

Abstract
Induction of the antiviral cytokine interferon α/β (IFN-α/β) is common in many viral infections. The impact of ongoing antiviral responses on subsequent bacterial infection is not well understood. In human disease, bacterial superinfection complicating a viral infection can result in significant morbidity and mortality. We injected mice with polyinosinic-polycytidylic (PIC) acid, a TLR3 ligand and known IFN-α/β inducer as well as nuclear factor κB (NF-κB) activator to simulate very early antiviral pathways. We then challenged mice with an in vivo septic shock model characterized by slowly evolving bacterial infection to simulate bacterial superinfection early during a viral infection. Our data demonstrated robust induction of IFN-α in serum within 24 h of PIC injection with IFN-α/β–dependent major histocompatibility antigen class II up-regulation on peritoneal macrophages. PIC pretreatment before septic shock resulted in augmented tumor necrosis factor alpha and interleukins 6 and 10 and heightened lethality compared with septic shock alone. Intact IFN-α/β signaling was necessary for augmentation of the inflammatory response to in vivo septic shock and to both TLR2 and TLR4 agonists in vitro. To assess the NF-κB contribution to PIC-modulated inflammatory responses to septic shock, we treated with parthenolide an NF-κB inhibitor before PIC and septic shock. Parthenolide did not inhibit IFN-α induction by PIC. Inhibition of NF-κB by parthenolide did reduce IFN-α–mediated potentiation of the cytokine response and lethality from septic shock. Our data demonstrate that pathways activated early during many viral infections can have a detrimental impact on the outcome of subsequent bacterial infection. These pathways may be critical to understanding the heightened morbidity and mortality from bacterial superinfection after viral infection in human disease.
Keywords: Rodent, endotoxin shock, cytokines, bacterial infection, viral infection
Viral infections induce a cascade of events, many of which are virus-specific, whereas others are more generalized. Induction of the antiviral cytokines interferon (IFN) α/β and IFN-γ are common in the response to most viral infections. The immune potentiating effects of IFN-γ have been well characterized. IFN-α/β is robustly induced early during most viral infections; however, its immune potentiating influence is less well characterized. Bacterial superinfection concurrent with an ongoing antiviral immune response is a relatively common event in human disease. Important examples of bacterial infections after viral infections resulting in serious morbidity are influenza, bacterial pneumonia and varicella, and staphylococcal/streptococcal toxic shock/necrotizing fasciitis (1–3). Although influenza and varicella disrupt protective barriers (respiratory epithelial and skin), increasing the risk for secondary bacterial infection, little is known about the impact of antiviral immune responses on the inflammatory response to secondary bacterial pathogens.
Some viral infections are known to induce a state of immune hyporesponsiveness such as measles virus, Epstein-Barr virus, cytomegalovirus, many pox viruses, some influenza strains, and hepatitis B and C viruses, leading to both disruption of protective mucosa/epithelium and a permissive state for bacterial infection (4–7). Other viruses such as lymphocytic choriomeningitis virus (LCMV),coxsackievirus, and other influenza strains have the ability to potentiate host responsiveness to subsequent inflammatory challenge (8–12). Very recently, adenovirus has also been shown to potentiate responses to lipopolysaccharide (LPS) without a precise mechanistic or mediator-based explanation (13).
Previously, we reported exaggerated cytokine responses and lethality in response to LPS in mice infected with LCMV (15). In that work, LPS was administered when high circulating levels of IFN-α/β were present in response to LCMV infection. Using mice deficient in elements of the IFN-α/β or IFN-γ signaling pathways, we demonstrated that enhanced sensitivity to LPS early during viral infection was IFN-α/β–dependent. Given these observations, we believe that IFN-α/β induced early during viral infection may play a significant role in determining the magnitude of inflammatory responses to subsequent bacterial challenges. High levels of IFN-α/β have been reported in serum from children with common viral infections such as parainfluenza, influenza, adenovirus, rotavirus, varicella, and herpes simplex at the time of their clinical diagnosis, indicating that IFN-α/β is a common event early in the response to ordinary viral infections in children (16, 17). Coupled with our previous data, it is possible that morbidity from bacterial superinfection encountered during viral infection may also be IFN-α/β–dependent. It is therefore possible that IFN-α/β, although critical for viral clearance, may predispose individuals, especially children who experience frequent viral infections, to more serious morbidity from subsequent invasive bacterial pathogens.
Having identified IFN-α/β as a mediator responsible for potentiating the LPS response, we sought to examine this effect in a more clinically relevant model of septic shock. The cecal ligation and puncture (CLP) model of peritonitis, bacteremia, tissue injury, and septic shock is a well-studied model that closely mimics human disease, making it more relevant to the study of septic shock compared with a bolus injection of LPS. The types of cytokines induced by LPS and CLP are quite similar; however, the most significant difference is the time over which this response progresses, the tissue injury involved, and the polymicrobial nature of the infection evolving from an initiating nidus (18). The cytokine response to CLP is much slower to evolve compared with the LPS model. The physiological response to activation of the inflammatory cascade during CLP is the evolution from a hyperdynamic metabolic state for approximately the first 12 h, followed by a hypodynamic state thereafter. These events, both inflammatory and physiological, closely mimic septic shock induced by a focal initiating nidus of infection, such as pneumonia, urinary tract infection, or intestinal injury such as perforation with peritonitis, which represent many of the causes of human septic shock (18–22). Conversely, the response to LPS is rapid and fulminant. Different from the LPS model, in the CLP model, multiple anti-inflammatory agents such as interleukin (IL) 10, IL-1Ra, or antitumor necrosis factor alpha antibodies are not always protective and can heighten and hasten mortality, indicating that CLP serves as a more complex and multifaceted challenge as is common in human disease (23–29). CLP provides not only a robust inflammatory challenge and tissue injury but also a live bacterial infection that requires an intact response to eradicate. It is therefore important to explore IFN-α/β–mediated effects on a live infection model such as CLP.
Previously, we used live viral infection followed by LPS challenge to demonstrate the impact of antiviral responses on a secondary bacterial challenge. We identified IFN-α/β as the critical cytokine responsible for potentiation of the inflammatory response to LPS. In this report we further examined the impact of IFN-α/β, which is common to many antiviral responses, by pretreating with dsRNA (polyinosinic-polycytidylic [PIC] acid), a potent inducer of high circulating levels of IFN-α/β. dsRNA is structurally similar to viral dsRNA intermediates released during replication (dsRNA and ssRNA viruses) and transcription (DNA viruses), and as such, mimics events common to many viral infections (30, 32). PIC is a well-described TLR3 agonist capable of activating nuclear factor κB (NF-κB) and inducing IFN-α/β (32–34).
The focus of this work was to further examine the potentiating impact of IFN-α/β specifically on subsequent inflammatory responses, given that the use of live viral infection with its type-specific, complex, and evolving immune response, including the production of many other cytokines, would make focusing on the IFN-α/β pathway very difficult. This is especially true when superimposing a live bacterial infection after initiation of live and evolving viral infection using survival as an end point. For instance, mice genetically deficient in key antiviral mediators/pathways die more readily to viral infection, making assessment of survival from CLP much less clear (35). PIC provides a short-lived and focused effect (32, 36), thereby providing a more controlled pretreatment. Our goal here was to examine the impact of preactivation of IFN-α/β and NF-κB signaling pathways on the outcome of a well-studied bacterial infection/septic shock model. PIC pretreatment allowed focusing on the limited early response to dsRNA, leading to IFN-α/β induction and NF-κB activation without the continued evolution of individual virus specific responses.
We hypothesized that PIC would exaggerate the inflammatory response and reduce survival from bacterial challenge through IFN-α/β–and NF-κB–dependent pathways. We report here that pretreatment with PIC induced IFN-α, and when CLP was performed concurrent with this induction, the cytokine response and lethality from CLP was enhanced. An intact IFN-α/β signaling pathway was required for this effect. In addition, blocking NF-κB abrogated potentiation by PIC and IFN-α. These data indicate that both the IFN-α/β and NF-κB pathways are critical for PIC-induced potentiation of the inflammatory response and lethality induced by CLP.
METHODS
Mice
All protocols involving mice were approved by the Institutional Animal Care and Use Committee. The mice used for most experiments were C57BL6 mice obtained from Taconic, Germantown, NY. All mice were age- and sex-matched. For all in vivo experiments, only males were used. The IFN-α/β receptor (R) −/− mice (129SvEv background) were obtained from Dr Loren Fast (Rhode Island Hospital). 129SvEv control mice were also obtained from Taconic. C3H/HeJ mice were obtained from Jackson Laboratories (Bar Harbor, ME), and C3H/HeN [wild-type (WT) background control for the C3H/HeJ] mice were obtained from Charles River Laboratories, Wilmington, MA.
Cells
J774 mouse macrophage cell line (ATCC, Manassas, Va) was used to confirm the absence of endotoxin contamination in muramyl dipeptide (MDP) by incubation with polymyxin B at 50 μg/mL before the addition of MDP or LPS, both at 1 μg/mL, in Dulbecco modified Eagle medium with 10% fetal bovine serum.
Surgery
Standard CLP was used to create polymicrobial sepsis (18). In brief, animals were anesthetized with isoflurane and prepared for a sterile laparotomy, including shaving and Betadine scrubbing of the abdomen. Then, a 1-cm laparotomy was performed, followed by identification of the cecum and ligating it approximately 0.75 cm proximal from the distal tip. For C57BL6 mice, one 22-gauge puncture was created in the ligated cecum, and in the 129SvEv mice, two 22-gauge punctures were created. The difference between the two strains was because of increased sensitivity observed in C57BL6 mice in early experiments compared with 129SvEv mice. Sham-CLP surgery consisted of a 1-cm laparotomy with exposure of the cecum without ligation and puncture followed by closure of the peritoneal fascia and skin. Serum and peritoneal fluid (peritoneal lavage with 1 mL sterile phosphate-buffered saline [PBS]) was harvested either 4 or 24 h after CLP was performed. For experiments involving ex vivo stimulation of peritoneal cells after priming with PIC or IFN-α in vivo, peritoneal cells were obtained by lavage with sterile cold PBS (5 mL × 2), then cultured at 5 × 105 cells/well in Dulbecco modified Eagle medium with 10% fetal bovine serum with increasing concentrations of LPS Escherichia coli strain 0111:B4 (Sigma ) and incubated overnight with supernatants harvested for tumor necrosis factor alpha (TNF-α) determination. Modified CLP was used for peritoneal bacterial culture comparison. Standard cecal exposure and ligation was performed; however, a standardized inoculum of a stool slurry was infused into the peritoneum before fascial closure to better control peritoneal bacterial inoculum compared with cecal puncture.
Bacterial culture
Peritoneal fluid was obtained with a sterile 1-mL PBS lavage at 24 h postmodified CLP. The peritoneal sample was serially diluted on blood agar, and aerobic colony-forming units were quantified after incubation at 37°C for 24 h.
Reagents
Gamma-irradiated sterile PIC (Sigma) was diluted in PBS administered to each mouse in vivo at 200 μg s.c (32). Alternatively, mice were injected subcutaneously with 2 × 105 U of recombinant human IFN-α A/D (IFN-α) active on mouse cells (PBL Biomedical Laboratories, New Brunswick, NJ). PBS injections of the same volume were used as control injections. IFN-α was determined by standard sandwich enzyme-linked immunosorbent assay (ELISA) from Antigenix America (Huntington Station, NY; lower limit of detection, 12.5 pg/mL), and TNF-α was measured by ELISA with reagents obtained from Endogen (Woburn, Mass) and BD Pharmingen (San Diego, Calif; lower limit of detection, 10 pg/mL). IL-12p40/70, IL-10, and IL-6 ELISA reagents were also obtained from BD Pharmingen (lower limit of detection, 40 and 80 pg/mL). For increased sensitivity we used a multiplex bead assay (Upstate Biotechnology (Charlottesville, Va) and Luminex Corporation (Austin, Tex) to measure IL-12p70, TNF-α, and IFN-γ after PIC injection. Parthenolide was dosed at 50 μg s.c. 1 h before PIC and 16 h before CLP for each mouse.
Flow cytometry was performed using 1 × 106 peritoneal macrophages obtained from mice pretreated with PIC (200 μg s.c.) 16 h prior. The cells were blocked (FcBlock, Pharmigen, San Diego, Calif) then labeled with fluorochrome-conjugated monoclonal antibodies to IAb (strain-specific major histocompatibility antigen class II [MHCII] for C57BL6 mice) (Pharmigen) and either F480 (Serotec, Raleigh, NC) or CD204 (Pharmigen), which are both specific for antigens expressed on mouse macrophages.
Statistics
All cytokine measurements are reported as mean ± SEM. Statistical difference between treatment groups (i.e., PIC versus PBS) was determined using the Student t test. Survival data were assessed for significant differences using Fisher exact analysis. We considered P < 0.05 to be statistically significant.
RESULTS
Cytokine induction by PIC
To delineate the nature of the cytokine milieu 24 h after PIC injection, we measured several inflammatory cytokines commonly induced early during many viral infections. At 16 and 24 h after PIC injection, a robust IFN-α response was seen (Fig. 1A). PIC induced a modest and transient increase in serum TNF-α at 6 h, which resolved by 16 h. No change in the baseline serum levels of IFN-γ or IL-12p70 was seen (Fig. 1B).
Fig. 1. C57BL6 mice were injected with PIC or PBS (both 200 μg s.c.), and serum IFN-α levels were assayed 16 and 24 h later.
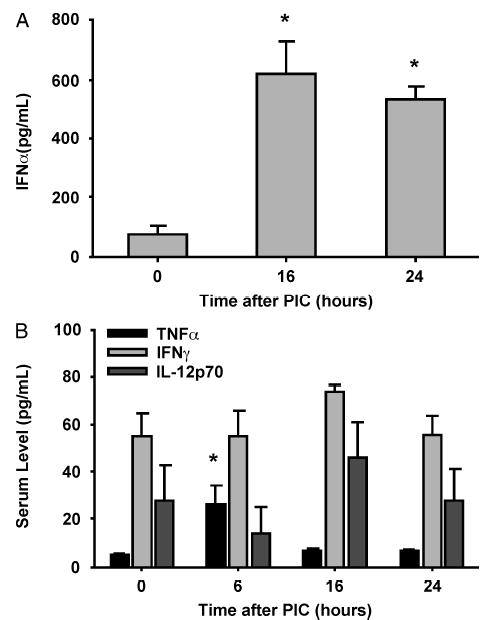
Serum was also collected at many time points and assayed for TNF-α, IFN-γ, and IL-12p70 (n = 3 mice/group). *P < 0.05 for IFN-α (A) or TNF-α (B) serum levels compared with baseline values (time 0).
Peritoneal leukocyte content after PIC and CLP
We sought to examine the peritoneal cellular content after PIC injection and after PIC + CLP to determine whether PIC alters the cellular content primarily or after CLP compared with PBS pretreatment. PIC did not alter the number of peritoneal macrophages present before CLP (> 95%) but significantly activated them as evidenced by grossly increased spreading and vacuolization (data not shown). The cellular content of the peritoneal exudate 24 h after CLP was also not significantly different between PIC- and PBS-pretreated mice with 22% to 24% macrophages and 75% neutrophils by Giemsa staining.
MHCII EXPRESSION ON PERITONEAL MACROPHAGES
Expression of MHCII on macrophages is a marker of activation (37). To confirm the activated state of these peritoneal macrophages they were harvested 16 h after treatment with PIC and assessed for MHCII (I-Ab) expression on F480+ peritoneal macrophages. Approximately 95% of the cells from the PBS- and PIC-pretreated mice were macrophages as defined by F480+ staining, roughly confirming the gross histological assessment above (data not shown). Treatment of WT mice with PIC resulted in higher numbers of F480+/I-Ab + macrophages by flow cytometry. In contrast, peritoneal cells from IFN-α/β R−/− mice did not show an increase in I-Ab+ cells, indicating that up-regulation in MHCII in the WT mice was dependent on PIC-induced v. The density per cell as indicated by mean fluorescence intensity did not change after PIC treatment in either group (Fig. 2, A and B).
Fig. 2. Peritoneal macrophages were obtained 24 h after injection with PIC (200 μg s.c.).
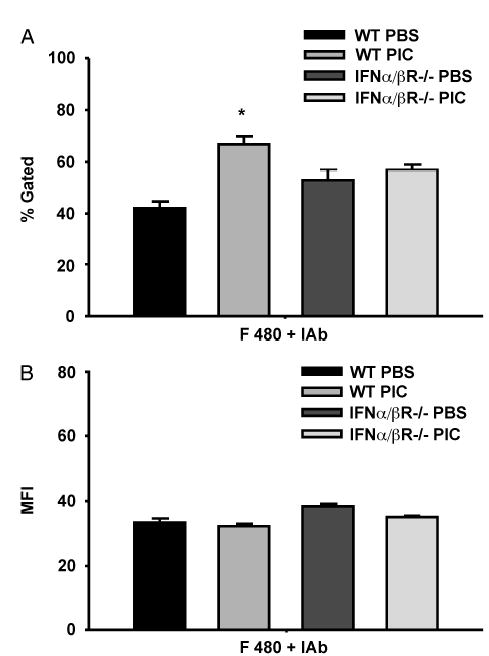
By flow cytometry, the number F480+ peritoneal macrophages also expressing MHCII (I-Ab) (percent gated), and the density of expression of MHCII (I-Ab) per cell (MFI) was determined (n = 3 mice/group). *P < 0.05 for WT PBS versus WT PIC groups.
Time course of PIC potentiation of the TNF- α response to LPS
To confirm that PIC would potentiate the TNF-α response to an inflammatory challenge and to assess the peak and duration of this potentiation, we treated mice with PIC 8, 16, 24, 48, 96, or 192 h before injection of LPS. We used serum TNF-α levels obtained 2 h after LPS injection as a marker of the impact of PIC on the response to LPS. PIC potentiated the TNF-α response to LPS with peak effect seen when LPS was administered 24 to 48 h after PIC injection (Fig. 3). Given the above data, we decided to administer PIC 16 h before CLP for all subsequent experiments. Although this time point is before peak potentiating activity as determined above, our goal was to induce sepsis from CLP, which requires several hours to evolve, simultaneous with the continuous presence of high levels of IFN-α, which occurred through the 16- to 24-h interval (Fig. 1A).
Fig. 3. C57BL6 mice were injected with PIC or PBS (both 200 μg s.c.), followed by 100 μg of LPS 8, 16, 24, 48, 96, and 192 h after PIC to determine the optimal interval between PIC and secondary inflammatory challenge.
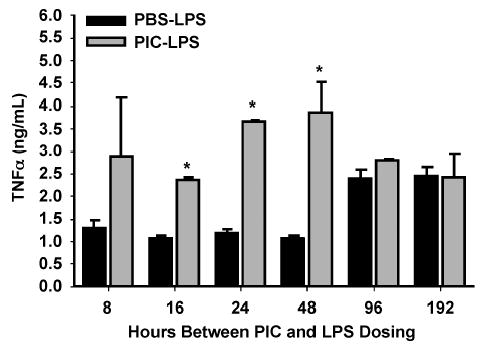
Serum was collected 2 h after LPS injection and assayed for TNF-α (n = 2 –3 mice/group). *P ≤ 0.05 for PBS-LPS versus PIC-LPS.
PIC enhances the inflammatory response to CLP and reduces survival
To explore the impact of PIC on the cytokine response to and survival from a live infection model of septic shock, we performed CLP 16 h after PIC treatment. Pretreatment with PIC before CLP resulted in potentiation of both serum and peritoneal proinflammatory and anti-inflammatory cytokine responses when harvested at 24 h postsurgery (Fig. 4A). In addition, PIC pretreatment diminished survival from CLP compared with controls (Fig. 4B).
Fig. 4. C57BL6 mice were injected with PIC or PBS (both 200 μg s.c.), followed by 22-gauge single-puncture CLP 16 h later.
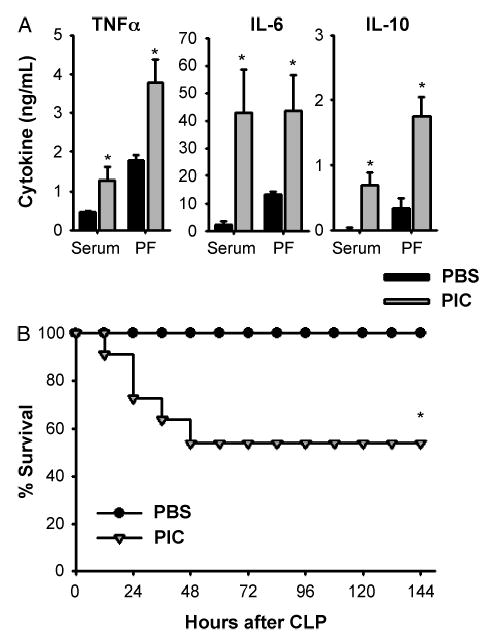
Twenty-four hours later serum and peritoneal fluid were collected for cytokine measurement (n = 3 – 4 mice/group). *P ≤ 0.05 for PIC versus PBS (A). Survival was followed for 7 days after CLP (n = 12 mice/group). *P ≤ 0.05 for PBS versus PIC pretreatment (B).
PIC-mediated potentiation of the response to CLP requires IFN- α/β
In a previous report, we demonstrated that IFN-α/β was essential to viral potentiation of the inflammatory response to LPS. Here, we sought to determine whether the PIC-mediated amplification of the inflammatory response was IFN-α/β–dependent in the CLP model of live infection. Because CLP consists of several stimuli, including tissue injury from laparotomy and cecal ligation as well as polymicrobial peritonitis and septic shock from cecal puncture, it was important to determine if IFN-α/β was required for potentiating the CLP response and to rule out a requirement for other cytokines induced by PIC such as IL-12, TNF-α, or IFN-γ (Fig. 1). PIC pretreatment of IFN-α/β R−/− mice followed by CLP failed to increase serum or peritoneal TNF-α compared with PIC pretreated WT mice (Fig. 5A). Survival was also improved in PIC-pretreated IFN-α/β R−/− mice compared with WT mice (Fig. 5B).
Fig. 5. WT and IFN-α/β R−/− mice were injected with PIC or PBS (both 200 μg s.c.), followed by 22-gauge two-puncture CLP or sham-CLP 16 h later.
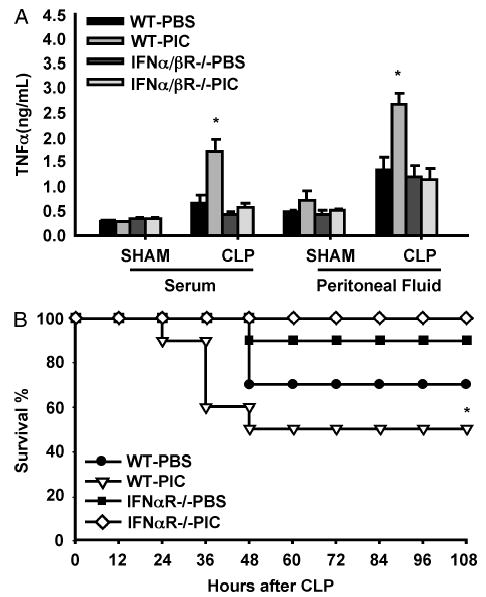
Serum and peritoneal fluid were collected 4 h after CLP for TNF-α determination (n = 3 – 4 mice/group). *P ≤ 0.05 for WT-PIC versus WT-PBS (A). Survival was followed for 7 days after CLP (n = 10 mice/group). *P ≤ 0.05 for PIC-WT versus PIC-IFN-α/β R−/− (B).
PIC potentiates the TNF- α response to TLR2 and TLR4 agonists
Having demonstrated that PIC can potentiate the response to LPS and CLP, we sought to determine whether this effect was specific to the TLR4 pathway or whether the response to other stimuli such as TLR2 agonists could also be potentiated. We used mice naturally LPS-resistant via TLR4 mutation (C3H/HeJ) to determine whether potentiation occurs by facilitation of TLR4 signaling only or by broader mechanisms. Although CLP elicits a much reduced TNF-α response in serum and peritoneal fluid in the C3H/HeJ mice compared with WT mice, PIC did potentiate this response in C3H/HeJ mice when compared with the C3H/HeJ PBS control group (Fig. 6A).
Fig. 6. C3H/HeJ mice were injected with PIC or PBS (both 200 μg s.c.), followed by 22-gauge single-puncture CLP 16 h later.
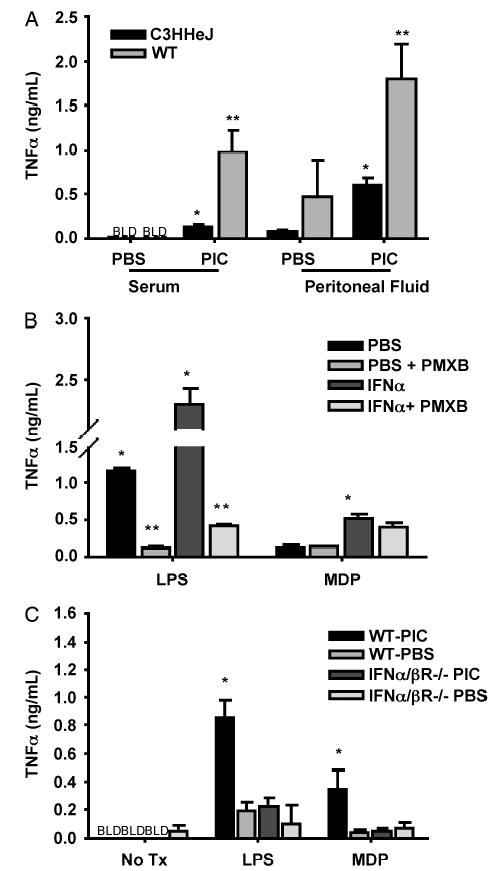
Serum and peritoneal fluid were collected 4 h later and assayed for TNF-α. WT mice treated in the same manner are displayed for comparison (n = 3 – 4 mice/group). *P ≤ 0.05 comparing PIC- versus PBS-pretreated C3H/HeJ mice; **P ≤ 0.05 comparing PIC- versus PBS-pretreated WT mice (A). J774 cells were incubated with or without IFN-α at 5000 U/mL for 16 h. Cells treated with ±IFN-α were then treated with either LPS (1 μg/mL) ± polymyxin B or MDP (1 μg/mL) ± polymyxin B. *P < 0.05 for IFN-α + LPS versus PBS + LPS and for IFN-α + MDP versus PBS + MDP. **P < 0.05 for PBS versus PBS polymyxin B and PIC versus PIC + polymyxin B (B). WT and IFN-α/β R−/− mice (129SvEv background) were injected with PIC or PBS (both 200 μg s.c.), followed by peritoneal cell collection 16 h later. Cells were cultured in the presence of 10 μg/mL LPS or 10 μg/mL MDP for 16 h with supernatants assayed for TNF-α (n = 3 – 4 mice/group). *P ≤ 0.05 comparing WT-PIC with IFN-α/β R−/− PIC (C).
The J774 macrophage cell line was incubated with 5000 U of IFN-α and exhibited an up-regulated TNF-α response to both LPS and MDP, which is a synthetic Gram-positive cell membrane component and TLR2 agonist. To verify that the response to MDP was not because of LPS contamination, we incubated cells with polymyxin B before the addition of MDP or LPS. The concentration of polymyxin B used eliminated most of the LPS effect but did not significantly affect signaling by MDP (Fig. 6B).
Peritoneal macrophages were harvested 16 h after PIC injection and exposed ex vivo to either LPS or MDP. PIC pretreatment also potentiated the TNF-α response to both TLR4 and TLR2 agonists in WT mice. Potentiation of the response to TLR2 and TLR4 agonists was blunted in IFN-α/β R−/− mice compared with WT. These data indicate that signaling through both TLR4 and TLR2 pathways in this model is altered by IFN-α/β (Fig. 6C).
PIC pretreatment effects on CLP can be interrupted by NF-κB inhibition
Another effect of PIC is activation of the NF-κB pathway (34). We sought to determine the contribution from NF-κB activation in PIC-mediated potentiation of the response to CLP. Parthenolide, a sesquiterpene lactone known to inhibit I kappa B kinase (IKK) α, thereby inhibiting NF-κB, was used to inhibit this pathway. Pretreatment with parthenolide 60 min before PIC abrogated PIC-mediated increased lethality from CLP (Fig. 7A.) The parthenolide dose used here was then optimized by selecting a dose that would inhibit PIC-mediated priming without significantly inhibiting cytokine/lethality from CLP alone.
Fig. 7. C57BL6 mice were injected with parthenolide (50 μg s.c.) 1 h before PIC followed by CLP 16 h later for survival assessment over 6 days (n = 10 – 12 mice/group).
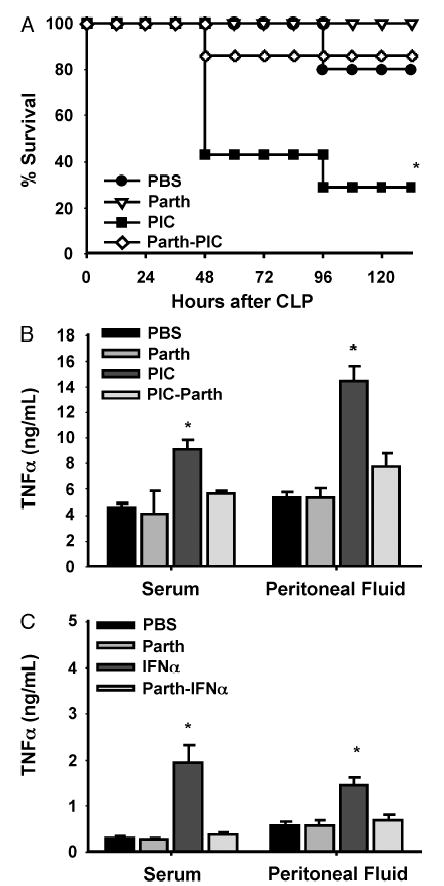
*P < 0.05 for PIC-parthenolide versus PIC (A). C57BL6 mice were injected with parthenolide (50 μg s.c.) 1 h before injection with either PIC (B) or IFN-α (C) followed by CLP 16 h later. Serum and peritoneal fluid were collected 24 h after CLP and assayed for TNF-α (n = 3 – 4 mice/group). *P < 0.05 for PIC versus parthenolide + PIC (B) or IFN-α versus parthenolide + IFN-α (C).
To determine whether parthenolide reduced IFN-α induced by PIC, possibly resulting in decreased lethality, we measured serum IFN-α levels 24 h after parthenolide + PIC injection. IFN-α levels were not reduced by pretreatment with parthenolide before PIC (657 ± 0 pg/mL in parthenolide + PIC vs. 522 ± 41 pg/mL in PIC alone). We also pretreated mice with parthenolide 1 h before treatment with recombinant IFN-α followed by CLP 16 h later. Figure 7, B and C, demonstrates that parthenolide blocked potentiation of TNF-α induced by CLP in both PIC and IFN-α pretreated mice. These data would suggest that IFN-α induced by PIC or injected directly could impact subsequent inflammatory responsiveness in apn NF-κB-dependent fashion.
Bacterial clearance
Peritoneal aerobic bacterial cultures were obtained 24 h after modified CLP. Although bacterial load was very high 24 h after modified CLP, PIC pretreatment resulted in 3-fold higher bacterial counts after 24 h (Fig. 8).
Fig. 8. C57BL6 mice injected with PIC (200 mg) or PBS were subjected to modified CLP 16 h later.
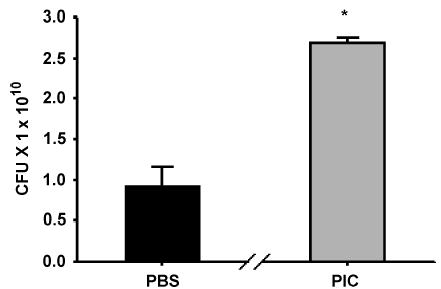
Aerobic bacterial colony-forming units (CFU) from peritoneal fluid were obtained 24 h post-CLP. *P < 0.05 for PIC versus PBS pretreatments.
DISCUSSION
Previously, we demonstrated that mice were sensitized to LPS when administered early during LCMV infection and that this effect was IFN-α/β–dependent (15). A major difference in this work is that the effects of PIC, a dsRNA synthetic viral mimetic, was used to activate many early antiviral pathways common to most viral infections, including IFN-α/β induction and NF-κB activation. In addition, rather than LPS, the secondary bacterial challenge used here was CLP, a live infection model that includes tissue injury to assess the impact of PIC on a complex, multifaceted, and slowly evolving challenge, which is more typical of human septic shock.
Here, we demonstrated that PIC induces a cytokine response that is similar to LCMV with the induction of primarily IFN-α/β within the first 24 h with very little TNF-α, IFN-γ, or IL-12p70 at that time. In fact, LCMV induced much higher IFN-α/β levels in serum compared with those induced by PIC in this report. In our previous work, we demonstrated the same potentiation with LCMV as we report here with PIC, indicating that the observed amount of IFN-α was sufficient to modulate inflammatory responsiveness to septic models (14, 32, 38). PIC induced a small amount of TNF-α but no significant IL-12p70 or IFN-γ. All 3 cytokines are important immunostimulants and antiviral cytokines. However, in this model, none seems to have a causative role in potentiation of the septic response after PIC treatment because their levels were minimal, and essentially all of the potentiation effects of PIC were abrogated in IFN-α/β R−/− mice. Our data further demonstrate that immune cross talk occurs between early events in the antiviral response and the response to bacterial infection.
PIC pretreatment did not change the number of macrophages and neutrophils in the peritoneum before or after CLP. In addition, when controlled numbers of peritoneal cells obtained from PIC-pretreated mice were stimulated with LPS ex vivo, potentiation was also seen, indicating that the quantity of cells responding to the bacterial stimulation was not a factor responsible for potentiation. These data also suggest that macrophage and neutrophil chemoattraction was not grossly altered in a significant way. Up-regulated MHCII expression is a marker of activation in antigen-presenting cells such as macrophages. We demonstrated up-regulated MHCII expression in peritoneal macrophages after PIC treatment, corroborating our microscopic observations of macrophage activation. These data suggest that PIC alters the phenotype of macrophages, potentially contributing to its potentiating effects.
PIC heightened the cytokine response and lethality from subsequent live bacterial challenge. PIC induces IFN-α/β, and these data demonstrate that intact IFN-α/β signaling is critical for augmentation of the inflammatory response to CLP. Unlike our previous work using LPS, these data show that augmentation of inflammatory responsiveness and lethality can occur when caused by a slowly evolving polymicrobial infection complicated by tissue injury—a common scenario in human septic shock.
We demonstrated that PIC-mediated potentiation occurs in LPS-resistant mice and that PIC potentiates the TNF-α response to both TLR2 and TLR4 agonists, indicating that this effect is not TLR4-specific. IFN-γ has been shown to induce TLR4 transcription, suggesting a possible mechanism for potentiation. It is not known whether IFN-α/β shares this property. Although we did not examine TLR4 gene activation, our data suggest that potentiation by PIC affects signaling through the TLR4 pathway and other pathways such as TLR2. TLR2 agonists can signal through NOD 2 to activate inflammatory responses. It is possible that this occurs in our model; however, identification of the elements in both the TLR4 and TLR2 pathways affected by IFN-α/β will require further investigation.
Pretreatment with PIC augments both the proinflammatory and anti-inflammatory cytokine responses to CLP. In this live infection model, augmenting the inflammatory response did not provide any survival benefit, suggesting that lethality may be because of the host inflammatory response to septic shock occurring in both the CLP and LPS models.
PIC signals through TLR3 with subsequent activation of TIR domain–containing adaptor protein–inducing interferon (TRIF), which activates IRF3, leading to the induction of IFN-β, followed by induction of multiple IFN-α subtypes. Signaling through TLR3 also activates NF-κB (34, 39, 40). The anti-inflammatory sesquiterpene lactones have been shown to inhibit NF-κB activation by a variety of mechanisms. The molecular target for the effects of parthenolide is inhibition of the IKK complex (subtypes α and β), which prevents IKK degradation and NF-κB activation. Parthenolide has been shown to inhibit the IKK complex without impacting TNF-α–mediated generation of reactive oxygen species or activation of JNK and p38, indicating specificity for the NF-κB pathway (41, 42). We demonstrated that pretreatment with parthenolide blocked PIC-mediated potentiation of the inflammatory response to CLP. NF-κB activation is important for IRF3 activation and subsequent IFN-β induction via noncanonical IKK-related kinases (and Tank-binding kinase 1) (43, 44). IFN-β is responsible for triggering and amplifying IFN-α release. Despite this, we did not find that parthenolide altered IFN-α serum levels induced by PIC. Because parthenolide abrogated the potentiating effects of IFN-α directly, our data suggest that IFN-α can impact regulation of NF-κB activation.
Viral components, including surface glycoproteins and intracellular viral proteins, are known to activate elements of the antiviral response. By limiting our pretreatment to PIC only, rather than an evolving viral infection, our data suggest that dsRNA, commonly released during viral replication, may also have direct immune modulating effects by induction of IFN-α/β.
These data expand the understanding of the important role of IFN-α/β in the inflammatory response to bacterial challenge. TLR4 activation leads to activation of the TRIF pathway, which leads to induction of an IFN-β, followed by subtypes of IFN-α (34, 39). In TRIF−/− mice, the TNF-α response and lethality from LPS were significantly diminished. Tyk2, a JAK family member, is necessary for LPS-induced IFN-α/β and lethality. Tyk2-deficient mice were also resistant to LPS significantly diminished IFN-α/β induction (45). These data demonstrate a central role for IFN-α/β in endotoxin-induced toxicity. The impact of IFN-α/β induced before challenge with TLR4 and TLR2 agonists or CLP has not been well defined. Our data suggest that elevated levels of IFN-α/β may have a role in determining the magnitude of the response to subsequent inflammatory challenge quite similar to bacterial superinfection during viral infection in human beings. In addition to modifying the inflammatory response, this model of early viral infection suggests that bacterial clearance may also be negatively affected by prior exposure to PIC. The impact on mortality of higher bacterial counts in PIC-pretreated mice is unclear at this point, and the mechanisms responsible for diminished bactericidal function after PIC pretreatment will require further investigation.
The morbidity and mortality from septic shock and consequent multiple organ failure have been related to the magnitude of the inflammatory response elicited early in the disease (46–48). Regulation of the magnitude of this response is poorly understood. There are many factors associated with suppressed inflammatory responsiveness leading to enhanced susceptibility and compromised pathogen clearance, including extremes of age, poor nutrition, severe systemic stress, trauma/surgery, genetic immunodeficiency syndromes, and a multitude of medications (49–54). However, very little is known about factors capable of potentiating the inflammatory response. The data presented here support the possibility that sources of IFN-α/β induction such as underlying antiviral responses can modulate the magnitude of the host inflammatory response to bacterial infection, possibly contributing to morbidity and mortality.
Footnotes
This work was supported by the National Institutes of Health (grant no. 1K08-GM71568-01 to L.A.D. and grant no. R01-46354-12 to A.A.).
References
- 1.Ray GC. Influenza, respiratory syncytial, adenovirus, and other respiratory viruses. In Ryan KJ (ed.): Sherris Medical Microbiology: An Introduction to Infectious Diseases East Norwalk, CT: Appleton and Lange Paramount Publishing Business and Professional Group, p 456, 1994.
- 2.MacDonald KL, Osterholm MT, Hedberg CW, Shrock CG, Peterson-Jensen JM, Leonard SA, Schlievert PM. Toxic shock syndrome: a newly recognized complication of influenza and influenza-like illness. JAMA. 1987;257:1053– 1058. doi: 10.1001/jama.257.8.1053. [DOI] [PubMed] [Google Scholar]
- 3.Degre M, Glasgow LA. Synergistic effect in viral-bacterial infection. I Combined infection of the respiratory tract in mice with parainfluenza virus and Haemophilus influenzae. J Infect Dis. 1968;118:449 – 462. doi: 10.1093/infdis/118.5.449. [DOI] [PubMed] [Google Scholar]
- 4.Large MK, Kittlesen DJ. Suppression of host immune response by the core protein of hepatitis C virus: possible implications for hepatitis C virus persistence. J Immunol. 1999;162:931 – 938. [PubMed] [Google Scholar]
- 5.Leung WC, Hashimoto K. Modification of susceptibility to K. pneumoniae during murine cytomegalovirus infection. Microbiol Immunol. 1986;30:761– 776. doi: 10.1111/j.1348-0421.1986.tb03003.x. [DOI] [PubMed] [Google Scholar]
- 6.Schneider-Schaulies S, Niewiesk S, Schneider-Schaulies J, Meulen VT. Measles virus induced immunosuppression: targets and effector mechanisms. Curr Mol Med. 2001;1:163 – 181. doi: 10.2174/1566524013363960. [DOI] [PubMed] [Google Scholar]
- 7.Yao ZQ, Ray S, Eisen-Vandervelde A, Waggoner S, Hahn YS. Hepatitis C virus: immunosuppression by complement regulatory pathway. Viral Immunol. 2001;14:277 – 295. doi: 10.1089/08828240152716547. [DOI] [PubMed] [Google Scholar]
- 8.Jones WT, Menna JH, Wennerstrom DE. Lethal synergism induced in mice by influenza type A and type Ia group B streptococci. Infect Immun. 1983;41:618– 621. doi: 10.1128/iai.41.2.618-623.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zerr DM, Alexander ER, Duchin JS, Koutsky LA, Rubens CE. A case-control study of necrotizing fasciitis during primary varicella. Pediatrics. 1999;103:783 – 790. doi: 10.1542/peds.103.4.783. [DOI] [PubMed] [Google Scholar]
- 10.Zhang WJ, Sarawar S, Nguyen P, Daly K, Rehg JE, Doherty PC, Woodland DL, Blackman MA. Lethal synergism between influenza infection and staphylococcal enterotoxin B in mice. J Immunol. 1996;157:5049– 5060. [PubMed] [Google Scholar]
- 11.Sarawar S, Blackman MA, Doherty PC. Superantigen shock in mice with inapparent viral infection. J Infect Dis. 1994;170:1189–1194. doi: 10.1093/infdis/170.5.1189. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen KB, Biron CA. Synergism for cytokine-mediated disease during concurrent endotoxin and viral challenges: roles for NK and T cell IFN-γ production. J Immunol. 1999;62:5238 – 5246. [PubMed] [Google Scholar]
- 13.Fejer G, Szalay K, Gyory I, Fejes M, Kusz E, Nedieanu S, Pali T, Schmidt T, Siklodi B, Lazar G, Jr, et al. Adenovirus infection dramatically augments lipopolysaccharide-induced TNF production and sensitizes to lethal shock. J Immunol. 2005;175:1498 – 1506. doi: 10.4049/jimmunol.175.3.1498. [DOI] [PubMed] [Google Scholar]
- 14.Nansen A, Christensen JP, Marker O, Thomsen AR. Sensitization to lipopolysaccharide in mice with asymptomatic viral infection: the role of T cell dependent production of interferon γ. J Infect Dis. 1997;176:151 – 157. doi: 10.1086/514017. [DOI] [PubMed] [Google Scholar]
- 15.Doughty L, Nguyen KB, Durbin JE, Biron CA. A role for IFNα/β in virus infection-induced sensitization to endotoxin. J Immunol. 2001;166:2658– 2664. doi: 10.4049/jimmunol.166.4.2658. [DOI] [PubMed] [Google Scholar]
- 16.Gendrel D, Raymond J, Coste J, Moulin F, Lorrot M, Guerin S, Ravilly S, Lefervre H, Royer C, Lacombe C, et al. Comparison of procalcitonin with C-reactive protein, interleukin 6 and interferon-alpha for differentiation of bacterial vs. viral infections. Pediatr Infect Dis J. 1999;18:875 – 881. doi: 10.1097/00006454-199910000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Moulin F, Raymond J, Iniguez J, Ravilly S, Lebon P, Gendrel D. Serum alpha-interferon in lower respiratory tract infections of children. Pediatr Infect Dis J. 1996;15:883 – 886. doi: 10.1097/00006454-199610000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Hubbard W, Choudhry M, Schwacha M, Kerby J, Rue L, Bland K, Chaudry I. Cecal ligation and puncture. Shock. 2005;24(suppl 1):52 – 57. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 19.Wichterman KA, Chaudry IH. Sepsis and septic shock—a review of laboratory models and a proposal. J Surg Res. 1980;29:189 – 201. doi: 10.1016/0022-4804(80)90037-2. [DOI] [PubMed] [Google Scholar]
- 20.Fink MP. Laboratory models of sepsis and septic shock. J Surg Res. 1990;49:186 – 196. doi: 10.1016/0022-4804(90)90260-9. [DOI] [PubMed] [Google Scholar]
- 21.Remick DG, Newcomb DE, Bolgos GE, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis. Shock. 2000;13:110 – 116. doi: 10.1097/00024382-200013020-00004. [DOI] [PubMed] [Google Scholar]
- 22.Ebong S, Call D, Nemzek J, Bolgos G, Remick DN. Immunopathologic alterations in murine models of sepsis of increasing severity. Infect Immun. 1999;67:6603 – 6610. doi: 10.1128/iai.67.12.6603-6610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869 – 871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- 24.Howard M, Muchamuel T, Andrade S, Menon S. Interleukin 10 protects mice from lethal endotoxemia. J Exp Med. 1993;177:1205 – 1208. doi: 10.1084/jem.177.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohlsson K, Bjork P, Bergenfeldt M, Hageman R, Thompson RC. Interleukin-1 receptor antagonist reduces mortality from endotoxin shock. Nature. 1990;348:550 – 552. doi: 10.1038/348550a0. [DOI] [PubMed] [Google Scholar]
- 26.Eskandari M, Bolgos G, Miller C, Nguyen D, DeForge LE, Remick DG. Anti–tumor necrosis factor antibody therapy fails to prevent lethality after cecal ligation and puncture or endotoxemia. J Immunol. 1992;148:2724– 2730. [PubMed] [Google Scholar]
- 27.Remick D, Garg SJ, Newcomb DE, Wollenberg G, Huie TK, Bolgos GL. Exogenous interleukin-10 fails to decrease the mortality or morbidity of sepsis. Crit Care Med. 1998;26:895 – 904. doi: 10.1097/00003246-199805000-00025. [DOI] [PubMed] [Google Scholar]
- 28.Garcia P, Mancilla J, Dinarello CA. The interleukin-1 receptor antagonist can either reduce or exaggerated lethality of Klebsiella pneumoniae sepsis in newborn rats. Infect Immun. 1993;61:926 – 932. doi: 10.1128/iai.61.3.926-932.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latifi SQ, O’Riordan MA, Levine AD. Interleukin-10 controls the onset of irreversible septic shock. Infect Immun. 2002;70:4441 – 4446. doi: 10.1128/IAI.70.8.4441-4446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majde JA. Viral double-stranded RNA, cytokines, and the flu. J Interferon Cytokine Res. 2000;20:259 – 272. doi: 10.1089/107999000312397. [DOI] [PubMed] [Google Scholar]
- 31.Malmgard L, Salazar-Mather T, Lewis C, Biron C. Promotion of alpha/beta interferon induction during in vivo viral infection through alpha/beta interferon receptor/STAT1 system-dependent and -independent pathways. J Virol. 2002;76:4520 – 4525. doi: 10.1128/JVI.76.9.4520-4525.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishikawa R, Biron CA. IFN induction and associated changes in splenic leukocyte distribution. J Immunol. 1993;150:3713 – 3727. [PubMed] [Google Scholar]
- 33.Maggi LB, Heitmeier MR, Scheuner D, Kaufman RJ, Buller RM, Corbett JA. Potential role of PKR in double-stranded RNA-induced macrophage activation. EMBO J. 2000;19:3630 – 3638. doi: 10.1093/emboj/19.14.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et al. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science. 2003;301:640 – 643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 35.Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443 – 450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- 36.Jacobs B, Langland J. When two strands are better than one: the mediators and modulators of the cellular response to double stranded RNA. Virology. 1996;219:339 – 349. doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- 37.Katze MG, HE Y, Gale M. Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2:675 – 687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 38.Biron CA. Interferons α and β as immune regulators—a new look. Immunity. 2001;14:661 – 664. doi: 10.1016/s1074-7613(01)00154-6. [DOI] [PubMed] [Google Scholar]
- 39.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743 – 748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NFκB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-β. Proc Natl Acad Sci U S A. 2004;101:3533 – 3538. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hehner SP, Heinrich M, Bork PM, Vogt M, Ratter F, Lehmann V, Schulze-Osthoff K, Droge W, Schmitz ML. Sesquiterpene lactones specifically inhibit activation of NFκB by preventing the degradation of IκBα and IκBβ. J Biol Chem. 1998;273:1288 – 1297. doi: 10.1074/jbc.273.3.1288. [DOI] [PubMed] [Google Scholar]
- 42.Hehner SP, Hofmann TG, Droge W, Schmitz ML. The anti-inflammatory sesquiterpene lactone parthenolide inhibits NFκB by targeting the IκB kinase complex. J Immunol. 1999;163:5617 – 5623. [PubMed] [Google Scholar]
- 43.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKα and TBK I are essential components of the IRF3 signaling pathway. Nature. 2003;4:491 – 496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 44.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148 – 1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 45.Karaghiosoff M, Steinborn R, Kovarik P, Kriegshauser G, Baccarini M, Donabauer B, Reichart U, Kolbe T, Bogdan C, Leanderson T, et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat Immunol. 2003;4:471 – 477. doi: 10.1038/ni910. [DOI] [PubMed] [Google Scholar]
- 46.Sunden-Cullberg J, Norrby-Teglund A, Rouhiainen A, Rauvala H, Herman G, Tracey K, Lee M, Andersson J, Tokics L, Treutiger C. Persistent elevation of high mobility group box-1 protein (HMGB-1) in patients with severe sepsis and septic shock. Crit Care Med. 2005;33:564 – 573. doi: 10.1097/01.ccm.0000155991.88802.4d. [DOI] [PubMed] [Google Scholar]
- 47.Panacek E, Marshall JC, Albertson TE, Johnson DH, Johnson S, MacArthur RD, Miller M, Barchuk WT, Fischkoff S, Kaul M, et al. Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody (Fab)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit Care Med. 2004;32:2173– 2182. doi: 10.1097/01.ccm.0000145229.59014.6c. [DOI] [PubMed] [Google Scholar]
- 48.Casey L, Balk R, Bone R. Plasma cytokine and endotoxin levels correlate with survival in patients with the sepsis syndrome. Ann Intern Med. 1993;119:771– 778. doi: 10.7326/0003-4819-119-8-199310150-00001. [DOI] [PubMed] [Google Scholar]
- 49.Agarwal SK, Marshall GD. Beta-adrenergic modulation of human type-1/type-2 cytokine balance. J Allergy Clin Immunol. 2000;105:91 – 98. doi: 10.1016/s0091-6749(00)90183-0. [DOI] [PubMed] [Google Scholar]
- 50.Bhaskaram P. Micronutrient, malnutrition, infection and immunity. Nutr Rev. 2002;60:S40 – S45. doi: 10.1301/00296640260130722. [DOI] [PubMed] [Google Scholar]
- 51.Chrousos G. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351 – 1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- 52.Lord G, Matarese G, Howard J, Baker R, Bloom S, Lechler R. Leptin modulates the T cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897 – 901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 53.Nelson C, Lysle D. Severity, time, and beta-adrenergic receptor involvement in surgery-induced immune alterations. J Surg Res. 1998;80:115 – 122. doi: 10.1006/jsre.1998.5429. [DOI] [PubMed] [Google Scholar]
- 54.Plata-Salaman C. Brain injury and immunosuppression. Nat Med. 1998;4:768 – 769. doi: 10.1038/nm0798-768. [DOI] [PubMed] [Google Scholar]