up-regulation in: |
| 1 | confluent biofilm time points (expression peak – 14 hrs) |
| 2 | confluent biofilm time points (expression peak – 48 hrs) |
| 3 | confluent biofilm time points and in SP planktonic culture |
| 4 | LP planktonic culture (expression peak) and in developing biofilms (8 hrs) |
| 5 | LP planktonic culture and in developing biofilms (8 hrs) (expression peak) |
| 6 | SP planktonic culture |
| 7 | LP planktonic culture, |
| 8 | developing biofilms (8 hrs) |
| 9 | LP and SP planktonic culture |
| 10 | SP planktonic culture and in developing biofilms (8 hrs) |
Confirmation of microarray transcription profiles by quantitative reverse transcriptase PCR (qRT-PCR)
In order to verify the results obtained using the P. aeruginosa GeneChip microarrays (Affymetrix), genes from four different clusters (PA0020, cluster 8; fliE, cluster 6; PA2184, cluster 1; PA5555, cluster 4) representing different transcriptional profiles were examined by qRT-PCR. This was performed on RNA samples taken from 4 conditions (LP planktonic culture, SP planktonic culture, 8 hr biofilm, and 48 hr biofilm) and the results are displayed relative to expression under LP planktonic culture conditions (Figure 2). The gene expression profiles were consistent with those obtained from the microarray profiling experiments. Therefore, data from qRT-PCR provides independent verification of the microarray results.
Figure 2.
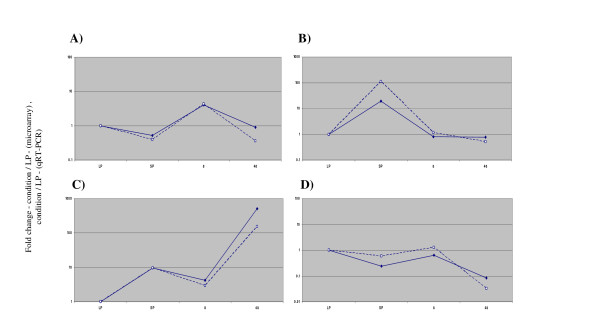
Comparison of microarray and quantitative reverse transcriptase PCR (qRT-PCR) transcription profiles. A) PA0020 (array cluster 8), B) fliE (array cluster 6), C) PA2184 – (array cluster 1), D) PA5555 – (array cluster 4). X-axis labels (expression conditions) – LP, LP planktonic culture; SP, SP planktonic culture; 8, 8 hr biofilm; 48, 48 hr biofilm. Microarray results – unbroken line, qRT-PCR results broken line. For both microarray (average expression value) and qRT-PCR (expression value) results, the value for each condition (SP, 8, and 48) was divided by the value obtained for LP.
Key functional classes in each cluster
The genes within each cluster were analysed and the percentage of twenty-six functional classes http://www.pseudomonas.com/ in each cluster was determined [see Additional file 1 – Supplementary Figure. S2]. Figure 3 shows the distribution of ten functional classes throughout the clusters.
Figure 3.
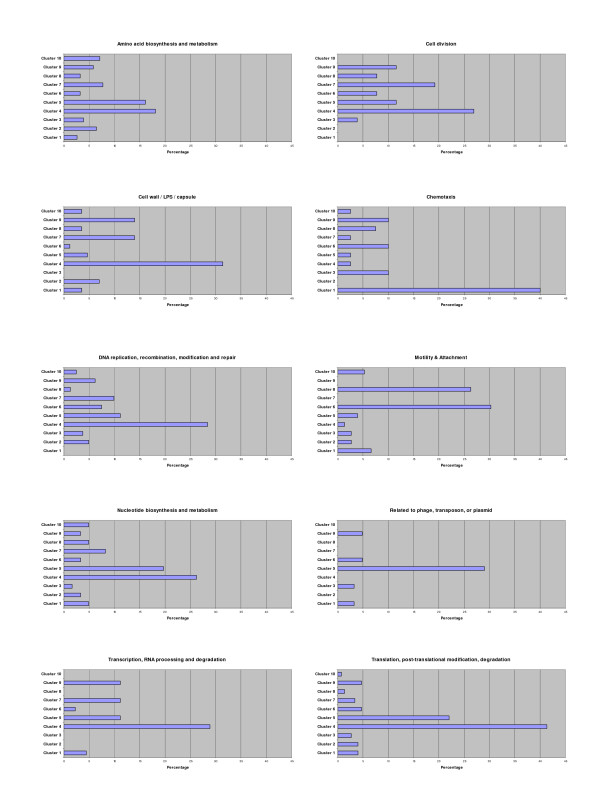
Distribution of ten functional classes throughout the clusters created by K-means clustering (K = 10). Percentages were obtained by dividing the number of genes of a functional class in each cluster, by the total number of genes in that functional class. For a representation of all twenty-six functional classes in all ten clusters see Additional File 1 (Figure. S2).
Of the functional classes, 'hypothetical, unclassified, unknown' was the most abundant, comprising at least 25% of the total number of genes in each cluster. As expected, clusters 4 and 5 (up-regulation in LP planktonic culture and developing biofilms) contain many genes in functional classes associated with growth (ie. 'transcription, RNA processing and degradation'; 'translation, post-translational modification, degradation', 'nucleotide biosynthesis and metabolism', 'DNA replication, recombination, modification and repair', 'cell division', 'amino acid biosynthesis and metabolism') (Figure 3). There is also a large percentage of genes from the classes 'transcription, RNA processing and degradation', 'cell wall/LPS/capsule' and 'cell division' in clusters 7 (up-regulation in LP planktonic culture) and 9 (up-regulation in LP and SP planktonic culture) (Figure 3). The 'motility and attachment' class features most prominently in clusters 6 (up-regulation in SP planktonic) and 8 (up-regulation in developing biofilms) (Figure 3); many genes in cluster 6 are involved in the biosynthesis and assembly of flagellar (essential for swimming and swarming motility) whereas cluster 8, contains many genes involved in biosynthesis and assembly of type-IV pili (essential for twitching motility). The regulators of biosynthesis of these motility structures are also present in these groups. Perhaps, the most interesting results are the appearance of a large percentage of genes involved in 'chemotaxis' and genes from the functional class 'phage, transposon, or plasmid' in clusters 1 (up-regulation in the confluent biofilm time points – expression peak 14 hrs) and 5 (up-regulation in LP planktonic culture and developing biofilms), respectively (discussed later) (Figure 3).
Gene lists for each cluster were compiled, together with their microarray expression results [see Additional file 3, Tables S3 – S12]. The following section describes some of the key genes found in the different expression profiles.
Genes up-regulated in confluent-biofilms, and genes up-regulated in confluent-biofilms and in SP planktonic culture
Cluster 1 and cluster 2 both contain genes up-regulated in the confluent biofilm time points with peak expression occurring at 14 hrs and 48 hrs, respectively [see Additional file 3, Supplementary Tables S3 and S4]. Cluster 3 contains genes up-regulated in SP planktonic culture as well as the confluent biofilm time points [see Additional file 3, Supplementary Table S5].
Although the previously identified QS and RpoS regulated genes were identified under planktonic growth conditions [11-13], it is known that these regulatory systems play an important role in biofilm maturation [14-16]. Therefore, we utilized the available QS and RpoS transcriptomic datasets [11-13] to determine the distribution of these genes throughout the 10 clusters. The majority of genes previously found to be regulated by QS and RpoS in planktonic culture were also expressed in confluent biofilms. 423 of the 504 genes found to be up-regulated by RpoS [12] were in the gene list used for the cluster analysis and 80.1% of these genes were found distributed between clusters 1, 2 and 3 (Figure 4a). 434 of the 534 genes (in total) shown to be up-regulated in two QS studies [11,13] were in the gene list used for the cluster analysis and 71% of these genes were found distributed amongst clusters 1, 2 and 3 (Figure 4b). This percentage would probably have been higher had full strength LB been used rather than the reduced strength LB broth (20%) used to culture planktonic and biofilm cells in this study. Taking into account the very different conditions we used when compared to previous QS and RpoS transcriptomic studies [11-13] and also considering the cell densities of the different growth states we used, the distribution of the QS and RpoS regulated genes over the 10 clusters that we obtained would be expected.
Figure 4.
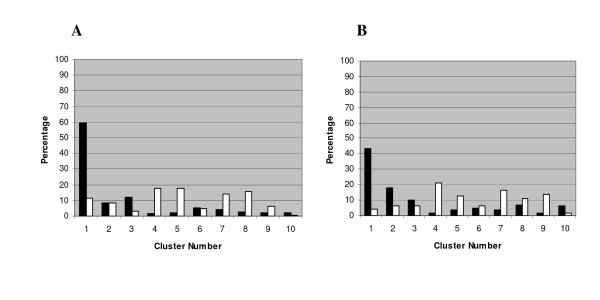
Percentage of a) RpoS and b) QS regulated genes in each cluster. a) Percentage obtained by dividing the number of RpoS activated or RpoS repressed genes in each cluster by the total number of RpoS activated or RpoS repressed genes. b) Percentage obtained by dividing the number of QS activated or QS repressed genes in each cluster by the total number of QS activated or QS repressed genes. Black columns – activated genes; white columns, repressed genes. QS and RpoS regulated genes were identified under planktonic growth conditions [11-13].
As many P. aeruginosa virulence determinants are regulated by QS, it is not surprising that the following are found in clusters 1, 2 and 3; elastase precursor (lasB, cluster 1), phenazine biosynthetic genes (PA1901 – PA1905, PA4209, PA4210-PA4211 cluster 1), and rhamnosyltransferases involved in rhamnolipid biosurfactant synthesis (rhlAB cluster 3, rhlC cluster 2). Many of the genes in clusters 1, 2 and 3 are activated in response to stress in P. aeruginosa or are orthologues of stress induced genes in other bacteria; general stress (PA1753, PA2190, cspD, rpoS, and rmf), nutrient stress (glgA, glgB, glgP – discussed in the next section), oxidative stress (katE – discussed in the next section), anaerobicity (anr), and osmolarity (osmC, osmE and osmY). Cluster 2 contains the gene encoding the transcriptional regulator – Anr, the anaerobic regulator of the arginine deiminase (ADI) and nitrate reductase pathways. Anr induces the ADI pathway in P. aeruginosa during oxygen limitation [17] enabling this organism to use arginine as an energy source. Three of the four genes of the ADI pathway (arcD, arcB, arcC) are present in clusters 1 and 3. Interestingly, cluster 1 includes a six ORF cluster napEFDABC (PA1172 – PA1177) and PA4130 which are thought to be involved in nitrate reduction, and nitrite or sulfite reduction, respectively. P. aeruginosa is known to use nitrate or nitrite as a terminal electron acceptor under anaerobic conditions [18]. Biofilms are known to be heterogeneous environments and it is known that even if biofilms are cultured under aerobic conditions there is little oxygen deep within mature biofilms [19] and in the CF lung [20]. Therefore, we can speculate that in our study cells at the surface of the biofilm are in an aerobic environment whereas cells nearest the filter surface after 48 hours biofilm growth are in a localised anaerobic environment. It is therefore not surprising that the gene encoding the anaerobic regulator Anr was found in cluster 2 with expression peaking after 48 hours biofilm growth.
Cluster 1 contains PA1753, a homologue of an E. coli universal stress protein (UspA). UspA is thought to have a general protective function, is one of most abundant proteins in growth arrested cells and is stimulated by numerous conditions such as SP conditions, nutrient limitation, and exposure to oxidants and antibiotics [21]. Cluster 2 contains PA2622 which is 71% similar to the cspD gene product of Escherichia coli, a member of the CspA cold shock stress adaptation family of proteins [22]. Although E. coli CspD is not induced by cold shock it is dramatically induced by SP growth and glucose starvation [22].
Other general stress genes in cluster 2 are rpoS, PA2190 (discussed in the next section) and rmf. rmf encodes a putative protein which is 66% similar to the characterised ribosome modulation factor of E. coli and was very highly expressed in the confluent biofilm time points. In E. coli this protein has been shown to be expressed during the transition from LP to SP growth and in slow growing cells and its role is to associate with 70S ribosomes, converting them into a dimeric form and reducing protein synthesis [23,24].
Cluster 1 contains PA0059, PA4876 and PA4739, homologues of three E. coli proteins (osmC, osmE, osmY) that are osmotically induced. Interestingly, in E. coli it has been previously found that bacteria within biofilms encounter higher osmolarity conditions than bacteria in a planktonic state [25].
In cluster 2 are two genes that encode lectins, lecA (PA2570 – galactophilic lectin) and lecB (fucose-binding lectin) which are very highly expressed in the three confluent biofilm time points (>2.5 fold up-regulated, when compared to the three other conditions, P < 0.05). Interestingly, a lecB deficient mutant has previously been found to be significantly impaired in biofilm formation [26].
Three genes of the mexGHI-opmD efflux system were found in cluster 2. This efflux pump is thought to be involved in acyl-homoserine lactone (AHL) homeostatis and resistance to vanadium, but mutants defective in this efflux system do not display decreased antibiotic resistance [27].
P. aeruginosa has five (I to V) 'clusters' (which will be called 'sets' to avoid confusion) of chemotaxis genes. Genes from sets I, II and III are found in cluster 1 and genes from set V is found in cluster 3. Why genes from these chemotaxis sets are up-regulated under biofilm conditions in which motility biosynthetic genes (flagellar and type IV pili) are down-regulated is not clear. One explanation could be that these chemotactic sets are involved in environmental sensing and responding to stress under biofilm conditions [12].
A genome region associated with stationary phase growth (planktonic and biofilm)
Whilst studying the genes in clusters 1 and 2 it became apparent that there was a region of the genome (PA2134 – PA2190; 60.69 Kbp from position 2349488 to 2410181) that had the same expression profile; up-regulation in the confluent biofilm time points (14, 24 and 48 hrs) (Figure 5).
Figure 5.
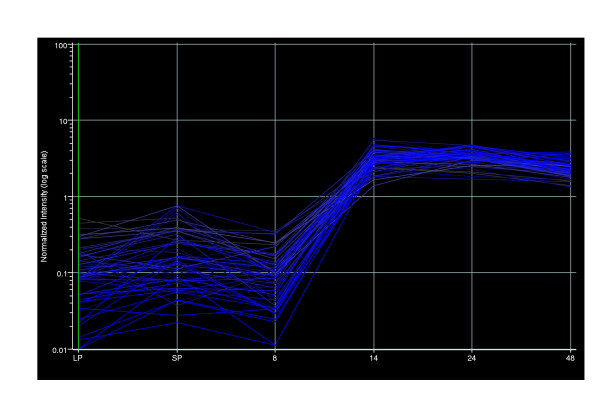
Gene expression profile of genome region PA2134-PA2190. X-axis labels – LP, LP planktonic culture; SP, SP planktonic culture; 8, 14, 24 & 48, biofilm time points.
32 of these 57 sequential genes have previously been found to be QS and/or RpoS regulated [11-13] using SP planktonic cultures of higher cell density than that of the SP planktonic cultures used in this study. Therefore, it is possible that this region could be associated with stationary phase growth (SP planktonic culture and/or confluent biofilms). There is no putative function assigned to the majority (40) of these genes. Interestingly this region contains genes with possible roles in the accumulation and breakdown of storage materials (glycogen, trehalose), oxidative stress protection (two catalases; katE, katN) and general stress (PA2190) (Table 2). glgA, glgB, glgP encode enzymes with putative roles in glycogen synthesis and degradation (GlgA – a probable glycogen synthase, GlgB – a 1, 4-alpha-glucan branching enzyme and GlgP – a putative glycogen phosphorylase). An E. coli glgA mutant was impaired in biofilm formation [28] and GlgP has been suggested to have a role in the slow degradation of endogenous glycogen during long stationary conditions [29]. This region also contains three probable glycosyl hydrolases (PA2160, PA2162, PA2164) which may also be involved in the hydrolysis of storage polysaccharides and PA2190 which is 62% similar to the glucose or phosphate starvation inducible gsiB general stress gene of Bacillus subtilis [30]. The two catalase genes present in this region; katE which encodes catalase HPII and katN which encodes a non-haem catalase, could aid the major housekeeping catalase KatA (PA4236) in SP conditions and in particular in the protection of biofilms against hydrogen peroxide.
Table 2.
Genes present in region PA2134 – PA2190 with an assigned putative functiona
| ORFb | Product Name | Gene Name |
| PA2135 | probable transporter | |
| PA2138 | probable ATP-dependent DNA ligase | |
| PA2140 | Probable metallothionein | |
| PA2142 | probable short-chain dehydrogenase | |
| PA2144 | glycogen phosphorylase | glgP |
| PA2147 | catalase HPII | katE |
| PA2152 | probable trehalose synthase | |
| PA2153 | 1, 4-alpha-glucan branching enzyme | glgB |
| PA2155 | probable phospholipase | |
| PA2158 | probable alcohol dehydrogenase (Zn-dependent) | |
| PA2160 | probable glycosyl hydrolase | glgX |
| PA2162 | probable glycosyl hydrolase | |
| PA2164 | probable glycosyl hydrolase | |
| PA2165 | probable glycogen synthase | glgA |
| PA2177 | Probable sensor/response regulator hybrid | |
| PA2185 | non-heme catalase KatN | katN |
| PA2188 | probable alcohol dehydrogenase (Zn-dependent) |
a Genes are identified by ORF designation, gene name and product name http://www.pseudomonas.com
b All other genes in this region are of the class 'hypothetical, unclassified, unknown'.
Given that amongst this region there are genes that appear to be involved in general stress, glycogen accumulation and breakdown, and oxidative stress survival, it would be interesting to determine whether there are any genomic or expression differences in this region between different isolates (clinical and environmental) and if so, whether there is any difference in survival of these isolates under long term stationary conditions (biofilm and planktonic).
Genes up-regulated in LP planktonic culture and 8 hrs developing biofilms
Clusters 4 and 5 both contain genes up-regulated in LP planktonic culture and in developing biofilms (8 hrs), with expression peaking in LP planktonic culture and developing biofilms, respectively [see Additional file 3, Supplementary Tables S6 and S7]. Genes involved in energy generation are well represented in clusters 4 and 5, for example: ATP synthase genes (PA5553 – PA5561, cluster 4), NADH dehydrogenase genes (PA2638 – PA2649, clusters 4 and 5), genes encoding enzymes involved the tri-carboxylic acid (TCA) cycle – succinate dehydrogenase, 2-oxoglutarate dehydrogenase, succinyl-CoA synthetase (PA1581 – PA1589, cluster 4). Also, present in cluster 5, are the chaperonins (groEL and groES) and a well characterised transcriptional regulator – Vfr, which regulates the las quorum sensing system [31] and twitching motility [32]. Many genes involved in protein secretion/export were present in clusters 4 and 5, for example; genes encoding the primary sec-dependent protein translocation pathway (secA, secB, – cluster 5; secF, secD, secY, secG – cluster 4), tatB (cluster 5)/tatC (cluster 4) of the twin arginine translocation system (Tat) and signal peptidase I encoded by lepB (cluster 4) which cleaves signal peptides off proteins translocated across biological membranes. Genes involved in lipopolysaccharide biosynthesis (PA3145 – PA3158) are found in cluster 4.
A previous P. aeruginosa strain PAO1 microarray study suggested phage induction to be important for gene transfer in biofilms, as a region of genes (PA0718 – PA0727) of the functional class 'phage, transposon, or plasmid' were found to be highly up-regulated in mature biofilms when compared to planktonic culture [15]. Therefore, it would be expected that these genes would appear in clusters 1 and 2 (ie. the late biofilm time points). However, only one gene from this region was found in cluster 1 (PA0723). In contrast, we found cluster 5 to contain the highest percentage of 'phage, transposon, or plasmid' functional class genes, all of which were in a different region (PA0616-PA0624, PA0632-PA0636, PA0643-PA0646) to the previous study. Many of these genes have homology to P. aeruginosa R pyocin-related phage family genes (e.g. phi CTX, PS17). Phi CTX is a temperate phage that produces a pore-forming cytotoxin [33] and R pyocin is a type of bacteriocin that has a structure similar to contractile phage tails [34,35]. PA0622 and PA0623 encode proteins with high homology (91 and 84%, respectively) to contractile tail proteins of phage PS17. Therefore, it is possible that these phage genes which have recently been shown to be induced in response to hydrogen peroxide stress [36] may play a role in virulence under actively growing conditions (LP planktonic culture and 8 hr biofilm) which could be encountered in vivo.
Genes up-regulated in SP planktonic culture
Cluster 6 contains genes up-regulated in SP planktonic culture [see Additional file 3, Supplementary Table S8]. As discussed above this cluster is unique, as many of the genes displaying this expression profile are involved in the biosynthesis, assembly and regulation of flagellar motility.
Other genes that display this profile suggest magnesium (phoQ, oprH, mgtA, mgtE) and potassium (KdpFABCD) may be limiting in the SP planktonic culture conditions used in this study. Genes known to be stimulated in response to magnesium starvation are; the oprH-phoPQ operon which encodes an outer-membrane protein (OprH) and a two component regulatory system (PhoP/Q) [37], mgtA (a ATP-dependent Mg2+ transporter homologue) [38], and mgtE (a probable Mg transporter). Whilst the kdp gene cluster (PA1632 – 1637) encodes a putative high affinity K+ ion transport system. However, the expression profile of these genes differs greatly to the kdpABC operon of Staphylococcus aureus, which has been found to be up-regulated in biofilms compared to planktonic culture [39].
In this study the general stress gene recA was found to be up-regulated in SP planktonic culture, unlike in E. coli in which recA has been found to be up-regulated in biofilms [40]. This cluster also contains algU the P. aeruginosa orthologue of E. coli extreme stress sigma factor σE.
Genes up-regulated in planktonic culture; LP, or LP and SP planktonic culture
Cluster 7 contains genes up-regulated in LP planktonic culture whereas cluster 9 contains genes up-regulated in LP and SP planktonic culture [see Additional file 3, Supplementary Tables S9 and S11]. These two clusters contain genes encoding two characterised multidrug efflux systems (MexAB-OprM, and MexXY) between them. Genes from two putative multidrug efflux systems (PA2527, PA2528, PA1541) were also present in these two clusters together with a two-component regulatory system (PmrA-PmrB) that regulates resistance to polymyxin B and cationic antimicrobial peptides. This two-component system is involved in the regulation of a putative LPS modification operon (PA3552 – PA3559) [41] which is also present in cluster 7. Biofilms are well known for their decreased susceptibility to antibiotics when compared to planktonic cells. However, no previously characterised antibiotic efflux system was found to be up-regulated in the static biofilm system used in this study. It is well known that other factors such as slow bacterial growth, oxygen limitation and decreased penetration of antibiotic through the exopolysaccharide matrix play a role in the decreased susceptibility of biofilms to antibiotic therapy [42,43]. However, recent studies have shown biofilm specific induction of the MexCD-OprJ efflux system in the presence of azithromycin [44] and biofilm specific induction of genes from two probable efflux systems upon exposure to tobramycin [15].
Genes up-regulated in 8 hrs developing biofilms
Cluster 8 contains genes up-regulated in developing biofilms (8 hrs) [see Additional file 3, Supplementary Table S10]. Given the importance of type-IV pili in microcolony formation [45] it is not surprising that many of the genes displaying this expression profile are involved in the biosynthesis, assembly and regulation of type-IV pili.
Although it is expressed highly under all conditions the transcriptional regulator mvaT was also included in this cluster as expression peaked in 8 hr biofilms. MvaT is a global regulator of gene expression [46] and negatively regulates the cupA gene cluster (chaperone-usher pathway) which has previously been found to be required for biofilm formation on abiotic surfaces [47,48] but is not expressed in our study. Other genes that display the cluster 8 profile are a chitin binding protein precursor (cbpD), bacterioferritin comigratory protein (bcp), a putative cold shock protein (PA1159) and the ecotin precursor which is thought to inhibit neutrophil elastase [49] and could play a protective role against the host immune system in developing biofilms.
Genes up-regulated in SP planktonic culture and in developing biofilms (8 hrs)
Three functional classes well represented in cluster 10 are those of amino acid biosynthesis and metabolism, fatty acid metabolism and carbon compound catabolism [see Additional file 3, Supplementary, Table S12]. Three gene clusters that illustrate this are; bkdA1A2B-lpdV, gnyDBHAL and soxBDAG. The bkd operon encodes a multi-enzyme complex branched-chain keto acid dehydrogenase involved in metabolism of valine, leucine and isoleucine [50]. Whereas the gny gene cluster encodes enzymes involved in degradation of acyclic isoprenoids [51] and the sox gene cluster encodes a putative sarcosine oxidase which catalyses the oxidative demethylation of sarcosine (N-methylglycine) to glycine, formaldehyde and hydrogen peroxide.
Conclusion
The three clustering methods employed in this study (K-means, K-means spectral and hierarchical clustering) allowed us to sort this large transcriptomic dataset into genes with similar expression profiles. This enabled direct visualisation of gene expression common to two or more culture conditions and unique to single conditions. Confidence in our results was gained from the fact that for equivalent settings of the parameters all three clustering methods gave broadly the same grouping of expression profiles. Thus, the distinct expression clusters obtained together with the genes with low expression and consistent expression [see Additional file 2, Table S1 and S2] over all conditions, gives a complete picture of gene expression for this organism under conditions relevant to its lifestyle.
Here we have discussed a selection of genes representative of each cluster but given the large number of hypothetical genes and un-characterised orthologues in each cluster, there is still much to understand about the molecular and genetic basis of planktonic and biofilm growth of Pseudomonas aeruginosa. However, we believe that the availability of this dataset as a resource for the research community will accelerate the determination of the roles of these genes.
Structuring this data in clusters provided a clear illustration of the main expression profiles of many novel genes, such as genes from the functional classes 'chemotaxis' and 'phage, transposon, or plasmid' which are found in clusters 1 and 5 respectively. The cluster analysis also enabled us to observe the up-regulation of many individual genes in confluent biofilms, for example; 1) un-characterised orthologues associated with general stress (PA1753, PA2190, cspD, rmf), nutrient stress (glgA, glgB, glgP), oxidative stress (katE), and osmolarity (osmC, osmE and osmY), 2) the characterised anaerobic transcriptional regulator Anr (encoded by anr). It also facilitated the identification of a region of 57 sequential genes (PA2134 – PA2190) which demonstrate the same expression profile (up-regulation in the confluent biofilm time points). 32 of these genes have previously been found to be QS and/or RpoS regulated [11-13].
Given the rise in antibiotic resistance, its ability to survive in the less antibiotic susceptible state – as a biofilm and the natural recalcitrance of this important human pathogen to many commonly used antibiotics, this dataset should aid in the pursuit of novel therapies against P. aeruginosa. Novel QS inhibitors have already been used with some success in vitro [52,53], and the increasing sophistication of proteomic and transcriptomic technology will accelerate the identification of other novel targets. For example this dataset could aid in the selection of new protein targets consistently expressed under all conditions (biofilm and planktonic) [see Additional file 2, Supplementary Table S2] or against protein targets expressed exclusively in the biofilm state. However, as biofilms are heterogeneous communities of cells, care should be taken when selecting biofilm specific targets and the spatial expression of these individual targets within biofilms should be studied. Other authors have found that different biofilm structures are formed when different media is used (uniform densely packed biofilms and mushroom shaped microcolonies) [54], which indicates that there is also a need to establish the difference in gene expression profiles between these different biofilm structures.
Methods
Bacterial strains
Pseudomonas aeruginosa wild-type strain PAO1 [55] was used for both biofilm and planktonic culture expression analysis.
Biofilm modelling, confocal imaging, planktonic phase culturing, RNA isolation, cDNA labelling and microarray scanning
Biofilm modelling, confocal imaging, planktonic phase culturing, RNA isolation, cDNA labelling and microarray scanning were performed as previously described [5]. The RNA samples used in this analysis were biological replicates, as they were derived from three independent cultures for each condition.
Quantitative reverse transcriptase PCR (qRT-PCR)
Planktonic and biofilm gene expression profiles generated using Affymetrix GeneChip arrays were verified using qRT-PCR. The same RNA samples used for the array analysis were also used for the qRT-PCR. 500 ng of RNA was reverse transcribed using the iScript cDNA Synthesis Kit as described by the manufacturer (BioRad Laboratories) and as a control the reaction was performed without reverse transcriptase (RT). Spectrophotometry was used to quantitate total cDNA and 67.5 ng of cDNA was used for each reaction using universal master mix (Applied Biosystems). Gene specific primers and probes were designed using Primer Express Software (Applied Biosystems), the sequences of which are shown in Table 3. Probes (purchased from Sigma-Genosys) were labeled 5' with 6-carboxyfluorescein (FAM) as the reporter and 3' with 6-carboxytetramethylrhodamine as the quencher (TAMRA). Each reaction was carried out in triplicate (in the presence of a no template control) using a model ABI 7900 HT thermal cycler (Applied Biosystems); thermal cycling parameters were 2 minutes at 50°C, 10 minutes at 95°C, and 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Calculation of expression value's for each gene (where cycle threshold is abbreviated as Ct): Ct (+RT) - Ct (-RT) = cCt (corrected CT). ΔCt = cCt (sample gene) - cCt (control gene, clpP). Expression value = 2-ΔCt. clpP was used as a control as it was found to be consistently expressed under all six conditions using microarray profiling [see Additional file 2, Supplementary Table S2].
Table 3.
Sequences of primers and TaqMan probes used for qRT-PCR
| Primer or probea | Oligonucleotide sequence 5'→3' |
| FliE F | GGAAATGCGTTCCATGCAA |
| FliE R | GGAGAGCATTTCCGAAAAGCT |
| FliE P | TTCAGGCGCCGGCCGAAG |
| PA0020 F | GCCGGCCTATGGCATCTT |
| PA0020 R | GCGTTGATGCCGAGGACTT |
| PA0020 P | CCAGGGCAAGGTCTACATCGATCCG |
| ClpP F | GGTGGTTGCTCAGTTGCTGTT |
| ClpP R | CGGCGAGTTGATGTAGAGATGA |
| ClpP P | CTGGAGGCTGAAAATCCCGAGAAGGA |
| PA2184 F | GCGCGGCTGGAACACTAT |
| PA2184 R | CCATGGGTTTCCTCGATGTG |
| PA2184 P | CGCAACTGAAGACGCGGATCGA |
| PA5555 F | CGCTGGTGGCCGATGA |
| PA5555 R | GGCGTCGGGTTCGTAGAGA |
| PA5555 P | TCAGGAGCTGAAGCACCACTGGGACT |
a. Forward primers (F), reverse primers (R) and TaqMan probes (P) are displayed.
Cluster analysis of microarray data
Before data processing, all microarray data was globally scaled to set the average signal intensity of each array to a target signal of 100.
We began the transcriptome analysis by filtering out 1429 genes showing very low expression for all six different conditions (under 50 signal units for all three replicates). A 1-way ANOVA (assuming unequal variances) was then performed which removed a further 805 genes that did not vary significantly between different conditions [see Additional file 2, Supplementary Table S2]. For the remaining 3315 genes, we averaged the values of the three replicates for each of the six conditions, and then normalized these averaged profiles to zero mean and unit variance. We used the correlation coefficient as distance measure between gene profiles, and we clustered them using K-means clustering [8], K-means spectral clustering [9,10] and hierarchical clustering [8]. The first two methods were applied using a value of K (i.e. number of clusters) of 4, 7, 8, 9, 10, 12, 13, 15, 16 and 19. For the hierarchical clustering we tried the complete, centroid and ward linkage metrics for building the hierarchical tree and different values of the threshold for cutting it. An important finding is that for equivalent settings of the parameters of these different algorithms we obtained broadly the same groupings of genes. This revealed the existence of distinct expression profiles in the transcriptomic data from these six conditions. As within the paper we discuss the results obtained using K-means clustering with K = 10, in the following section we discuss some details of K-means clustering. K-means is a standard algorithm for partitioning N data points into K disjoint subsets Sj containing Nj data points. The algorithm consists of a re-estimation procedure that attempts to minimize the sum-of-squares criterion:
where xn is a vector representing the nth data point and μj is the mean of the data points in Sj.
When using this algorithm for clustering gene expression profiles, each profile was considered a data point, and the distance in (A) was the correlation coefficient between xn and μj. This amounts to say that we are learning the expression profiles which are the centers of the clusters (the thick yellow line Figure 1b).
The clearest representation of the expression profiles within the dataset was obtained with a K = 10 (Figure 1a and 1b). The 10 clusters are defined in Table 1. The clusters found were quite stable with respect to different random initialization of the algorithm. Gene lists for each cluster were compiled, together with their microarray expression results [see Additional file 3, Tables S3 – S12]. We also performed the analysis directly on the experimental data, without averaging the replicates. As expected, the results obtained [see Additional file 1, Supplementary Figure S3] were similar to the ones presented in Figure 1 although more noisy.
Authors' contributions
RDW performed biofilm and planktonic culturing, microarray experiments, quantitative RT-PCR, data analysis and drafted the manuscript. AlP performed the cluster analysis and drafted the manuscript. JH performed data analysis. AnP and MS supervised the study and reviewed the manuscript. EL initiated the study. MC initiated and supervised the study, and reviewed the manuscript.
Supplementary Material
Figure S1. Results of clustering the expression data with K-means spectral clustering (K = 10) and hierarchical clustering with ward linkage. Shows the expression profiles obtained through these two methods of clustering. It also compares these two clustering methods with K-means clustering (K = 10) which is shown in the paper. For this analysis the average of replicates for each condition was used. Figure S2. Functional class representation in the ten clusters created by K-means clustering. From this figure it can be determined what percentage of genes in each of twenty-six functional classes are present in each cluster. Figure S3. Results of K-means clustering (K = 10) of expression data from all replicates from all six conditions. Shows an Eisen diagram of the expression profiles grouped according to the results of the clustering and a graphical plot of the profiles in each cluster.
Table S1. Contains a list of all genes that display low expression in all six conditions studied. This list contains genes showing very low expression (under 50 signal units for all three replicates) for all six different conditions. These 1429 genes were not used in the clustering analysis. The table displays the average microarray expression results for each gene for each condition. Table S2. Contains a list of genes in which gene expression did not vary significantly between the six different conditions. This list was obtained using a 1-way ANOVA (assuming unequal variances) and genes in this list were not used in the clustering analysis. The table displays the average microarray expression results for each gene for each condition.
Tables S3 – S12. Genes present in the clusters obtained by K-means clustering (K = 10). The genes in clusters 1 -10 are displayed together with their average microarray expression results for each condition.
Acknowledgments
Acknowledgements
Thanks also to Tracy Chaplin (Medical Oncology Unit, Barts and The London, Queen Mary's School of Medicine and Dentistry) for hybridisation & scanning of the arrays. The P. aeruginosa GeneChip arrays (Affymetrix) were subsidised by Cystic Fibrosis Foundation Therapeutics, Inc. RDW, AlP and JH were funded by was funded by the DTI/MRC Link Programme in Applied Genomics (G0100109) in collaboration with Medivir UK.
Contributor Information
Richard D Waite, Email: R.D.Waite@qmul.ac.uk.
Alberto Paccanaro, Email: alberto.paccanaro@rhul.ac.uk.
Anastasia Papakonstantinopoulou, Email: a.papakonsta@qmul.ac.uk.
Jacob M Hurst, Email: jacob@biochemistry.ucl.ac.uk.
Mansoor Saqi, Email: m.saqi@qmul.ac.uk.
Eddie Littler, Email: Eddy.Littler@medivir.com.
Michael A Curtis, Email: m.a.curtis@qmul.ac.uk.
References
- Bodey GP, Bolivar R, Fainstein V, Jadeja L. Infections caused by Pseudomonas aeruginosa. Rev Infect Dis. 1983;5:279–313. doi: 10.1093/clinids/5.2.279. [DOI] [PubMed] [Google Scholar]
- Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- Drenkard E. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 2003;5:1213–1219. doi: 10.1016/j.micinf.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Waite RD, Papakonstantinopoulou A, Littler E, Curtis MA. Transcriptome analysis of Pseudomonas aeruginosa growth: comparison of gene expression in planktonic cultures and developing and mature biofilms. J Bacteriol. 2005;187:6571–6576. doi: 10.1128/JB.187.18.6571-6576.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagge N, Schuster M, Hentzer M, Ciofu O, Givskov M, Greenberg EP, Hoiby N. Pseudomonas aeruginosa biofilms exposed to imipenem exhibit changes in global gene expression and beta-lactamase and alginate production. Antimicrob Agents Chemother. 2004;48:1175–1187. doi: 10.1128/AAC.48.4.1175-1187.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma M, DeLuca D, Worgall S, Quadri LE. Transcriptome analysis of the response of Pseudomonas aeruginosa to hydrogen peroxide. J Bacteriol. 2004;186:248–252. doi: 10.1128/JB.186.1.248-252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt BS. Cluster Analysis. 3. London: Edward Arnold; 1993. [Google Scholar]
- Ng A, Jordan M, Weiss Y. On spectral clustering: analysis and an algorithm. In: Dietterich TG, Becker S, Ghahramani Z, editor. NIPS 14, Advances in Neural Information Processing Systems. 2001. pp. 849–856. [Google Scholar]
- Paccanaro A, Casbon JA, Saqi MA. Spectral clustering of protein sequences. Nucleic Acids Res. 2006;34:1571–1580. doi: 10.1093/nar/gkj515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M, Lostroh CP, Ogi T, Greenberg EP. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J Bacteriol. 2003;185:2066–2079. doi: 10.1128/JB.185.7.2066-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M, Hawkins AC, Harwood CS, Greenberg EP. The Pseudomonas aeruginosa RpoS regulon and its relationship to quorum sensing. Mol Microbiol. 2004;51:973–985. doi: 10.1046/j.1365-2958.2003.03886.x. [DOI] [PubMed] [Google Scholar]
- Wagner VE, Bushnell D, Passador L, Brooks AI, Iglewski BH. Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J Bacteriol. 2003;185:2080–2095. doi: 10.1128/JB.185.7.2080-2095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science. 1998;280:295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- Whiteley M, Bangera MG, Bumgarner RE, Parsek MR, Teitzel GM, Lory S, Greenberg EP. Gene expression in Pseudomonas aeruginosa biofilms. Nature. 2001;413:860–864. doi: 10.1038/35101627. [DOI] [PubMed] [Google Scholar]
- Heydorn A, Ersboll B, Kato J, Hentzer M, Parsek MR, Tolker-Nielsen T, Givskov M, Molin S. Statistical analysis of Pseudomonas aeruginosa biofilm development: impact of mutations in genes involved in twitching motility, cell-to-cell signaling, and stationary-phase sigma factor expression. Appl Environ Microbiol. 2002;68:2008–2017. doi: 10.1128/AEM.68.4.2008-2017.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winteler HV, Haas D. The homologous regulators ANR of Pseudomonas aeruginosa and FNR of Escherichia coli have overlapping but distinct specificities for anaerobically inducible promoters. Microbiology. 1996;142:685–693. doi: 10.1099/13500872-142-3-685. [DOI] [PubMed] [Google Scholar]
- Hassett DJ. Anaerobic production of alginate by Pseudomonas aeruginosa : alginate restricts diffusion of oxygen. J Bacteriol. 1996;178:7322–7325. doi: 10.1128/jb.178.24.7322-7325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu KD, Stewart PS, Xia F, Huang CT, McFeters GA. Spatial physiological heterogeneity in Pseudomonas aeruginosa biofilm is determined by oxygen availability. Appl Environ Microbiol. 1998;64:4035–4039. doi: 10.1128/aem.64.10.4035-4039.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiss T, Botzenhart K, Yankaskas JR, Randell S, Boucher RC, Doring G. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest. 2002;109:317–325. doi: 10.1172/JCI200213870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvint K, Nachin L, Diez A, Nystrom T. The bacterial universal stress protein: function and regulation. Curr Opin Microbiol. 2003;6:140–145. doi: 10.1016/S1369-5274(03)00025-0. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Inouye M. Growth-phase-dependent expression of cspD, encoding a member of the CspA family in Escherichia coli. J Bacteriol. 1997;179:5126–5130. doi: 10.1128/jb.179.16.5126-5130.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada A, Yamazaki Y, Fujita N, Ishihama A. Structure and probable genetic location of a "ribosome modulation factor" associated with 100S ribosomes in stationary-phase Escherichia coli cells. Proc Natl Acad Sci USA. 1990;87:2657–2661. doi: 10.1073/pnas.87.7.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada A, Igarashi K, Yoshimura S, Aimoto S, Ishihama A. Ribosome modulation factor: stationary growth phase-specific inhibitor of ribosome functions from Escherichia coli. Biochem Biophys Res Commun. 1995;214:410–417. doi: 10.1006/bbrc.1995.2302. [DOI] [PubMed] [Google Scholar]
- Prigent-Combaret C, Vidal O, Dorel C, Lejeune P. Abiotic surface sensing and biofilm-dependent regulation of gene expression in Escherichia coli. J Bacteriol. 1999;181:5993–6002. doi: 10.1128/jb.181.19.5993-6002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loris R, Tielker D, Jaeger KE, Wyns L. Structural basis of carbohydrate recognition by the lectin LecB from Pseudomonas aeruginosa. J Mol Biol. 2003;331:861–870. doi: 10.1016/S0022-2836(03)00754-X. [DOI] [PubMed] [Google Scholar]
- Aendekerk S, Ghysels B, Cornelis P, Baysse C. Characterization of a new efflux pump, MexGHI-OpmD, from Pseudomonas aeruginosa that confers resistance to vanadium. Microbiology. 2002;148:2371–2381. doi: 10.1099/00221287-148-8-2371. [DOI] [PubMed] [Google Scholar]
- Jackson DW, Suzuki K, Oakford L, Simecka JW, Hart ME, Romeo T. Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli. J Bacteriol. 2002;184:290–301. doi: 10.1128/JB.184.1.290-301.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Jen Y, Takeuchi E, Inouye M, Nakayama H, Tagaya M, Fukui T. Alpha-glucan phosphorylase from Escherichia coli. Cloning of the gene, and purification and characterization of the protein. J Biol Chem. 1988;263:13706–13711. [PubMed] [Google Scholar]
- Mueller JP, Bukusoglu G, Sonenshein AL. Transcriptional regulation of Bacillus subtilis glucose starvation-inducible genes: control of gsiA by the ComP-ComA signal transduction system. J Bacteriol. 1992;174:4361–4373. doi: 10.1128/jb.174.13.4361-4373.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albus AM, Pesci EC, Runyen-Janecky LJ, West SE, Iglewski BH. Vfr controls quorum sensing in Pseudomonas aeruginosa. J Bacteriol. 1997;179:3928–3935. doi: 10.1128/jb.179.12.3928-3935.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatson SA, Whitchurch CB, Sargent JL, Levesque RC, Mattick JS. Differential regulation of twitching motility and elastase production by Vfr in Pseudomonas aeruginosa. J Bacteriol. 2002;184:3605–3613. doi: 10.1128/JB.184.13.3605-3613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Matsumoto H, Ohnishi M, Yokota S, Shinomiya T, Kageyama M, Terawaki Y. Cytotoxin-converting phages, phi CTX and PS21, are R pyocin-related phages. FEMS Microbiol Lett. 1994;122:239–244. doi: 10.1111/j.1574-6968.1994.tb07174.x. [DOI] [PubMed] [Google Scholar]
- Kageyama M. Studies of a Pyocin. I. Physical and Chemical Properties. J Biochem (Tokyo) 1964;55:49–53. doi: 10.1093/oxfordjournals.jbchem.a127839. [DOI] [PubMed] [Google Scholar]
- Kageyama M, Shinomiya T, Aihara Y, Kobayashi M. Characterization of a bacteriophage related to R-type pyocins. J Virol. 1979;32:951–957. doi: 10.1128/jvi.32.3.951-957.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W, Small DA, Toghrol F, Bentley WE. Microarray analysis of Pseudomonas aeruginosa reveals induction of pyocin genes in response to hydrogen peroxide. BMC Genomics. 2005;6:115. doi: 10.1186/1471-2164-6-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane EL, Kwasnicka A, Ochs MM, Hancock RE. PhoP-PhoQ homologues in Pseudomonas aeruginosa regulate expression of the outer-membrane protein OprH and polymyxin B resistance. Mol Microbiol. 1999;34:305–316. doi: 10.1046/j.1365-2958.1999.01600.x. [DOI] [PubMed] [Google Scholar]
- Kato A, Tanabe H, Utsumi R. Molecular characterization of the PhoP-PhoQ two-component system in Escherichia coli K-12: identification of extracellular Mg2+-responsive promoters. J Bacteriol. 1999;181:5516–5520. doi: 10.1128/jb.181.17.5516-5520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenken KE, Dunman PM, McAleese F, Macapagal D, Murphy E, Projan SJ, Blevins JS, Smeltzer MS. Global gene expression in Staphylococcus aureus biofilms. J Bacteriol. 2004;186:4665–4684. doi: 10.1128/JB.186.14.4665-4684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloin C, Valle J, Latour-Lambert P, Faure P, Kzreminski M, Balestrino D, Haagensen JA, Molin S, Prensier G, Arbeille B, Ghigo JM. Global impact of mature biofilm lifestyle on Escherichia coli K-12 gene expression. Mol Microbiol. 2004;51:659–674. doi: 10.1046/j.1365-2958.2003.03865.x. [DOI] [PubMed] [Google Scholar]
- McPhee JB, Lewenza S, Hancock RE. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol Microbiol. 2003;50:205–217. doi: 10.1046/j.1365-2958.2003.03673.x. [DOI] [PubMed] [Google Scholar]
- Mah TF, O'Toole GA. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001;9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- Borriello G, Werner E, Roe F, Kim AM, Ehrlich GD, Stewart PS. Oxygen limitation contributes to antibiotic tolerance of Pseudomonas aeruginosa in biofilms. Antimicrob Agents Chemother. 2004;48:2659–2664. doi: 10.1128/AAC.48.7.2659-2664.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis RJ, White KG, Choi KH, Wagner VE, Schweizer HP, Iglewski BH. Molecular basis of azithromycin-resistant Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother. 2005;49:3858–3867. doi: 10.1128/AAC.49.9.3858-3867.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- Diggle SP, Winzer K, Lazdunski A, Williams P, Camara M. Advancing the quorum in Pseudomonas aeruginosa : MvaT and the regulation of N-acylhomoserine lactone production and virulence gene expression. J Bacteriol. 2002;184:2576–2586. doi: 10.1128/JB.184.10.2576-2586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet I, Olson JW, Lory S, Lazdunski A, Filloux A. The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Proc Natl Acad Sci USA. 2001;98:6911–6916. doi: 10.1073/pnas.111551898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet I, Diggle SP, Stacey RE, Camara M, Ventre I, Lory S, Lazdunski A, Williams P, Filloux A. Biofilm formation in Pseudomonas aeruginosa: fimbrial cup gene clusters are controlled by the transcriptional regulator MvaT. J Bacteriol. 2004;186:2880–2890. doi: 10.1128/JB.186.9.2880-2890.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers CT, Murray IA, Delmar VA, Day AG, Craik CS. The periplasmic serine protease inhibitor ecotin protects bacteria against neutrophil elastase. Biochem J. 2004;379:107–118. doi: 10.1042/BJ20031790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hester KL, Lehman J, Najar F, Song L, Roe BA, MacGregor CH, Hager PW, Phibbs PV, Jr, Sokatch JR. Crc is involved in catabolite repression control of the bkd operons of Pseudomonas putida and Pseudomonas aeruginosa. J Bacteriol. 2000;182:1144–1149. doi: 10.1128/JB.182.4.1144-1149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Perez AL, Zavala-Hernandez AN, Cervantes C, Campos-Garcia J. The gnyRDBHAL cluster is involved in acyclic isoprenoid degradation in Pseudomonas aeruginosa. Appl Environ Microbiol. 2004;70:5102–5110. doi: 10.1128/AEM.70.9.5102-5110.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentzer M, Riedel K, Rasmussen TB, Heydorn A, Andersen JB, Parsek MR, Rice SA, Eberl L, Molin S, Hoiby N, Kjelleberg S, Givskov M. Inhibition of quorum sensing in Pseudomonas aeruginosa biofilm bacteria by a halogenated furanone compound. Microbiology. 2002;148:87–102. doi: 10.1099/00221287-148-1-87. [DOI] [PubMed] [Google Scholar]
- Smith KM, Bu Y, Suga H. Induction and inhibition of Pseudomonas aeruginosa quorum sensing by synthetic autoinducer analogs. Chem Biol. 2003;10:81–89. doi: 10.1016/S1074-5521(03)00002-4. [DOI] [PubMed] [Google Scholar]
- Klausen M, Heydorn A, Ragas P, Lambertsen L, Aaes-Jorgensen A, Molin S, Tolker-Nielsen T. Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol Microbiol. 2003;48:1511–1524. doi: 10.1046/j.1365-2958.2003.03525.x. [DOI] [PubMed] [Google Scholar]
- Holloway BW. Genetic recombination in Pseudomonas aeruginosa. J Gen Microbiol. 1955;13:572–581. doi: 10.1099/00221287-13-3-572. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Results of clustering the expression data with K-means spectral clustering (K = 10) and hierarchical clustering with ward linkage. Shows the expression profiles obtained through these two methods of clustering. It also compares these two clustering methods with K-means clustering (K = 10) which is shown in the paper. For this analysis the average of replicates for each condition was used. Figure S2. Functional class representation in the ten clusters created by K-means clustering. From this figure it can be determined what percentage of genes in each of twenty-six functional classes are present in each cluster. Figure S3. Results of K-means clustering (K = 10) of expression data from all replicates from all six conditions. Shows an Eisen diagram of the expression profiles grouped according to the results of the clustering and a graphical plot of the profiles in each cluster.
Table S1. Contains a list of all genes that display low expression in all six conditions studied. This list contains genes showing very low expression (under 50 signal units for all three replicates) for all six different conditions. These 1429 genes were not used in the clustering analysis. The table displays the average microarray expression results for each gene for each condition. Table S2. Contains a list of genes in which gene expression did not vary significantly between the six different conditions. This list was obtained using a 1-way ANOVA (assuming unequal variances) and genes in this list were not used in the clustering analysis. The table displays the average microarray expression results for each gene for each condition.
Tables S3 – S12. Genes present in the clusters obtained by K-means clustering (K = 10). The genes in clusters 1 -10 are displayed together with their average microarray expression results for each condition.