

Abstract
Clinically and experimentally, primary tumor formation and metastasis are distinct processes — locally growing tumors can progress without the development of metastases. This observation prompted the hypothesis that the molecular processes regulating tumorigenicity and metastasis are distinguishable and could be targeted therapeutically. During the process of transformation and subsequent progression to a malignant phenotype, both genetic and epigenetic alterations alter a cell’s ability to perceive and respond to signals that regulate normal tissue homeostasis. A minority of tumorigenic cells accrue the full complement of alterations that enables them to disseminate from the primary tumor, survive insults from the immune system and biophysical forces, and respond to growth-promoting and/or inhibitory signals from the distant tissues and thrive there. Identification of genes and proteins that specifically inhibit the ability of cells to form metastases (e.g., metastasis suppressors) is providing new insights into the molecular mechanisms that regulate this complex process. This review will highlight: (a) the functional identification of metastasis suppressors, (b) the signaling cascades and cellular phenotypes which are controlled or modulated by metastasis suppressors, and (c) op portunities for translation and clinical trials that are based on mechanistic studies regarding metastasis suppressors.
Functional Identification of Metastasis Suppressors
The identification of nm23, the first metastasis suppressor gene, provided functional evidence for the existence of genes that specifically regulate metastasis (1, 2). Subsequent to this initial discovery, researchers used in vivo studies to identify additional metastasis suppressors. These pioneering studies used an unbiased approach to identify such candidates by demonstrating that ectopic expression of the putative suppressor gene inhibited the development of spontaneous macroscopic metastases without significantly affecting primary tumor growth (3-5). Recently, this definition has been extended to include genes which specifically inhibit metastatic colonization (i.e., experimental metastasis formation using i.v. injection). The use of in vivo assays is required because in vitro assays are often of inadequate complexity to sufficiently model the entire process of metastasis. Furthermore, there are currently no in vitro models that allow the study of preferential growth within different target tissues. Table 1 lists the proteins which have bona fide metastasis suppressor activity in vivo (i.e., suppression of metastasis following ectopic expression into metastatic cell lines). It is interesting to note that metastasis suppressor activity for many of these genes would not have been predicted a priori based on their known cellular function(s). Furthermore, the unbiased, functional strategy identified novel genes for which no cellular function was known at the time of discovery.
Table 1.
Summary of metastasis suppressor proteins
| Protein | Function | Reference |
|---|---|---|
| Cytoskeletal signaling | ||
| Cadherin-11 | Type II (mesenchymal) cadherin | (49, 51) |
| Caspase-8 | Proapoptotic enzyme; potentiates integrin-mediated death | (36-38) |
| CD44 | Transmembrane glycoprotein which binds hyaluronic acid | (43-47) |
| Claudin-1, claudin-4 | Structural components of tight junctions | (40, 41) |
| CRSP3 | Transcriptional coactivator/corepressor | (11) |
| Connective tissue growth factor | Integrin binding | (33) |
| Gelsolin | Actin-regulatory protein which controls the length of actin polymers | (24) |
| E-cadherin, N-cadherin | Integral membrane protein involved in Ca2+-dependent cell adhesion | (49, 50, 52, 53) |
| KAI1 | Integrin interaction, EGFR desensitization | (34, 35) |
| KISS1 | G-coupled protein receptor ligand | (19) |
| RECK | GPI-anchored glycoprotein; negatively regulates MMPs | (42) |
| RhoGDI2 | Regulates Rho and Rac function | (86) |
| Src-suppressed C kinase substrate | Scaffold proteinmodulates Rho signaling | (10) |
| Stress-activated signaling | ||
| JNKK1/MKK4 | MAP2K in stress-activated protein kinase cascade; activates p38 and/orJNK | (8,12, 55, 56) |
| MKK6 | MAP2K in stress-activated protein kinase cascade; activates p38 | (12) |
| MKK7 | MAP2K in stress-activated protein kinase cascade; activatesJNK | (55) |
| ERK signaling | ||
| nm23-Hi | Histidine kinase; phosphorylates KSR, can reduce ERK1/2 activation | (2, 75) |
| RKIP | Inhibits RAF-mediated MAP/ERK kinase phosphorylation | (63) |
| PI3K/AKTsignaling | ||
| BRMS1 | Chromatin remodeling, cell-cell signaling, PI3K signaling | (67, 68) |
| Drg-1 | Function unknown | (69) |
Metastasis suppressors can impart their suppressive activity at one or more of the steps in the metastatic cascade (6). For example, in vivo studies showed that metastatic cancer cells which express ectopic KISS1, JNKK1/MKK4, MKK6, MKK7, TXNIP, nm23-H1, or SSeCKS proteins could successfully disseminate and lodge at secondary sites, but are suppressed in their ability to colonize (i.e., form overt metastases) target tissues (7-12). After lodging at secondary sites, disseminated cells may die, persist as nondividing cells, or initiate growth (13). Such pivotal cellular decisions depend on both the expression of a specific gene profile as well as the activation status of key signaling pathways and the cumulative inputs of timing, amplitude, and duration of signaling responses. In short, cells expressing metastasis suppressors grow at primary sites, but fail to proliferate at secondary or metastatic sites, suggesting differential responses to site-specific external signals. Although the observation that a gene of interest functions as a metastasis suppressor is an excellent starting point, research is now focused on the biochemical and molecular mechanisms by which metastasis suppressor proteins execute their in vivo functions.
Biochemical and Cellular Functions of Metastasis Suppressors
Metastasis suppressors vary widely in their cellular locations and biochemical functions. Such proteins could display either extracellular (e.g., KISS1) or intracellular localization patterns. Within the cell, they are located in various cellular compartments, from the plasma membrane (e.g., cadherin, KAI1, CD44), cytoskeleton (e.g., RhoGDI2, gelsolin), cytosol (e.g., JNKK1/MKK4, nm23-H1, RKIP), mitochondria (e.g., caspase 8), and nucleus (e.g., BRMS1, CRSP3, TXNIP) (14-21).
Cells respond to external stimuli by using a limited number of signaling pathways. Signaling specificity is achieved, at least in part, by combinatorial spatiotemporal activation of signaling proteins. The summation of these signaling events, enabled by a cell-specific gene expression profile, is a tailored, situation-appropriate response. During the process of transformation and progression to a malignant phenotype, both genetic and epigenetic alterations influence a cell’s ability to perceive and respond to signals which regulate normal tissue homeostasis. The accumulation of such alterations during progressive rounds of cell division could endow a minority of tumorigenic cells with the ability to disseminate from the primary tumor. It is likely that as a result of these changes, metastatic cells are no longer bound by tissue-of- origin-derived signaling specificity and acquire the ability to modulate their responses to the changing environments encountered throughout the metastatic cascade. Current data supports a model in which ectopic expression of metastasis suppressor proteins may restore, at least in part, the endogenous signaling repertoire of earlier, more benign cellular generations, thereby blocking metastasis formation. In this light, metastasis formation can be viewed as the result of a cell’s ability to respond to multiple growth milieus as opposed to being restricted to growth in the microenvironment of the tissue of origin.
Defining pathways regulating metastatic growth requires the integration and interpretation of data obtained over experimental settings ranging from the molecular interactions of specific proteins, to communication between signaling networks within single cells, and ultimately cellular interactions among populations of cells that yield a particular disease state. This line of inquiry is a particular challenge in studies of metastasis regulation because of the complexity and diversity of downstream events that take place in metastasis formation. The majority of metastasis suppressors identified participate in highly conserved eukaryotic signal transduction pathways. Based on their known biochemical functions, we have divided the metastasis suppressors into four general signaling categories: cytoskeletal signaling, mitogenic pathways, stress-activated pathways, and survival path-ways (Fig. 1). Although it may seem straightforward to place a metastatic suppressor into a linear signaling pathway, it is important to be mindful that these pathways are, in fact, dynamic networks that exist in series and parallel leading to crosstalk and interplay; phenomena known as emergent properties.
Fig. 1.
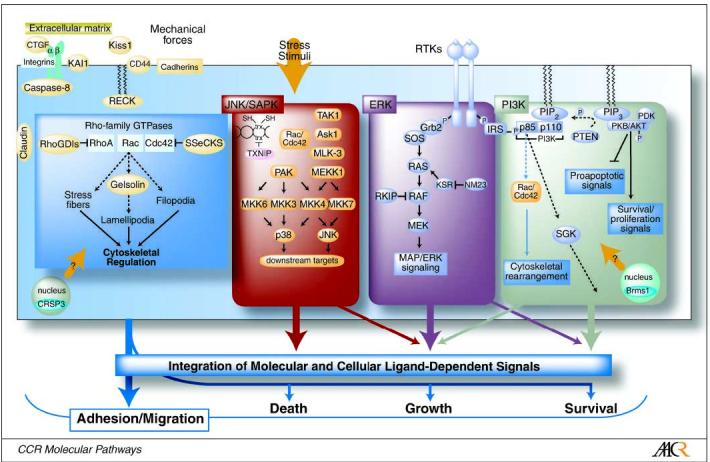
Metastasis suppressor proteins function in conserved signaling pathways. Determination of metastasis suppressor function requires integration of information; from gene identification, to protein function within signaling pathways, to functional consequences within populations of cells.
Cytoskeletal and Extracellular Matrix-Related Signaling
The cytoskeleton is a dynamic structure that enables motility, deformability, and flexibility, while maintaining cellular architecture. For most nonhematopoietic cells, motility is an ancillary function; however, as a nonhematopoietic cell transitions from the benign to the invasive to the metastatic state, motility becomes an increasingly paramount ability. Without a dynamic actin cytoskeleton, a tumor cell would be incapable of intravasation or extravasation, and the cellular deformability is necessary to complete the metastatic cascade. Not surprisingly, several proteins involved in cellular motility have been implicated as metastasis suppressors.
The Rho family of GTP-binding proteins, which include Rho, Rac, and Cdc42, are central players in the regulation of actin dynamics. The Rho-GTPase family consists of small 20 to 30 kDa monomeric GTP-binding proteins that bind GDP/GTP and hydrolyze GTP, leading to the activation of downstream effector molecules (22). In this signaling scheme, metastasis suppressors have been identified as upstream inhibitors of the Rho family (23), or downstream effectors (24). RhoGDIs inhibit Rho family members by impeding the dissociation of GDP from Rho proteins, thereby locking the Rho protein in its inactive state, preventing Rho proteins from interacting with effector targets, and/or sequestering Rho-GTPases in the cytosol (25). RHOGDI2 was shown to suppress experimental lung metastasis but not affect in vitro growth or in vivo tumorigenicity.
The Src-suppressed C kinase substrate is a protein kinase C substrate with protein scaffolding properties that have been shown to be a negative regulator of Rho family members (26). Reexpression of Src-suppressed C kinase substrate suppressed secondary lung metastases in nude mice and increased cell-cell adhesion, yet had little effect on primary tumor growth (10). Furthermore, Src-suppressed C kinase substrate reexpression at physiologic levels suppresses podosome formation and correlates with the induction of normal actin cytoskeletal structures and cell morphology, but not with the inhibition of Src kinase activity in cells (26).
The finding that RhoGDI displayed the ability to suppress metastasis suggested that the downstream molecules it inhibits may have metastasis-promoting activities. This hypothesis seems to hold true in that mice deficient in RhoC, a target of RhoGDI, displayed a marked decrease in metastasis potential (27). Furthermore, specific inhibition of a downstream effector of Rho, ROCK, decreased tumor cell invasiveness in vitro and reduced the dissemination of tumor cells implanted in the peritoneal cavity in vivo (28). RhoGDI2 has also been shown to regulate secreted growth factors such as endothelin 1 (ET-1), which contribute to metastasis and will be discussed below (29). This prometastasic activation of effectors downstream of the Rho family is not universal. Gelsolin is a downstream effector of Rac that regulates the length of actin via its filament severing and capping activities. Overexpression of gelsolin has been shown to inhibit metastasis in vivo (24). Counterintuitively, gelsolin is an indispensable protein of podosomes, which are thought to play an active role in tissue invasion and matrix remodeling through their regulation of matrix metalloprotease (MMP) activity (30, 31).
Cell interactions with the extracellular matrix and with neighboring cells trigger numerous responses that have essential roles in the regulation of behavior and fate. Integrin-mediated cell adhesions provide bidirectional links between the microenvironment and the cytoskeleton. This crosstalk between a cell and its local environment occurs whether the cell is benign, malignant, or metastatic. In this light, the cytoskeleton serves as a conduit for the integration and processing of intracellular and extracellular information; however, changes in the cytoskeletal program during pathologic progression may alter how this information is integrated and processed. Two metastasis suppressors have been shown to have interactions with integrins. Connective tissue growth factor is a 38-kDa cysteine-rich heparin-binding protein which is a secreted growth factor that can bind to integrins on the cell surface (32). Ectopic expression of connective tissue growth factor inhibited metastatic colonization by lung cancer cells (33). In contrast, the KAI1 metastasis suppressor protein is a member of the tetraspanin family of proteins and has been shown to interact with a myriad of cell surface proteins including other tetraspans (i.e., CD9, CD81), integrins β1 and β2, MHCII growth factor receptors, and intracellular signaling proteins such as protein kinase C (34). It has been hypothesized that KAI1 modulates integrin and growth receptor signaling by accelerating the rate of internalization via a protein kinase C-dependent pathway, thereby modulating cell adhesion and cell migration (34). Furthermore, KAI1 has been shown to decrease the integrin- and ligand-induced activation of the receptor tyrosine kinase c-Met, and independently decreases integrin-induced Src activation. Both c-Met and Src are thought to be required for invasion (35). A final observation with respect to the involvement of integrins in metastasis suppressor activity concerns caspase-8 function. Loss of caspase-8 in neuroblastoma cells leads to increased survival and an increased incidence of metastasis. The induction of caspase-8-mediated apoptosis seems to be due to unligated integrins because decreased expression of unligated integrins enhanced cell survival (36-38).
Claudin-4 is a component of tight junctions which form the most apical component of intercellular junctional complexes where they establish cell polarity and cellular functions such as permeability (39, 40). These intercellular junctions not only carry out adhesive functions but also contain crucial components of signaling pathways that regulate epithelial proliferation and differentiation. Complementary in vitro and in vivo studies identified claudin-4 as an inhibitor of invasion and metastasis in pancreatic cancer cells and a target of transforming growth factor-h and extracellular signal-regulated kinase (ERK) signaling (40, 41).
RECK, a membrane glycoprotein that encodes a GPI-anchored glycoprotein harboring three protease inhibitor-like domains, has been shown to negatively regulate MMP-2, MMP-9, and MT1-MMPs in vitro, suggesting that it is an important regulator of extracellular matrix remodeling. Furthermore, RECK-null mouse embryos displayed markedly elevated MMP activity. Interestingly, RECK is down-regulated by Ras signaling, a commonly altered pathway in many malignant cells (42).
The role of some putative metastasis suppressors is more complex. There are several examples of a protein functioning as a metastasis suppressor in one model and a potential tumor suppressor in another. For example, CD44 is a transmembrane glycoprotein that mediates responses of the cell to their cellular microenvironment. Although it has been reported in the literature that overexpression of CD44 has been shown to suppress metastasis in the AT3.1 prostate cancer system (43, 44), other reports have indicated that CD44 may augment metastatic activity (45, 46). Notably, a splice variant of CD44, variant v7-v10, facilitated the invasion and up-regulation of CD44 and has been suggested to be a poor prognostic marker (47).
CD44 is not the only protein to have an enigmatic role in metastasis. Cadherins are a superfamily of transmembrane glycoproteins that mediate cell-cell adhesion in various tissues in a calcium ion-dependent manner (48). Cadherin function is controlled by its cytoplasmic tail. Both N-cadherin and cadherin-11 seem to inhibit cell migration and the in vivo metastatic potential of LM8 osteosarcoma cells (49). However, other studies have suggested that cadherins may promote invasion (50-53). It is important to note that cancer cells of differing tissue origins were used in several of the studies. The tissue specificity of metastasis suppressor function is consistent with the tissue-specific response to, and integration of, information through signal transduction pathways.
KISS1 is a secreted protein and is hypothesized to undergo processing by prohormone convertases (19). The resulting processed products are known as kisspeptins. Whether or not KISS1 must be secreted in order to exert its metastatic suppressor activity has yet to be determined. Studies aimed at the identification of upstream regulators of KISS1 identified a metastasis suppressor activity for CRSP3, a part of the vitamin D receptor coactivator complexes. Transfection of cells with CRSP3 suppressed metastasis and was correlated with an up-regulation of KISS1 expression, providing a potential link between these vitamin D receptors and metastasis regulation (11).
Stress-Activated Pathways
The stress-activated protein kinase/c-Jun-NH2-kinase (JNK) and p38 pathways operate in parallel to the mitogen-activated protein kinase (MAPK)/ERK pathway. In contrast to the association of ERK with proliferation, JNK and p38 have been classically associated with cell cycle arrest and apoptosis in response to environmental stresses and cytokines, pH changes, UV irradiation, hypoxia, and growth factor deprivation (54). Several MAP2Ks specifically activate either p38- or JNK-mediated signaling. The JNKK1/MKK4 protein is a dual-specificity kinase that has been shown to phosphorylate and activate the JNK and p38 MAPKs in response to a variety of extracellular stimuli. JNKK1/MKK4 can function as a metastasis suppressor in both prostate and ovarian cancer models. Complementary in vitro (biochemical) and in vivo (metastasis) assays showed that the kinase activity of JNKK1/MKK4 is required for the suppression of overt metastases and is sufficient to prolong animal survival. Subsequent studies identified metastasis suppressor activities for MKK7 and MKK6 in prostate and ovarian cancers, respectively. Of interest is the finding that in ovarian cancer models, JNKK1/MKK4 signals through the p38 arm of the pathway to suppress metastasis (12), whereas in prostate cancer models, JNKK1/MKK4 signals through the JNK arm of the pathway to suppress metastasis (55). in vivo studies show that both JNKK1/MKK4 and MKK7 suppress the formation of overt metastases by inhibiting the ability of disseminated cells to colonize the lung (8, 12, 55, 56).
The identification of a metastasis suppressor function for thioredoxin-interacting protein (TXNIP, also called vitamin D up-regulating protein-1, or thioredoxin-binding protein 2) represents another mechanistic link between the cells’ response to cellular stress and modulation of metastatic ability. TXNIP is an endogenous inhibitor of cytoplasmic thioredoxin, a component of a ubiquitous thiol-reducing system that has been implicated in cancer progression (57). The levels of reactive oxygen species in cells are critically regulated by endogenous antioxidants (such as the thioredoxin system) in order to prevent oxidative damage to organelles and macromolecules, including DNA (58). The primary source of cellular reactive oxygen species is the mitochondrial electron transport chain. If electron transport is blocked, through disruption of mitochondrial membrane integrity, chemical inhibition, or in response to hypoxia, electrons escape to react with molecular oxygen or water; thereby generating highly reactive superoxide anions or hydroxyl radicals (58), setting forth a chain of events that result in necrotic cell death, inflammation, and potentially promotion of metastasis. At this time, how, and if, ectopic expression of TXNIP modulates these events to suppress metastasis formation is currently unknown.
Mitogenic Pathways
The MAPK/ERK pathway is activated by mitogenic ligands such as platelet-derived growth factor, epidermal growth factor, or insulin, which activate receptor tyrosine and serine-threonine kinases. The binding of mitogenic factors to cell surface receptors initiates a series of phosphorylation/activation reactions that culminates with the activation of various transcription factors and DNA-binding proteins (59, 60). Signal amplification and regulation may occur at each step in this pathway. Specificity is conferred through both the affinity of a kinase for a given substrate, as well as protein expression levels. Additional levels of regulation are conferred by intracellular localization and interaction with scaffolding or adaptor proteins. These events may be important targets for regulating metastatic progression. In the case of metastasis suppression, the nm23-H1 protein interacts with the MAPK module by binding to and phosphorylating the kinase suppressor of Ras (KSR) protein, a scaffold protein that has a role in the regulation of Ras activity and activation of the MAPK/ERK pathway (61). KSR has been shown to positively regulate Ras-mediated MAPK signaling, suggesting that nm23′s metastasis suppression activity may be mediated by the inhibition of KSR-enabled MAPK signaling (62). Raf kinase inhibitor protein (RKIP) was identified as a metastasis suppressor gene in human prostate cancers (63). RKIP functions as a negative upstream regulator of MAPK/ERK signaling (20, 64). Raf and MAP/ERK kinase interact with RKIP at overlapping sites, and binding of either molecule inhibits binding of the other. Both binding domains must be destroyed in order to alleviate RKIP-mediated MAPK inhibition (64). Thus, RKIP may suppress metastasis by inhibiting Raf-mediated phosphorylation of MAP/ERK kinase.
Survival Pathways
Two metastasis suppressors, Brms1 and Drg1, are associated with the phosphoinositide 3-kinase (PI3K)/AKT signaling module. The PI3K signaling pathway plays a key role in many aspects of cell survival and growth. Class 1 PI3Ks are heterodimers composed of an inhibitory adaptor/regulatory (p85) and a catalytic (p110) subunit. p85 binds and integrates signals from various cellular proteins such as RTKs, and provides an integration point for the activation of p110 and downstream molecules. Activated, PI3Ks phosphorylate phosphatidylinositol-4,5,bisphosphate (PIP2) to produce phosphatidylinositol-3,4,5,bisphosphate P3 (PIP3), a second messenger that binds a subset of downstream targets, predominantly the serine-threonine kinase, AKT and PDK1. Fully activated AKT also regulates a wide range of target proteins that control cell proliferation and survival (65). The levels of PIP3 are strictly regulated and several lipid phosphatases rapidly remove it. Of particular interest is the 3′-phosphatase PTEN, which converts PIP3 to PIP2, inhibiting PI3K signaling (66).
The Brms1 metastasis suppressor protein (67, 68) has been functionally associated with the modulation of phospholipid metabolism. Specifically, Brms1-expressing cells showed significant decreases in endogenous levels of PIP2 (i.e., <10% of controls). This finding is consistent with a model in which Brms1-associated changes in PI3K signaling impair the ability of disseminated cells to survive in the circulation and progressively grow at secondary sites. In contrast, expression of the metastasis suppressor Drg-1 is apparently modulated by PTEN. Of clinical relevance is the finding that PTEN expression correlates significantly with Drg-1 in both prostate and breast cancer cases and that the two markers in combination emerged as a significantly better predictor of prostate and breast cancer patient survival than either marker alone (69).
The Feasibility of Suppressing Metastasis as a Therapeutic Objective
The evidence acquired from the cell molecular and genetic studies outlined above indicates that the metastatic process requires many steps to complete. The impairment of only one step in this chain of in vivo events thwarts the overall process and produces a clinically beneficial result. This principle enabled the identification of metastasis suppressor proteins. Despite the simplicity of the concept, however, the application is not as straightforward because deciding which signaling nodes in the metastatic process are “therapeutically viable” is not immediately obvious.
Clinical observations have yielded insights into this problem by defining metastatic colonization as the step in the metastatic cascade that is tractable from a therapeutic point of view. A clinical vignette will best illustrate this point. Up to 60% of invasive bladder cancers are associated with metastatic recurrence and death (70), despite aggressive treatment (i.e., radical cystectomy, chemotherapy). These results clearly indicate that many patients with invasive disease harbor microscopic disseminated cancer cells at diagnosis. Therefore, preventing the outgrowth of disseminated cells into the lethal clinically detectable metastatic lesions (i.e., the process of “metastatic colonization”) would constitute a major step forward for cancer therapy. This approach offers considerably more hope than efforts aimed at preventing cancer cells from escaping the primary tumor for the simple fact that this latter process has already taken place when the patient is first diagnosed with advanced cancer. In fortunate cases where this process has not yet occurred, conventional local therapies such as surgery and radiotherapy can effectively cure the patient, eliminating the need for additional therapy aimed at the metastasis.
In summary, herein lies an exceptional opportunity for metastasis prevention: preventing the growth of disseminated tumor cells into an overt clinical metastasis or directly eliminating the microscopic metastases. Even if the latter approach were not possible, the former could, in essence, hold microscopic disease in check, providing increased patient survival. One of the continuing challenges associated with both of these approaches is the identification of patients at risk for disease recurrence, i.e., those that harbor microscopic disease, so they may receive adjuvant therapies aimed at metastatic disease. Predictive tools that can be applied to this problem include both high-sensitivity tumor imaging (71), as well as molecular evaluation of the primary tumor to help predict the development of metastasis (72).
Clinical-Translational Advances
From a theoretical standpoint, it has been difficult to envision “growth” of the disseminated tumor cells at a secondary site (such as the bone, liver, lung, and brain) as being distinct from that at the primary site, particularly with reference to the tumor cell. However, tumor cells encounter distinct microenvironments in the primary and secondary sites, and these interactions can facilitate or repress growth. Examples include locally produced chemokines and cytokines, as well as cell-cell interactions in specialized environments such as the bone, liver, lung, and brain (73, 74). By applying these concepts together with the basic biology of metastasis suppressor protein function, one can begin to formulate therapeutically viable approaches.
In essence, there are two ways to functionally restore metastasis suppressor proteins. The selection of these approaches hinges on knowledge of the fundamental defect leading to loss of metastasis suppressor protein expression. For example, if the gene is mutated, then it follows that replacement of a normal gene would be the appropriate intervention. In contrast, if loss was secondary to suppressed transcriptional expression, for example, then approaches aimed at reexpressing the endogenous gene seem warranted. Below, we will provide an example of each approach from recent literature that have exploited the basic science biology of the first identified metastasis suppressor, nm23-H1 (2, 75), towards a translational application.
Restoration of metastasis suppressor protein function in disseminated tumor cells. In many cases, ovarian cancer is associated with metastasis that occurs before diagnosis. A recent study (76) used adeno-associated virus- mediated gene transfer to reexpress nm23-H1 and evaluate whether this would prevent ovarian cancer metastasis in an animal model of this disease. A cell line of high metastatic potential to the liver derived by in vivo selection was used to evaluate the biological significance of nm23-H1 reexpression on liver metastasis. Following orthotopic implantation of these cells, an adeno-associated virus construct expressing nm23-H1 was injected i.p. This therapy resulted in expression of the transgene gene in most of the tumor cells in situ in mice, and was associated with a 60% reduction in the number of animals developing liver metastases and a 35-day prolongation of median survival. This work provides proof of principle of the value of nm23-H1 induction as a therapeutic approach towards ovarian cancer. The advantages of this approach are also related to the biology of ovarian cancer, which is a disease that rarely spreads outside the abdominal cavity. In addition, high-dose i.p. inoculation with gene therapy vectors is feasible and avoids the problems associated with i.v. inoculation (77, 78).
A recent development from the Steeg laboratory also exploited the characteristics of the nm23-H1 metastasis gene but applied the second approach mentioned above, i.e., reexpression of the endogenous gene as a therapeutic tool. Here, the objective was to find a therapy that would induce nm23-H1 expression and allow patients at high risk to go on lifelong maintenance therapy aimed at elevation of the nm23 expression. This type of therapy would require a drug that is relatively nontoxic and with an easily tolerated dose and schedule. Several agents have been reported to elevate nm23-H1 expression. For example, all-trans retinoic acid was reported to elevate nm23-H1 expression in a hepatocellular carcinoma cell line (79), whereas g-linolenic acid elevated the nm23-H1 expression of MDA-MB-231 breast carcinoma cells (80). By carrying out an analysis of the nm23-H1 promoter, the Steeg laboratory noted that dexamethasone (glucocorticoid receptor-mediated) elevated the nm23-H1 expression of metastatic breast carcinoma cell lines (81, 82). Medroxyprogesterone acetate (MPA) is a progestin used at low concentrations in birth control and hormone replacement therapy, as well as at high concentrations in breast cancer chemotherapy (83, 84). Furthermore, MPA elevated nm23-H1 expression and inhibited soft agar colonization (82), and this effect was mediated through glucocorticoid receptor. The effect of MPA on the elevation of nm23-H1 expression was tested in an in vivo model system for the outgrowth of metastases (85). Using the MDA-MB-231 breast carcinoma cell line to generate lung metastases, mice were randomized to vehicle control or MPA. Metastases developed in 100% of control mice versus 64% to 73% of MPA-treated mice. The mean number of gross metastases per mouse was reduced by 33% to 62% depending on the MPA dose. Large metastases (> 3 mm in any diameter) were reduced by 55% to 75% in the MPA-treated mice. Thus, MPA inhibited the incidence, number and size of metastases in this model. This reduction in metastases was associated with nm23-H1 reexpression in the lung tumors. Importantly, a phase II trial of MPA in patients with lymph node metastases and estrogen- and progesterone-negative tumors is planned.
Identification of “druggable” gene expression alterations associated with loss of metastasis suppressor protein expression. A natural expectation would be that functional loss of metastasis suppressor proteins is associated with gene expression changes, and consequently, cellular phenotypic alterations. Recently, this premise, coupled with the idea that loss of metastasis suppressor proteins is associated with the overexpression of prometastatic genes, was used to describe a novel therapeutic approach. These studies focused on RhoGDI2, a suppressor of metastasis in an isogenic model of bladder cancer metastasis (86), whose reduced expression was found to be associated with decreased survival for patients with bladder cancer (87). Titus et al. (29) postulated that RhoGDI2 works as a brake on the expression of prometastatic genes and sought to identify such genes in a combined analysis of the isogenic xenograft lung metastasis model and primary human bladder carcinomas. Because clinical reconstitution of RhoGDI2 protein is currently impractical, they sought to discover clinically druggable proteins or pathways downstream of RhoGDI2. These candidate genes were required to be up-regulated following the loss of RhoGDI2 protein from the cells, and be capable of being inhibited by small-molecule antagonists or functionally antagonistic antibodies.
Using DNA microarrays to monitor the changes in gene expression following restoration of RhoGDI2 expression in metastatic cells, several potentially targetable proteins were identified, including ET-1. This finding was confirmed by examining the relationship between RhoGDI2 expression levels and those of ET-1 in human tumor samples (29). Furthermore, inhibition of the endothelin axis with orally bioavailable small-molecule antagonists of the endothelin receptor A, such as Atrasentan, lead to metastatic suppression in cells deficient for RhoGDI2 (29). The endothelin ligand has been shown to regulate vascular tone, tissue differentiation, cell proliferation, hormone production, cell invasion, angiogenesis, and bone remodeling (88). ET-1 also has potent effects on cells in the skeleton (88) and lung (89), two important sites of bladder cancer metastasis (70). In addition to such direct effects, ET-1 may indirectly increase vascular endothelial growth factor and induce hypoxia-inducing factor 1-α (90). ET-1 and vascular endothelial growth factor stimulate one another (91), resulting in the proliferation of endothelial and vascular smooth muscle cells thus promoting tumor growth. Previous work also shows a role for tumor-secreted ET-1 in skeletal metastases from breast and prostate cancers (92) and supports a model in which tumor-secreted ET-1 stimulates bone cells, in turn, providing a fertile microenvironment for metastases.
Collectively, these findings indicate that adjuvant trials with endothelin antagonists may be considered for patients with advanced bladder cancer following therapy of the primary lesion. The rationale for its use in the adjuvant setting stems from the remarkable effect these drugs have had in preventing experimental metastasis. These findings, however, do not exclude the utility of these drugs in established early metastatic disease, but this is yet to be tested experimentally. The molecular relationship shown between ET-1 and RhoGDI2 is significant because of clinically tested, orally active, small-molecule endothelin receptor A antagonists such as Atrasentan with good clinical safety profiles (92). In fact, in patients with prostate cancer, treated in a recently reported dose escalation trial, the most common adverse events were rhinitis, headache, and peripheral edema, indicating that Atrasentan is well tolerated. In this study, there was no observed dose-limiting adverse event observed at doses up to 95 mg (93), whereas in other studies, quality of life was not adversely affected by this drug (94). Finally, the application of this effector discovery approach to other metastasis suppressor proteins could identify potential novel targets for therapy in other common malignancies (95, 96).
Acknowledgments
We thank Dr. Kay Macleod for her considerable assistance and advice during the preparation of key sections of this manuscript.
Footnotes
Grant support: CA88728, CA89019, National Foundation for Cancer Research Center for Metastasis Research and U.S. Army Medical Research and Materiel Command grant DAMD17-02-1-0541 (D.R. Welch), WB1XWH-06-1-0041 (J.P. O’Keefe, C.W. Rinker-Schaeffer); CA 89569 (C.W. Rinker-Schaeffer),The University of Chicago Research Cure and Education Fund (J.P. O’Keefe, C.W. Rinker-Schaeffer); CA104106, CA075115 (D. Theodorescu).
References
- 1.Steeg PS, Cohn KH, Leone A. Tumor metastasis and nm23: current concepts. Cancer Cell. 1991;3:257–62. [PubMed] [Google Scholar]
- 2.Steeg PS, Bevilacqua G, Kopper L, et al. Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst. 1988;80:200–4. doi: 10.1093/jnci/80.3.200. [DOI] [PubMed] [Google Scholar]
- 3.Kauffman EC, Robinson VL, Stadler WM, Sokoloff MH, Rinker-Schaeffer CW. Metastasis suppression: the evolving role of metastasis suppressor genes for regulating cancer cell growth at the secondary site [comment] J Urol. 2003;169:1122–33. doi: 10.1097/01.ju.0000051580.89109.4b. [DOI] [PubMed] [Google Scholar]
- 4.Shevde LA, Welch DR. Metastasis suppressor pathways-Ean evolving paradigm. Cancer Lett. 2003;198:1–20. doi: 10.1016/s0304-3835(03)00304-5. [DOI] [PubMed] [Google Scholar]
- 5.Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. 2003;3:55–63. doi: 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- 6.Robinson VL, Kauffman EC, Sokoloff MH, Rinker-Schaeffer CW. The basic biology of metastasis. CancerTreat Res. 2004;118:1–21. doi: 10.1007/978-1-4419-9129-4_1. [DOI] [PubMed] [Google Scholar]
- 7.Leone A, Flatow U, King CR, et al. Reduced tumor incidence, metastatic potential, and cytokine responsiveness of nm23-transfected melanoma cells. Cell. 1991;65:25–35. doi: 10.1016/0092-8674(91)90404-m. [DOI] [PubMed] [Google Scholar]
- 8.Yoshida BA, Dubauskas Z, Chekmareva MA, Christiano TR, Stadler WM, Rinker-Schaeffer CW. Mitogen-activated protein kinase kinase 4/stress-activated protein/Erk kinase 1 (MKK4/SEK1), a prostate cancer metastasis suppressorgeneencoded by human chromo-some 17. Cancer Res. 1999;59:5483–7. [PubMed] [Google Scholar]
- 9.Vander Griend DJ, Kocherginsky M, Hickson JA, Stadler WM, Lin A, Rinker-Schaeffer CW. Suppression of metastatic colonization by the context-dependent activation of the c-Jun NH2-terminal kinase kinases JNKK1/MKK4 and MKK7. Cancer Res. 2005;65:10984–91. doi: 10.1158/0008-5472.CAN-05-2382. [DOI] [PubMed] [Google Scholar]
- 10.Xia W, Unger P, Miller L, Nelson J, Gelman IH. The Src-suppressed C kinase substrate, SSeCKS, is a potential metastasis inhibitor in prostate cancer. Cancer Res. 2001;61:5644–51. [PubMed] [Google Scholar]
- 11.Goldberg SF, Miele ME, Hatta N, et al. Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003;63:432–40. [PubMed] [Google Scholar]
- 12.Hickson JA, Huo D, Vander Griend DJ, Lin A, Rinker-Schaeffer CW, Yamada SD. The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res. 2006;66:2264–70. doi: 10.1158/0008-5472.CAN-05-3676. [DOI] [PubMed] [Google Scholar]
- 13.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 14.Dong JT, Lamb PW, Rinker-Schaeffer CW, et al. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science. 1995;268:884–6. doi: 10.1126/science.7754374. [DOI] [PubMed] [Google Scholar]
- 15.Yin HL, Stossel TP. Control of cytoplasmic actin gelsol transformation by gelsolin, a calcium-dependent regulatory protein. Nature. 1979;281:583–6. doi: 10.1038/281583a0. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu K, Kaibuchi K, Nonaka H, Yamamoto J, Takai Y. Tissue and subcellular distributions of an inhibitory GDP/GTP exchange protein (GDI) for the rho proteins by use of its specific antibody. Biochem Biophys Res Commun. 1991;175:199–206. doi: 10.1016/s0006-291x(05)81220-3. [DOI] [PubMed] [Google Scholar]
- 17.Boller K, Vestweber D, Kemler R. Cell-adhesion molecule uvomorulin is localized in the intermediate junctions of adult intestinal epithelial cells. J Cell Biol. 1985;100:327–32. doi: 10.1083/jcb.100.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Girgrah N, Ackerley CA, Moscarello MA. Localization of CD44 (P80) on the external surface of a human astrocytoma cell. Neuroreport. 1991;2:441–4. doi: 10.1097/00001756-199108000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Nash KT, Welch DR. The KISS1 metastasis suppressor: mechanistic insights and clinical utility. Front Biosci. 2006;11:647–59. doi: 10.2741/1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeung K, Seitz T, Li S, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–7. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 21.Bosnar MH, De Gunzburg J, Bago R, Brecevic L, Weber I, Pavelic J. Subcellular localization of A and B Nm23/NDPK subunits. Exp Cell Res. 2004;298:275–84. doi: 10.1016/j.yexcr.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 22.Lin M, van Golen KL. Rho-regulatory proteins in breast cancer cell motility and invasion. Breast Cancer Res Treat. 2004;84:49–60. doi: 10.1023/B:BREA.0000018424.43445.f3. [DOI] [PubMed] [Google Scholar]
- 23.Gildea JJ, Seraj MJ, Oxford G, et al. RhoGDI2 is an invasion and metastasis suppressor gene in human cancer. Cancer Res. 2002;62:6418–23. [PubMed] [Google Scholar]
- 24.Fujita H, Okada F, Hamada J, et al. Gelsolin functions as a metastasis suppressor in B16-BL6 mouse melanoma cells and requirement of the carboxyl-terminus for its effect. Int J Cancer. 2001;93:773–80. doi: 10.1002/ijc.1413. [DOI] [PubMed] [Google Scholar]
- 25.DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15:356–63. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Gelman IH, Gao L. SSeCKS/Gravin/AKAP12 metastasis suppressor inhibits podosome formation via RhoA- and Cdc42-dependent pathways. Mol Cancer Res. 2006;4:151–8. doi: 10.1158/1541-7786.MCR-05-0252. [DOI] [PubMed] [Google Scholar]
- 27.Hakem A, Sanchez-Sweatman O, You-Ten A, et al. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005;19:1974–9. doi: 10.1101/gad.1310805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itoh K, Yoshioka K, Akedo H, Uehata M, Ishizaki T, Narumiya S. An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat Med. 1999;5:221–5. doi: 10.1038/5587. [DOI] [PubMed] [Google Scholar]
- 29.Titus B, Frierson HF, Jr., Conaway M, et al. Endothelin axis is a target of the lung metastasis suppressor gene RhoGDI2. Cancer Res. 2005;65:7320–7. doi: 10.1158/0008-5472.CAN-05-1403. [DOI] [PubMed] [Google Scholar]
- 30.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 31.Chellaiah M, Kizer N, Silva M, Alvarez U, Kwiatkowski D, Hruska KA. Gelsolin deficiency blocks podosome assembly and produces increased bone mass and strength. J Cell Biol. 2000;148:665–78. doi: 10.1083/jcb.148.4.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moussad EE, Brigstock DR. Connective tissue growth factor: what’s in a name. Mol Genet Metab. 2000;71:276–92. doi: 10.1006/mgme.2000.3059. [DOI] [PubMed] [Google Scholar]
- 33.Chang CC, Shih JY, Jeng YM, et al. Connective tissue growth factor and its role in lung adenocarcinoma invasion and metastasis. J Natl Cancer Inst. 2004;96:364–75. doi: 10.1093/jnci/djh059. [DOI] [PubMed] [Google Scholar]
- 34.Liu WM, Zhang XA. KAI1/CD82, a tumor metastasis suppressor. http://www.doi:10.1016/j.Canlet.2005.08.018. Cancer Lett. 2005 doi: 10.1016/j.canlet.2005.08.018. [DOI] [PubMed]
- 35.Sridhar SC, Miranti CK. Tetraspanin KAI1/CD82 suppresses invasion by inhibiting integrin-dependent crosstalk with c-Met receptor and Src kinases. Oncogene. 2006;25:2367–78. doi: 10.1038/sj.onc.1209269. [DOI] [PubMed] [Google Scholar]
- 36.Stupack DG, Teitz T, Potter MD, et al. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006;439:95–9. doi: 10.1038/nature04323. [DOI] [PubMed] [Google Scholar]
- 37.Stupack DG, Puente XS, Boutsaboualoy S, Storgard CM, Cheresh DA. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J Cell Biol. 2001;155:459–70. doi: 10.1083/jcb.200106070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao H, Ross FP, Teitelbaum SL. Unoccupied α(v)β3 integrin regulates osteoclast apoptosis by transmitting a positive death signal. Mol Endocrinol. 2005;19:771–80. doi: 10.1210/me.2004-0161. [DOI] [PubMed] [Google Scholar]
- 39.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–93. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 40.Michl P, Barth C, Buchholz M, et al. Claudin-4 expression decreases invasiveness and metastatic potential of pancreatic cancer. Cancer Res. 2003;63:6265–71. [PubMed] [Google Scholar]
- 41.Howe KL, Reardon C, Wang A, Nazli A, McKay DM. Transforming growth factor-β regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157:H7-induced increased permeability. Am J Pathol. 2005;167:1587–97. doi: 10.1016/s0002-9440(10)61243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noda M, Oh J, Takahashi R, Kondo S, Kitayama H, Takahashi C. RECK: a novel suppressor of malignancy linking oncogenic signaling to extracellular matrix remodeling. Cancer Metastasis Rev. 2003;22:167–75. doi: 10.1023/a:1023043315031. [DOI] [PubMed] [Google Scholar]
- 43.Gao AC, Lou W, Dong JT, Isaacs JT. CD44 is a metastasis suppressor gene for prostatic cancer located on human chromosome 11p13. Cancer Res. 1997;57:846–9. [PubMed] [Google Scholar]
- 44.Gao AC, Lou W, Sleeman JP, Isaacs JT. Metastasis suppression by the standard CD44 isoform does not require the binding of prostate cancer cells to hyaluronate. Cancer Res. 1998;58:2350–2. [PubMed] [Google Scholar]
- 45.Sleeman J, Moll J, Sherman L, et al. The role of CD44 splice variants in human metastatic cancer. Ciba Found Symp. 1995;189:142–51. doi: 10.1002/9780470514719.ch11. discussion 151-46, 174-46. [DOI] [PubMed] [Google Scholar]
- 46.Ponta H, Sleeman J, Dall P, Moll J, Sherman L, Herrlich P. CD44 isoforms in metastatic cancer. Invasion Metastasis. 1994;14:82–6. [PubMed] [Google Scholar]
- 47.Tkach T, Li E, Bestagno M, Burrone OR. Expression of CD44 splice variants in metastatic and non-meta-static mouse tumour cell lines. Immunol Lett. 1996;52:81–7. doi: 10.1016/0165-2478(96)02588-6. [DOI] [PubMed] [Google Scholar]
- 48.McGary EC, Lev DC, Bar-Eli M. Cellular adhesion pathways and metastatic potential of human melanoma. Cancer Biol Ther. 2002;1:459–65. doi: 10.4161/cbt.1.5.158. [DOI] [PubMed] [Google Scholar]
- 49.Kashima T, Nakamura K, Kawaguchi J, et al. Overexpression of cadherins suppresses pulmonary metastasis of osteosarcoma in vivo. Int J Cancer. 2003;104:147–54. doi: 10.1002/ijc.10931. [DOI] [PubMed] [Google Scholar]
- 50.Suyama K, Shapiro I, Guttman M, Hazan RB. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell. 2002;2:301–14. doi: 10.1016/s1535-6108(02)00150-2. [DOI] [PubMed] [Google Scholar]
- 51.Feltes CM, Kudo A, Blaschuk O, Byers SW. An alternatively spliced cadherin-11 enhances human breast cancer cell invasion. Cancer Res. 2002;62:6688–97. [PubMed] [Google Scholar]
- 52.Qi J, Chen N, Wang J, Siu CH. Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the h-catenin signaling pathway. Mol Biol Cell. 2005;16:4386–97. doi: 10.1091/mbc.E05-03-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li G, Satyamoorthy K, Herlyn M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001;61:3819–25. [PubMed] [Google Scholar]
- 54.Kyriakis JM, Banerjee P, Nikolakaki E, et al. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–60. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 55.Vander Griend DJ, Kocherginsky M, Hickson J, Stadler W, Lin A, Rinker-Schaeffer C. Suppression of Metastatic Colonization by the Context-Dependent Activation of the JNK Kinases JNKK1/MKK4 and MKK7. Cancer Res. 2005;65:10984–91. doi: 10.1158/0008-5472.CAN-05-2382. [DOI] [PubMed] [Google Scholar]
- 56.Yamada SD, Hickson JA, Hrobowski Y, et al. Mitogen-activated protein kinase kinase 4 (MKK4) acts as a metastasis suppressor gene in human ovarian carcinoma. Cancer Res. 2002;62:6717–23. [PubMed] [Google Scholar]
- 57.Cheng GC, Schulze PC, Lee RT, Sylvan J, Zetter BR, Huang H. Oxidative stress and thioredoxin-interacting protein promote intravasation of melanoma cells. Exp Cell Res. 2004;300:297–307. doi: 10.1016/j.yexcr.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 58.Finkel T. Neutrophils with a license to kill: permeabilized, not stirred. Dev Cell. 2003;4:146–8. doi: 10.1016/s1534-5807(03)00023-6. [DOI] [PubMed] [Google Scholar]
- 59.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–69. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 60.Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 61.Morrison DK. KSR: a MAPK scaffold of the Ras pathway. J Cell Sci. 2001;114:1609–12. doi: 10.1242/jcs.114.9.1609. [DOI] [PubMed] [Google Scholar]
- 62.Lozano J, Xing R, Cai Z, et al. Deficiency of kinase suppressor of Ras1prevents oncogenic ras signaling in mice. Cancer Res. 2003;63:4232–8. [PubMed] [Google Scholar]
- 63.Fu Z, Smith PC, Zhang L, et al. Effects of Raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J Natl Cancer Inst. 2003;95:878–89. doi: 10.1093/jnci/95.12.878. [DOI] [PubMed] [Google Scholar]
- 64.Yeung K, Janosch P, McFerran B, et al. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the Raf kinase inhibitor protein. Mol Cell Biol. 2000;20:3079–85. doi: 10.1128/mcb.20.9.3079-3085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 66.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–63. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 67.Shevde LA, Samant RS, Goldberg SF, et al. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp Cell Res. 2002;273:229–39. doi: 10.1006/excr.2001.5452. [DOI] [PubMed] [Google Scholar]
- 68.Zhang S, Lin QD, Di W. Suppression of human ovarian carcinoma metastasis by the metastasis-suppressor gene, BRMS1. Int J Gynecol Cancer. 2006;16:522–31. doi: 10.1111/j.1525-1438.2006.00547.x. [DOI] [PubMed] [Google Scholar]
- 69.Bandyopadhyay S, Pai SK, Hirota S, et al. PTEN upregulates the tumor metastasis suppressor gene Drg-1 in prostate and breast cancer. Cancer Res. 2004;64:7655–60. doi: 10.1158/0008-5472.CAN-04-1623. [DOI] [PubMed] [Google Scholar]
- 70.Stein JP, Lieskovsky G, Cote R, et al. Radical cystectomy in the treatment of invasive bladder cancer: long-term results in 1,054 patients. J Clin Oncol. 2001;19:666–75. doi: 10.1200/JCO.2001.19.3.666. [DOI] [PubMed] [Google Scholar]
- 71.Pomper MG. Translational molecular imaging for cancer. Cancer Imaging. 2005;5 doi: 10.1102/1470-7330.2005.0101. Spec No A:S16-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006;24:778–89. doi: 10.1200/JCO.2005.03.2375. [DOI] [PubMed] [Google Scholar]
- 73.Rennebeck G, Martelli M, Kyprianou N. Anoikis and survival connections in the tumor microenvironment: is there a role in prostate cancer metastasis. Cancer Res. 2005;65:11230–5. doi: 10.1158/0008-5472.CAN-05-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kopfstein L, Christofori G. Metastasis: cell-autonomous mechanisms versus contributions by the tumor microenvironment. Cell Mol Life Sci. 2006;63:449–68. doi: 10.1007/s00018-005-5296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Steeg PS, Bevilacqua G, Pozzatti R, Liotta LA. Sobel ME. Altered expression of NM23, a gene associated with low tumor metastatic potential, during adenovirus 2 Ela inhibition of experimental metastasis. Cancer Res. 1988;48:6550–4. [PubMed] [Google Scholar]
- 76.Li J, Zhou J, Chen G, et al. Inhibition of ovarian cancer metastasis by adeno-associated virus-mediated gene transfer of nm23H1in an orthotopic implantation model. Cancer GeneTher. 2006;13:266–72. doi: 10.1038/sj.cgt.7700899. [DOI] [PubMed] [Google Scholar]
- 77.Williams DA, Baum C. Medicine. Gene therapy-new challenges ahead. Science. 2003;302:400–1. doi: 10.1126/science.1091258. [DOI] [PubMed] [Google Scholar]
- 78.Chuah M. Challenges in advancing the field of gene therapy—American Society of Gene Therapy Stake-holders’ Meeting A critical review of the science, medicine and regulation. IDrugs. 2005;8:483–6. [PubMed] [Google Scholar]
- 79.Liu LT, Chang HC, Chiang LC, Hung WC. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Res. 2003;63:3069–72. [PubMed] [Google Scholar]
- 80.Jiang W, Hiscox S, Bryce R, Horrobin D, Mansel R. The effects of n-6 polyunsaturated fatty acids on the expression of nm-23 in human cancer cells. Br J Cancer. 1988;77:731–8. doi: 10.1038/bjc.1998.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ouatas T, Clare S, Hartsough M, DeLaRosa A, Steeg P. MMTV-associated transcription factor binding sites increase nm23-1 metastasis suppressor gene expression in human breast carcinoma cell lines. Clin Exp Metastasis. 2002;19:35–42. doi: 10.1023/a:1013897022827. [DOI] [PubMed] [Google Scholar]
- 82.Ouatas T, Halverson D, Steeg P. Dexamethasone and medroxyprogesterone acetate elevate Nm23-1 metastasis suppressor expression in metastatic human breast carcinoma cells: new uses for old compounds. Clin Cancer Res. 2003;9:3763–72. [PubMed] [Google Scholar]
- 83.Zaucha R, Sosinska-Mielcarek K, Jassem J. Long-term survival of a patient with primarily chemo-resistant metastatic breast cancer treated with medroxyprogesterone acetate. Breast. 2004;13:321–4. doi: 10.1016/j.breast.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 84.Focan C, Beauduin M, Majois F, et al. High-dose oral medroxyprogesterone acetate or tamoxifen as adjuvant hormone therapy for node-negative early-stage breast cancer: randomized trial with 7-year up-date. Clin Breast Cancer. 2004;5:136–41. doi: 10.3816/cbc.2004.n.018. [DOI] [PubMed] [Google Scholar]
- 85.Palmieri D, Halverson D, Ouatas T, et al. Medroxyprogesterone acetate elevation of Nm23-1 metastasis suppressor expression in hormone receptor-negative breast cancer. 2005;97:632–42. doi: 10.1093/jnci/dji111. [DOI] [PubMed] [Google Scholar]
- 86.Gildea J, Seraj M, Oxford G, et al. RhoGDI2 is an invasion and metastasis suppressor gene in human cancer. 2002;62:6418–23. [PubMed] [Google Scholar]
- 87.Theodorescu D, Sapinoso LM, Conaway MR, Oxford G, Hampton GM, Frierson HF, Jr. Reduced expression of metastasis suppressor RhoGDI2 is associated with decreased survival for patients with bladder cancer. 2004;10:3800–6. doi: 10.1158/1078-0432.CCR-03-0653. [DOI] [PubMed] [Google Scholar]
- 88.Masaki T. Historical review: Endothelin. 2004;25:219–24. doi: 10.1016/j.tips.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 89.Fagan KA, McMurtry IF, Rodman DM. Role of endothelin-1 in lung disease. 2001;2:90–101. doi: 10.1186/rr44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Spinella F, Rosano L, Di Castro V, Natali PG, Bagnato A. Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-inducible factor1ain ovarian carcinoma cells. 2002;277:27850–5. doi: 10.1074/jbc.M202421200. [DOI] [PubMed] [Google Scholar]
- 91.Okuda Y, Tsurumaru K, Suzuki S, et al. Hypoxia and endothelin-1induceVEGF production in human vascular smooth muscle cells. 1998;63:477–84. doi: 10.1016/s0024-3205(98)00296-3. [DOI] [PubMed] [Google Scholar]
- 92.Nelson J, Bagnato A, Battistini B, Nisen P. The endothelin axis: emerging role in cancer. 2003;3:110–6. doi: 10.1038/nrc990. [DOI] [PubMed] [Google Scholar]
- 93.Zonnenberg BA, Groenewegen G, Janus TJ, et al. Phase I dose-escalation study of the safety and pharmacokinetics of Atrasentan: an endothelin receptor antagonist for refractory prostate cancer. 2003;9:2965–72. [PubMed] [Google Scholar]
- 94.Carducci MA, Padley RJ, Breul J, et al. Effect of endothelin-A receptor blockade with Atrasentan on tumor progression in men with hormone-refractory prostate cancer: a randomized, phase II, placebo-controlledtrial. JClinOncol. 2003;21:679–89. doi: 10.1200/JCO.2003.04.176. [DOI] [PubMed] [Google Scholar]
- 95.Steeg PS, Ouatas T, Halverson D, Palmieri D, Salerno M. Metastasis suppressor genes: basic biology and potential clinical use. 2003;4:51–62. doi: 10.3816/cbc.2003.n.012. [DOI] [PubMed] [Google Scholar]
- 96.Welch DR. Microarrays bring new insights into understanding of breast cancer metastasis to bone. 2004;6:61–4. doi: 10.1186/bcr736. [DOI] [PMC free article] [PubMed] [Google Scholar]