

Abstract
During a complex macromolecular reaction multiple changes in molecular conformation and interactions with ligands may occur. X-ray crystallography may provide only a limited set of snapshots of these changes. Solution methods can augment such structural information to provide a more complete picture of a macromolecular reaction. We analyzed the changes in protein conformation and protein:nucleic acid interactions which occur during transcription initiation by using a chemical nuclease tethered to cysteines introduced site-specifically into the RNA polymerase of bacteriophage T7 (T7 RNAP). Changes in cleavage patterns as the polymerase steps through transcription reveal a series of structural transitions which mediate transcription initiation. Cleavage by tethered chemical nucleases is seen to be a powerful method for revealing the conformational dynamics of macromolecular reactions, and has certain advantages over cross-linking or energy transfer approaches.
Keywords: DNA-Directed RNA Polymerases; Transcription, Genetic; Protein Conformation
Introduction
As increasing numbers of atomic coordinate data sets are deposited in our macromolecular structure databases, attention begins to shift to efforts to describe the conformational dynamics of the reactions mediated by these molecules, i.e. to develop ‘movies’ of these molecules to augment the static snapshots provided by crystal structures. Progression through a transcription reaction is one example of a process expected to be characterized by extensive and continuous changes in protein conformation and protein:nucleic acid interactions. For example, comparison of crystal structures of the T7RNA polymerase alone (2), in complexes with promoter or a transcriptional inhibitor (3, 5), in a transcription complex which has not yet released the promoter (an initial transcription complex or ITC; 4) or a complex which has released promoter (an elongation complex or EC; 6, 7) reveal that this enzyme undergoes significant conformational changes as it progresses through transcription or interacts with different ligands. The solution dynamics of such processes can be probed with cross-linking or energy transfer approaches. However, each of these approaches has limitations. The presence of cross-linking cannot be interpreted simply in terms of amino acid:nucleic acid proximity because the efficiency of cross-linking is influenced by differences in the chemical reactivity of different side-chains. Energy transfer (FRET or LRET) allows for rigorous interpretation of changes in distance between acceptor-donor groups, but requires specialized equipment which may not be available in every lab. Cleavage by chemical nucleases tethered the sulfhydryl groups of introduced cysteines can also be used to assess protein:nucleic acid proximity in protein:DNA and protein:RNA complexes. The technique requires no specialized equipment other than that available in a standard molecular biology lab, and allows cleavage patterns to be interpreted largely in terms of proximity and accessibility to the chemical nuclease because the cutting agent (OH . ) displays minimal chemical specificity. One of the more commonly used nucleases of this type is Fe-BABE [iron (S)-1-(p-Bromoacetamidobenzyl)EDTA; Fig. 1].
Fig. 1.
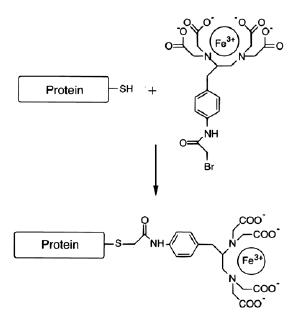
Incubation of Fe(III)-BABE with a protein containing a free and accessible sulfhydryl group leads to covalent attachment of the Fe-BABE to the protein as shown.
Reduction of the EDTA-chelated Fe (III) ion with ascorbic acid and hydrogen peroxide produces reactive free radical (OH×) by the Fenton reaction. While Fe-BABE cleavage has been used previously to map protein:nucleic acid proximity in protein:DNA or protein:RNA complexes, there are few examples of using this reagent to monitor structural changes during the course of a multi-step reaction. Recently, we prepared a set of mutant of T7RNAPs bearing single cysteine substitutions. The mutant enzymes were conjugated with Fe-BABE and the cleavage patterns of the conjugates on the template (T) and non-template (NT) DNA strands and on the RNA were monitored as the polymerase was stepped through transcription initiation and then transited to elongation (8). The changes in cleavage patterns reveal a series of concerted structural transitions, and illustrate the potential of this method for monitoring the conformational dynamics of complex macromolecular reactions.
Materials and Methods
Mutagenesis of T7 RNA polymerase
Point mutants were generated by PCR using the Quick ChangeTM Site-Directed Mutagenesis Kit (Promega) and synthetic oligonucleotides. The plasmid template for mutagenesis (pDPT7; 9) was purified using the Qiagen maxi-prep kit.
Purification of cysteine mutants
Plasmids expressing mutant RNAPs were transformed into competent BL21 cells (Stratagene). Transformed cells were plated out on 4-6 LB-agar plates with 100 mg/ml ampicillin at a cell density of 500-2000 colonies per plate. In the morning, the colonies were resuspended in LB and grown in 12 liters of LB containing 100ug/ml ampicillin and protein expression was induced by adding IPTG; 4-6 hours later the cells were harvested by centrifugation and stored at -80oC. For purification, cells were thawed and resuspended in Buffer A (20mM sodium phosphate pH8.0, 1 mM DTT, 1 mM EDTA, 5% glycerol, 0.1mM phenylmethylsulfonylfluoride) and lysed by sonication; NaCl was added to 0.5 M, followed by addition of polymin P (pH 8.0) to precipitate nucleic acid. The cell lysate was clarified by centrifugation and then 4 grams of ammonium sulfate were added for every 10 ml of supernatant. The precipitated protein was recovered by centrifugation and resuspended with enough buffer A to give a final conductivity equal to 180 mM NaCl. The protein solution was loaded onto a 100 ml Whatman P11 phosphocellulose column equilibrated with buffer A+150 mM NaCl, and RNAP was eluted with a linear gradient of 150-950 mM NaCl in Buffer A. T7 RNAP eluted at 300-400 mM NaCl. The fractions containing T7RNAP were identified, pooled, and dialyzed against Buffer A. The sample was then loaded onto a 50 ml Q-sepharose column (Amersham Pharmacia Biotech) equilibrated with buffer A, washed with buffer A+20 mM NaCl, and eluted with a linear gradient of 20-500mM NaCl in Buffer A. Fractions containing T7RNAP eluted at 150-250 mM NaCl and were concentrated by dialysis against storage buffer (20 mM Tris-HCl pH8.0, 0.5 M NaCl, 5 mM DTT, 1 mM EDTA, 50% glycerol). The protein concentration was calculated from its absorbance at 280nm using the molar extinction coefficient of 1.4x105 M-1 cm-1 (10). Typical yields were 10 mls of 4-6 mg/ml protein. Purified proteins were stored as aliquots at -80˚C until needed. Specific activity of all RNAPs was determined as described (10).
Conjugation with FeBABE
Prior to conjugation the polymerase was exchanged into storage buffer without DTT either by dialysis or by using microSpin TM G-25 columns (Amersham Pharmacia Biotech). The polymerase was then incubated with 5 fold molar excess of FeBABE at 37oC for 1 hr. Excess unreacted FeBABE was removed either by dialysis, or by using microspin columns, into storage buffer with 1mM DTT. RNAPs conjugated in this way can be stored for up to 6 months in aliquots at -80oC without loss of activity or conjugate. The transcription activity of each conjugated mutant was verified by its ability to synthesize run-off transcripts from a linearized plasmid template.
Free thiol group assay of FeBABE conjugated enzyme
The number of reactive sulfhydryl groups in each mutant was determined both before and after conjugation by measuring its reactivity with DTNB (Ellman’s reagent; 11). Conjugation efficiency averaged 60%.
Cleavage reaction
Transcription complexes were cleaved by addition of ascorbic acid and hydrogen peroxide from freshly prepared solutions of 100 mM and 200mM, respectively. Cleavage reactions were initially carried out with varying concentrations of ascorbic acid and H2O2 to define optimal cleavage conditions, which were obtained at final concentrations of 10 mM and 25 mM, respectively. Cleavage was allowed to proceed for 2 min at 37˚C and was quenched with equal volumes of 95% formamide, 20 mM EDTA and 0.01% xylene cyanol.
DNA foot printing by Fe-BABE
The synthetic 42 bp template used was designed to allow RNA extension to be halted at lengths of 1, 4, 6, 7, or 13 nt either by omission of certain nucleotides from the transcription reaction, or by inclusion of 3’-dNTPS. The template sequence from -17 to +6 corresponded to the consensus T7 promoter, and the transcript sequence from +1 to +25 was GGGAGACCGGAAUUCGAGGGGCUCG. The 5'-end of either the T or NT strand was phosphorylated with γ-32PATP by T4 polynucleotide kinase. Labeled DNA was purified with the Qiaquick Nucleotide Removal kit (Qiagen). To prepare double stranded 5’-labeled DNA, labeled NT or T strands were mixed 1:1 with unlabeled T or NT strands, respectively, and annealed in 10 mM Tris-HCl pH8.0, 50m M NaCl, 1 mM EDTA. Marker G+A ladders were prepared according to Maxam and Gilbert (12). Transcription reactions were carried out by mixing DNA and RNAPs at 0.1 and 1 μM, respectively in transcription buffer with NTPs (from Trilink Biotechnologies) added to 0.5 mM. Reactions were incubated for 3 min at 37oC and then split into 2 aliquots. Ascorbic acid and H2O2 were added to one aliquot and the other served as the uncleaved control. Identical reactions carried out with unconjugated mutants were also done. After 2 min, cleavage reactions were quenched with 2X formamide stop solution, boiled for 5 min, placed on ice, and resolved by electrophoresis on 17.5% polyacrylamide, 0.8% bis-acrylamide, 7M urea gels in 1 X TBE. Gels were analyzed by phosphorimaging.
RNA Cleavage By Fe-BABE
To detect RNA cleavage in the initial transcription complexes we used supercoiled pT7-7 as template (13). The +1 to +8 sequence of the transcript from this template is GGGAGCUU. RNAP and template were mixed in transcription buffer and incubated for 10 min at room temperature, followed by addition of GTP and ATP, and a further 3 min incubation. Reactions were placed on ice for 2 min, followed by addition of 3'-dUTP and α32P-CTP. Reactions were then split in 2. Ascorbic acid and H2O2 were added to one aliquot, reactions were placed at 37oC for 1 minute, quenched, and mixed with formamide loading buffer. To detect RNA cleavage in the EC we used Bgl-II linearized pPK7 (RNA seq.: GAGGGAGGGAGGGAGGGAGACU; 14). Template and RNAP in transcription buffer were incubated at room temperature for 10 minutes; GTP, ATP, α32P-CTP, and 3'-dUTP were added and reactions were incubated for 4 min at 37oC. Reactions were split in 2, and one aliquot was reacted with ascorbate and H2O2 for 2 min at 37oC, then quenched and resolved as described above. Reactions were also done with heparin added to 0.1mg/ml after NTP addition to limit initiation to a single round and to prevent any nonspecific binding of RNAP to DNA or RNA. Heparin addition had no effect on the cleavage patterns (not shown).
Results and Discussion
Use of a molecule with endogenous cysteines for Fe-BABE cleavage
T7 RNAP has 12 cysteines. Though previous studies have emphasized the importance of working with proteins which contain only a single site-specifically introduced cysteine so as to allow for unambiguous interpretation of cleavage patterns, we found that only 7 of the 12 cysteines (C530, C540, C510, C515, C347, C723 and C125) in T7RNAP could be substituted with serine without causing large reductions in enzyme activity (the resulting enzyme was dubbed “7-”). Reactivity with DTNB corresponds to ~2 cysteines per native w.t. polymerase (~12 cysteines per SDS-denatured polymerase) and less than 0.2 cysteines per 7- polymerase. We reasoned that this 7- mutant could nevertheless be a suitable background for our experiments if it gave minimal cleavage of RNA or DNA upon conjugation with Fe-BABE, so we compared cleavage patterns of Fe-BABE conjugated wild type (w.t.) and 7- enzymes (Fig. 2).
Fig. 2.
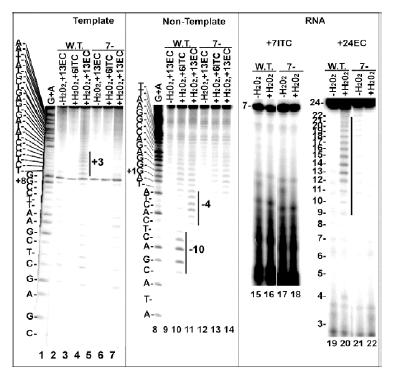
DNA/RNA cleavage by Fe-BABE conjugated w.t. and cys-minimized (7-) enzyme. Lanes 1-14: ECs halted at +13 (lanes 2, 4, 5, 7, 9, 11, 12, 14) or ITCs halted at +6 (lanes 3, 6, 10, 13) were formed with DNAs in which either the 5'-end of the T strand (lanes 2-7) or NT strand (lanes 9-14) was labeled with γ32P-ATP, and with either Fe-BABE conjugated w.t. (lanes 2-4, 9-11) or 7- (lanes 5-7, 12-14) enzyme H202 was added to lanes 2, 3, 6, 7, 10, 11, 13 and 14, and cleavage was mapped by reference to G+A ladders (lanes 1, 8). Cleavages are highlighted by vertical bars. Lanes 15-22: ITCs halted at +7 (lanes 15-18) or ECs halted at +24 (lanes 19-22) were formed with 3'-end labeled RNAs, and with either Fe-BABE conjugated w.t. (lanes 15, 16, 19, 20) or 7- (lanes 17, 18, 21, 22) enzyme. H202 was added to lanes 16, 18, 20, 22. Templates were a duplex T7 promoter from -17 to +25 in lanes 1-14,supercoiled pT7-7 plasmid in lanes 15-18 and a linearized plasmid (pPK7) containing a T7 promoter in lanes 19-22.
The w.t. enzyme cleaved the T strand near +3 in an EC halted at +13 (lane 4). It cleaved the NT strand in an ITC and an EC near -10 and -4, respectively (lanes 10, 11). The w.t. enzyme also cleaved the 5'-region of the RNA in an EC halted at +24 (lane 20). The 7- enzyme showed little cleavage in any of these complexes and we therefore concluded that it would provide a suitable background in which to observe DNA or RNA cleavage by Fe-BABE conjugated to single cysteines introduced into this mutant.
Cleavage patterns of Fe-BABE tethered to introduced cysteines
T7RNAP is organized around a large cleft which contains the active site and in which the T strand binds (the “Template binding cleft”). Surrounding this cleft are structural elements – designated thumb, fingers, intercalating hairpin, N-terminal domain, and promoter recognition loop (Fig. 3D) – which have been observed crystallographically to change conformation during the course of the transcription reaction. A total of 10 cysteines were individually introduced into these structures (at position 153, 239, 303, 385, 388, 393, 394, 644, 745, and 764) and used to map cleavage positions on the T and NT strands and RNA in transcription complexes bearing RNAs 1, 4, 6, 7, or 13 nt in length (an RNA length of ‘1’ corresponds to adding only 3’-dGTP to the reaction). RNAs of length 1-7 are produced during the abortive or initial phase of transcription, during which the RNAP is believed to retain interactions with the promoter; while an RNA length of 13nt corresponds to the elongation phase of transcription which follows release (or ‘clearance’) of the promoter. The full set of data from all these conjugates is described in (8). Here we limit our description to examples illustrative of this approach (cleavage by a particular conjugate is referred to by the number of the amino acid to which it is attached).
Fig. 3.
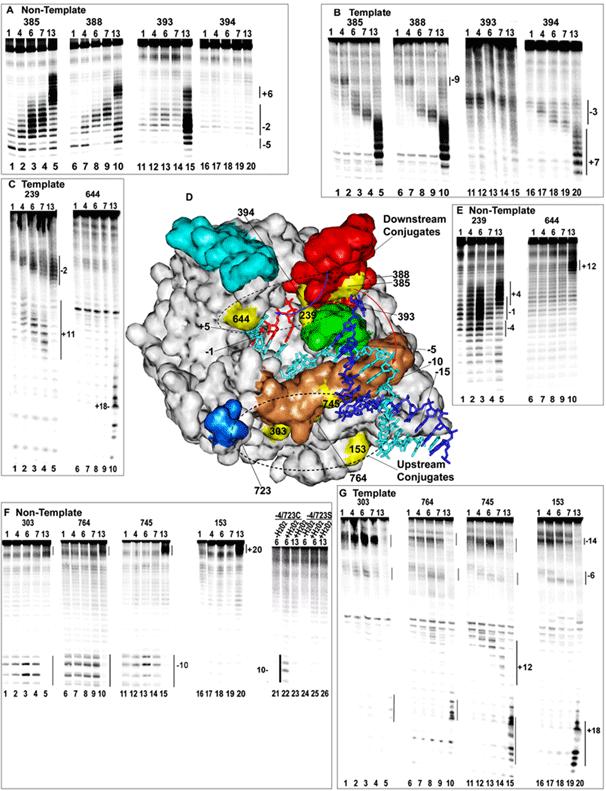
DNA cleavage by Fe-BABE tethered to introduced cysteines. A: Cleavage of transcription complexes with 5'-end labeled NT strand by conjugates at aa positions 385 (lanes 1-5), 388 (lanes 6-10), 393 (lanes 11-15), or 394 (lanes 16-20). The RNA length in the complex is 1, 4, 6, 7, or 13 nt as indicated at the top of each lane (a length of 1 corresponds to adding only 3'-dGTP). B: As in A but with 5'-labeled T strand. C: As in B, but with conjugates at either 239 (lanes 1-5) or 644 (lanes 1-10) D: Crystal structure of a T7RNAP ITC with a 3 nt RNA (pdb # 1QLN). NT strand is dark blue; T strand is cyan; RNA is red; the promoter recognition loop is tan; the thumb subdomain (aa 330-410) is red; the intercalating hairpin (aa 232-242) is green; the fingers 'flap' (aa 586-620) is cyan; and the 'positive patch' (K711/713/714) is blue. Positions of Fe-BABE conjugation are yellow and are labeled (aa 723 is hidden; 764 and 393 are largely obscured). Numbering of the DNA is relative to the +1 start site, and of the RNA is relative to the RNA 3'-nt. E: As in C, but with 5'-labeled NT strand. F: As in A but with conjugates at 303 (lanes 1-5), 764 (lanes 6-10), 745 (lanes 11-15), and 153 (lanes 16-2-0). Also shown is cleavage by an Fe-BABE conjugated enzyme from which 4 cysteines have been removed, but which retains a cys at 723 (lanes 21-23), and an enzyme in which C723 was mutated to ser (lanes 24-26). H2O2 was added to all reactions except those in lanes 21 and 24.. G: As in F but with 5'-labeled T strand.
Enzymes carrying Fe-BABE conjugated at positions 385, 388, 393 or 394 were used to cleave T and NT strand in transcription complexes with RNAs of varying length. These 4 residues are close together on helix N of the thumb subdomain of the RNAP (Fig. 3D) but sit on different faces of this helix. Residues 385 and 388 face away from the T strand binding cleft, while 394 faces into the T strand binding cleft and 393 is in an intermediate position. The crystal structure of an ITC with a 3 nt RNA (4) indicates that the NT strand lies outside the T strand binding cleft and, during initial transcription, is expected to be most accessible to cleavage by 385/388, less accessible to 393, and inaccessible to 394. The pattern of NT strand cleavage by these conjugates during initial transcription conforms to this expectation, since it is strongest for 385 and 388 (Fig. 3A, lanes 1-4, 6-9), is weaker for 393 (Fig. 3A, lanes 11-14), and is not cleaved at all by 394 (Fig. 3A, lanes 16-19). These cleavage patterns emphasize the importance of accessibility over proximity in determining whether cleavage is detected. Though cleavage is stronger if the conjugate is close to the nucleic acid, we consistently observed that the interposition of protein or nucleic acid between a conjugate and a cleavage target blocked cleavage of the target.
On the T strand conjugates 385/388 cleave weakly near -9 when the RNA is 1 or 4 nt in length (Fig. 3B; lanes 1, 2, 6, 7). This cleavage pattern is consistent with the structure of an ITC with a 3 nt RNA (Fig. 3D). Since 385/388 face outside of the T strand binding cleft they cannot cleave the T strand downstream of -5 since it lies inside the cleft. T strand cleavage by 385/388 between -5 and -7 would be blocked by the intercalating hairpin, and the NT strand would block T strand cleavage upstream of -10. Thus, as suggested by the red arrow in Fig. 3D, only the -8 to -10 region of the T strand would be accessible to OH . generated at 385/388, and the distance between 385/388 and -9 would account for the relative weakness of this cleavage. Remarkably, it is seen that upon extension of the RNA to 6 or 7 nt, T strand cleavage by 385/388 shifts downstream to -2 (Fig. 3D; lanes 3, 4, 8, 9). This 7-nt shift in cleavage position (from -9 to -2) accompanying an extension of the RNA by only 2 nt (from 4 to 6 nt) seems difficult to account for in terms of polymerase translocation. In addition to the discrepancy between the size of the RNA extension and the shift in the cleavage position, we note also that it moves T strand cleavage from a site outside of the T strand binding cleft, to a site inside the T strand binding cleft. Furthermore, cleavage by conjugates at other positions does not show comparable shifts in position upon extension of the RNA from 4 to 6 nt (8). For all these reasons, we conclude that the shift in T strand cleavage position by conjugates at 385/388 probably reflects a conformational change in the polymerase, for example the thumb might bend towards the RNA, as suggested by the blue arrow in Fig. 3D.
In contrast to the changes seen with the ‘thumb’ conjugates as the RNA is extended during initial transcription, conjugates at 153, 303, 723, 745, and 764 show static cleavage patterns as the RNA is extended from 1 to 7 nt (lanes 1-4, 6-9, 11-14, and 22 of Fig. 3F and lanes 1-4, 6-9, 11-14, and 16-19 of Fig. 3G). For example, these conjugates always cleave the NT strand between -9 and -12, even as the RNA is extended from 1 to 7 nt. This upstream portion of the promoter is known to be responsible for sequence specific binding by the RNAP and it remains base-paired during initial transcription (4, 13, 15). The conjugates at 303, 153, 745 and 764 are all located on elements (the N-terminal domain and promoter recognition loop) which participate in sequence specific binding to this upstream, recognition half of the promoter. The static nature of the cleavage patterns by these conjugates in this region of the promoter is therefore consistent with the idea that the sequence specific interaction made with the upstream part of the promoter remains intact as the RNA is extended from 1 to 7 nt (13, 15).
In contrast, the elongation complex with a 13 nt RNA is expected to have released the promoter and, consistent with this, we observe loss of cleavage by 153, 303, 745, and 764 in the upstream region of the promoter in the complex with the 13 nt RNA (lanes 5, 10. 15, and 20 of Figs. 3F and 3G). However, if all that happens during promoter clearance is that the RNAP translocates downstream, then we would expect 153, 303, 745, and 764 to cleave DNA a similar distance upstream of the RNA 3’-end in both the EC and the ITC (i.e. 303 cleaves the NT strand around -10, or 10-17 nt upstream of the RNA3’-end, in ITCs 1-7; so we might expect 303 to cleave the NT strand between -4 and +2 in EC13). Instead we observe that in EC13 these conjugates do not cleave upstream of the RNA 3’-end, but instead cleave ~6 nt downstream of the RNA, near +19 (lanes 5, 10. 15, and 20 of Figs. 3D and 3E). Figure 4 summarizes the changes in cleavage patterns occurring in the DNA and RNA during transcription initiation. These are detailed at greater length in Reference 8. All together these observations indicate that the transition to elongation is associated not simply with movement of the polymerase away from the promoter, but also with changes in protein conformation and in the disposition of DNA within the transcription complex. Both of these conclusions were recently confirmed by the crystal structure of a T7 RNAP elongation complex, which reveals a large conformational change in the polymerase and a rearrangement of RNAP:DNA contacts in comparison to the structure of an initial transcription complex (6, 7).
Fig. 4.
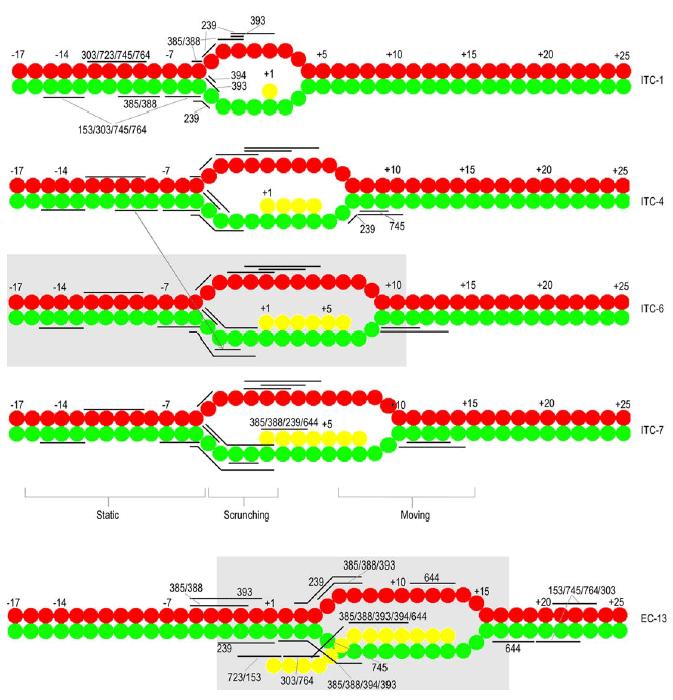
Summary of cleavage patterns. NT, T, and RNA are in red, green, and yellow, respectively. Cleavage ranges are indicated by horizontal bars and labeled by conjugate number in ITC-1 and EC13. In ITCs-4, -6, and -7 the bars indicating cleavage positions maintain the same vertical registry relative to the DNA as in ITC-1. The arrow connecting ITCs4 and 6 indicates a shift in the T strand cleavage site of 385/388 at this point. Grey boxes in ITC6 and EC13 indicate extent of the RNAP footprint in those complexes.
The results presented here and in Reference 8 demonstrate the power of using tethered chemical nucleases to reveal and monitor structural transitions during the course of a macromolecular reaction. Our experience with this methodology suggests a number of modifications which facilitate its application. Previously, this technology has depended on introducing single cysteines into a cysteine free protein, but our results indicate that the presence of multiple cysteines is not a barrier to use of this method. In the case of T7 RNAP we were able to eliminate almost all detectable cleavage due to Fe-BABE conjugation by removing 7 of the 12 endogenous cysteines (Fig. 2). Further, even if conjugates at endogenous cysteines will cleave nucleic acid, this should not preclude use of this method. However, in such cases (which may occur when endogenous cysteines cannot be removed without loss of activity), it is critical that experiments be appropriately controlled; i.e. the cleavage patterns of proteins conjugated in parallel and differing only in the presence of a single, introduced cysteine must be clearly distinguishable so that cleavage due to the conjugate at the introduced cysteine can be unambiguously identified.
The preparation of the conjugates themselves can be time-consuming since it requires a buffer transfer step to remove reducing agents (which are usually present in the storage buffer of a protein which contains free cysteines), following which the protein must be incubated with the conjugate for 1 or more hours, and then a 2nd buffer exchange must be carried out to remove unreacted conjugate. For this reason, we were anxious to find conditions that would allow long-term storage of conjugated proteins without loss of either transcriptional or cleavage activities, and since T7 RNAP precipitates if salt concentrations are less than ~0.1 M, we also sought buffer conditions for conjugation which would maintain protein solubility and activity. We found that we could carry out the conjugation reaction at pH 8.0 with 50% glycerol and 0.5 M NaCl present, both of which helped stabilize the polymerase. Indeed, except for omission of reducing agents, there should be no real restrictions on buffer conditions during conjugation. We also knew that T7RNAP is inactivated upon long-term storage without reducing agents, however we found that, once conjugation was complete, DTT could be added back to the protein at 1-2 mM without causing loss of the conjugate. We therefore store conjugated proteins at -80o C for up to 6 months in 50 mM Tris pH 8.0, 1mM EDTA, 1 mM DTT, 0.5 M NaCl, 50% glycerol without appreciable loss of transcription or cleavage activities. Proteins are stored as small aliquots, and thawed only once.
In addition to identifying procedural changes which facilitate use of this method we also learned some lessons in how to analyze the cleavage data. It is important to prepare conjugates at a large number of sites on the protein. This makes it possible to evaluate the internal consistency and resolution of the cleavage patterns (conjugates at closely apposed amino acids should show very similar—but not identical—cleavage patterns; e.g. compare cleavage by 388 and 385 in Fig. 3). This can also help to distinguish between changes in cleavage patterns which are due to relative movements of the protein and nucleic acid vs. protein conformational changes. The latter are distinguished by the fact that they cause cleavage by conjugates on one part of the protein to change while cleavage by conjugates on other parts of the protein change very little or not at all (i.e., the change in T strand cleavage by 385/388 from -9 to -2 upon extension of the RNA from 4 to 6 nt in Fig. 3A is ascribed to a conformational change in the thumb because cleavage by other conjugates changes very little at this point). The other important lesson was that accessibility is more important than proximity in determining whether cleavage is observed. Though cleavage diminishes with distance, the diffusible nature of the OH . allows cleavage to occur tens of angstroms from the site of OH . generation (see also 16). However, interposition of protein or nucleic acid between the site of OH . generation (the conjugate) and a cleavage target effectively blocks cleavage at the target. This method is therefore particularly useful for detecting conformational changes that alter the accessibility of a potential cleavage site to a particular conjugate. However, there are significant limitations to this methodology. If a protein has a large number of accessible cysteines which cannot be removed without loss of structural integrity or activity, and if Fe-BABE conjugation to such a protein leads to high background cleavage which interferes with detection of cleavage by conjugates at introduced cysteines, this may preclude use of this method. However, such a situation will also preclude most cross-linking or energy transfer approaches, which also rely on sulfhydryl chemistry to introduce reactants onto a protein surface. It is also almost essential to have at least some direct (X-ray or NMR) structural information on the complexes in question if one is to try to infer structural changes from changes in cleavage patterns. Without such information, the method is primarily useful for identifying proximity between parts of a protein and the macromolecules (DNA/RNA/protein) to which it binds, but is potentially misleading since absence of cleavage in such a case could simply reflect the presence of an obstruction between target and conjugate, even if the target and conjugate are close in space. Finally, it must be remembered that the interpretation of the changes in cleavage patterns is rather subjective and is guided by the principle that the change in a cleavage pattern is ascribed due to the simplest (smallest) conformational change which can explain the altered cleavage. This is distinct from energy transfer approaches which allow rigorous interpretation in terms of distance changes and, through a process of triangulation with multiple donor-acceptor pairs, allow absolute distances and orientation between donor and acceptor positions to be determined. It also means that if a particular step in a reaction is associated with a very large conformational change (i.e., refolding of a large part of a protein) then the Fe-BABE approach is very likely to detect such a change, but is less likely to reveal the detailed nature of the change.
In the case of T7RNAP we were able to study the progress of transcription initiation by using NTP limitation or 3’-dNMP incorporation to prepare intermediates in this reaction. However, this is not a generally applicable strategy, so an important question is whether this method can be used in time-resolved studies. Ongoing experiments in our laboratory indicate that this should be possible. In manual mixing experiments we have found that the amount of cleavage obtained 5 sec after initiation of cleavage is identical to that obtained in 16 min, and that addition of formamide to 50% v/v immediately quenches the reaction (though the ‘quenching’ may be due to disruption of the transcription complex rather than quenching of the chemistry). This indicates that this method can be used to monitor transitions occurring in the 1-10 sec range. To achieve even higher time resolution will require quench-flow instrumentation to monitor shorter reaction times than can be achieved by manual mixing.
Acknowledgments
Supported by NIHGMS 52522 (to R.S.) and funds from the Welch Foundation.
Appendix
Protocols
Purification of Mutant T7RNAPs
Solutions for purification are as follows:
Buffer A: 20mM sodium phosphate (pH 8.0), 1 mM DTT, 1 mM EDTA, 5% glycerol and 0.1mM phenylmethylsulfonylfluoride.
Storage buffer: 20 mM Tris-HCl (pH 8.0), 0.5 M NaCl, 5 mM DTT, 1 mM EDTA, 50% glycerol.
10% Polymin P, pH 8.0: Mix 1 part 100% polymin P with 6 parts H2O. Adjust pH to 8.0 with 10 N NaOH and HCl. Adjust final volume with H2O to give 10% v/v polymin P.
The night before protein expression plate out transformed cells on 4-6 LB agar plates with 100 μg/ml ampicillin.
In morning resuspend colonies from each plate in 10ml LB and use this to inoculate 12 liters of LB containing 100ug/ml ampicillin.
Grow cells at 37oC until OD600 reaches 0.4- 0.6.
Induce expression by adding isopropyl-b-D-thiogalactopyranoside to 1 mM.
Harvest cells 4-6 hours after induction by centrifugation at 7000xg for 10 min and store at -80˚C.
For purification thaw and resuspend cells in 200 ml Buffer A + 0.1 mM PMSF. Lyse cells by sonication for 60 sec on a Sonic Dismembrator (Fisher Scientific) with macrotip probe.
Add NaCl to 0.5 M, followed by 1/20th volume of a 10% v/v polymin P (pH 8.0) to precipitate nucleic acids. Stir at 4˚C for 5-10 min.
Centrifuge cell lysate at 20,000xg for 15 min at 4˚C. Add 4 grams of ammonium sulfate for every 10 ml of clarified cell lysate and stir for 20 min at 4oC to dissolve the salt. Keep on ice for additional 30 min for protein precipitation and centrifuge at 20,000xg for 20 min.
Resuspend the precipitate in enough buffer A to give a final conductivity equal to 180 mM NaCl (typically 150-200 mls of buffer A).
Load the protein solution onto a 100ml phosphocellulose P11 (Whatman) column equilibrated with buffer A+150 mM NaCl. Wash the column with 500ml of Buffer A+150 mM NaCl and elute RNAP with a 750ml linear gradient of 150-950 mM NaCl in Buffer A.
The fractions containing T7RNAP are identified by SDS-PAGE, pooled, and dialyzed against Buffer A for 3-6 hrs until the conductivity of the solution is < BufferA+20 mM NaCl.
Load sample onto a 50ml Q-sepharose column (Amersham Pharmacia Biotech) equilibrated with buffer A, wash with 100 ml buffer A+20 mM NaCl, and elute with a 500 ml linear gradient of 20-500 mM NaCl in Buffer A.
Pool the fractions containing T7RNAP based on their purity on SDS gel, concentrate to 1-4 mg/ml and dialyze overnight against storage buffer.
Store purified proteins as aliquots at -80˚C until needed.
Conjugation with FeBABE
Thiol containing reducing agents such as 2 mercaptoethanol or DTT are removed before conjugation with FeBABE. Each purified mutant protein in storage buffer is exchanged to the same buffer without DTT either by dialysis or by using microSpin TM G-25 columns (Amersham Pharmacia Biotech).
Equilibrate microSpin TM G-25 columns with storage buffer without DTT.
Add 140ul of protein solution diluted in storage buffer without DTT to the column and centrifuge at 3000rpm for 2 min. Recover DTT-free eluate containing T7RNAP.
Incubate protein with 5 fold molar excess of FeBABE (20 mM in DMSO; Dojindo Laboratories, Japan) at 37oC for 1 hr. The final DMSO concentration is kept <5% to prevent protein precipitation.
Remove unreacted FeBABE by using a microspin column to transfer the protein into storage buffer with 1mM DTT.
Store conjugated protein in aliquots at -80˚C.
Free thiol group assay of FeBABE conjugated enzyme
The concentration of free sulfhydryl group (solvent accessibility) of each single cysteine mutant was determined by DTNB assay (Ellman’s reagent).
Prepare 4mg/ml DTNB solution in 0.1M sodium phosphate pH 8.0, 1 mM EDTA.
Add 850ul 0.1 M sodium phosphate pH 8.0, 1mM EDTA to cuvette and adjust A412 to zero.
Add 100ul of DTNB solution. Record A412 of DTNB solution.
Add 50ul of protein sample and mix. Record A412 until the reaction is complete (~10 min).
Calculate the concentration of reactive sulfhydryl groups according to: ε412=1.36x104M-1cm-1
Preparation of G+A Ladder
Add the following in a sterile micro centrifuge tube:
32P-labeled DNA (3-6ng): 1-8ul
Calf thymus or carrier DNA (0.5ug/ul): 2ul
H2O: 0-7ul
Total volume: 10ul
Add 1ul of 4% Formic acid and incubate at 37oC for 25 min (21ul of stock formic acid in 500ul of water).
During the incubation add 15ul of stock piperidine to 135ul of water to prepare a 1M piperidine solution.
Place the tube containing the reaction on ice, add 150 ul of diluted piperidine and incubate at 90oC for 30 min.
Place the reaction on ice for 5 min and add 3M Na-Acetate (pH5.2) to a final concentration of 0.3M.
Add 2 volumes of ice cold ethanol and vortex. Place at -80oC for 30 min.
Centrifuge at 10,000rpm for 10 min. Carefully remove the supernatant.
Add 70% Ethanol. Centrifuge 2 min at 12,000g. Repeat twice.
Air dry the DNA and re-suspend in 1x formamide stop solution.
DNA Cleavage by Fe-BABE
Transcription buffer : 10mM Tris-HCl (pH 8.0), 10mM NaCl, 6mM MgCl2, 5mM DTT, 2mM spermidine.
2x formamide stop solution: 95% formamide, 20mM EDTA, 0.01% xylene cyanol.
TBE buffer: 89mM Tris-borate, 2mM EDTA (pH 8.0).
To synthesize different lengths of RNA, different NTP combination were used:
1mer RNA: 3'd GTP
4mer RNA: GTP, 3'd ATP
6mer RNA: GTP, ATP
7mer RNA: GTP, ATP, 3'd CTP
13mer RNA: GTP, ATP, CTP, 3'd UTP
Mix 32P-labeled DNA (1x10-7M), RNAP (1x10-6 M), transcription buffer, nucleotides (0.5mM) together and incubate at 37oC for 3 min and split into 2 aliquots.
Add 100mM ascorbic acid to 10mM, and 200mM hydrogen peroxide to 25mM to one aliquot and incubate at 37oC for 2 min.
Quench with an equal volume of 2x formamide stop solution.
Heat reactions for 5 min in boiling water bath and immediately place on ice.
Resolve reactions by electrophoresis at 70 W for 2 hrs. on pre-warmed 17.5% polyacrylamide, 0.8% bis-acrylamide, 7M urea gels in 1xTBE.
Visualize and analyze gels with a Molecular Dynamics Phosphorimager.
RNA Cleavage By Fe-BABE
In Initial Transcription Complex
Mix T7RNAP and supercoiled pT7-7 plasmid at 1x10-7 M and 3x10-7 M, respectively, in transcription buffer and incubate at room temperature for 10 min.
Add 0.5 mM GTP and ATP and incubate further for 3 min.
Cool on ice for 2min.
Add 0.5 mM 3'd UTP and 1% v/v α-32PCTP (800 Ci/mM and 10mCi/ml) and immediately split reaction into 2 aliquots.
Add 100mM ascorbic acid to 10mM, and 200mM hydrogen peroxide to 25mM to one aliquot and incubate at 37oC for 1 min. The other aliquot is kept as control.
Stop reaction with an equal volume of 2x formamide stop solution and resolve on 20% polyacrylamide, 1% bis-acrylamide, 6 M urea gels.
Visualize and analyze gels with a Molecular Dynamics Phosphorimager.
In Elongation Complex
Mix T7RNAP and pPK7 template at 0.1 and 0.3uM respectively in transcription buffer and incubate at room temperature for 10 min.
Add 0.5 mM GTP, ATP, 3’d UTP and 1% v/v α-32PCTP (800 Ci/mM at 10 mCi/ml), incubate for 4 min at 37oC and split into 2 aliquots.
Add 100mM ascorbic acid to 10mM and 200mM hydrogen peroxide to 25mM to one aliquot and incubate at 37oC for 1 min. The other aliquot is kept as a control.
Stop reaction with an equal volume of 2x formamide stop solution and resolve on 20% polyacrylamide, 1% bis-acrylamide, 6 M urea gels.
References
- Greiner DP, Miyake R, Moran JK, Jones AD, Negishi T, Ishihama A, Meares CF. Synthesis of the protein cutting reagent iron (S)-1-(p-bromoacetamidobenzyl) ethylenediamine-tetraacetate and conjugation to cysteine side chains. Bioconjug Chem. 1997;8:44–48. doi: 10.1021/bc9600731. [DOI] [PubMed] [Google Scholar]
- Sousa R, Chung YJ, Rose JP, Wang BC. Crystal structure of bacteriophage T7 RNA polymerase at 3.3 Å resolution. Nature. 1993;364:593–599. doi: 10.1038/364593a0. [DOI] [PubMed] [Google Scholar]
- Cheetham GM, Jeruzalmi D, Steitz TA. Structural basis for initiation of transcription from an RNA polymerase-promoter complex. Nature. 1999;399:80–83. doi: 10.1038/19999. [DOI] [PubMed] [Google Scholar]
- Cheetham G, Steitz TA. Structure of a transcribing T7 RNA polymerase-promoter complex. Science. 1999;286:2305–2309. doi: 10.1126/science.286.5448.2305. [DOI] [PubMed] [Google Scholar]
- Cheetham G, Steitz TA. Structure of T7 RNA polymerase complexed to the transcriptional inhibitor T7 lysozyme. EMBO J. 1998;17:4101–4113. doi: 10.1093/emboj/17.14.4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin YW, Steitz TA. Structural Basis for the Transition from Initiation to Elongation Transcription in T7 RNA Polymerase. Science. 2002;298:1387–1395. doi: 10.1126/science.1077464. [DOI] [PubMed] [Google Scholar]
- Tahirov TH, Temiakov D, Anikin M, Patlan V, Mcallister WT, Vassylyev DG, Yokoyama S. Structure of a T7 RNA polymerase elongation complex at 2.9 Å resolution. Nature. 2002;420:43–50. doi: 10.1038/nature01129. [DOI] [PubMed] [Google Scholar]
- Muhkerjee S, Brieba LG, Sousa R. Structural Transitions Mediating Transcription Initiation by T7 RNA Polymerase. Cell. 2002;110:81–91. doi: 10.1016/s0092-8674(02)00815-2. [DOI] [PubMed] [Google Scholar]
- Bonner G, Patra D, Lafer EM, Sousa R. Mutations in T7 RNA Polymerase that Support the Proposal for a Common Polymerase Active Site Structure. EMBO J. 1992;11:3767–3775. doi: 10.1002/j.1460-2075.1992.tb05462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GC, Martin CT, Pham TT, Coleman JE. Transcription by T7 RNA polymerase is not zinc-dependent and is abolished on amidomethylation of cysteine-347. Biochemistry. 1986;25:36–40. doi: 10.1021/bi00349a006. [DOI] [PubMed] [Google Scholar]
- Reiner CK, Kada G, Gruber HJ. Quick measurement of protein sulfhydryls with Ellman's reagent and with 4,4'-dithiodipyridine. Anal Bioanal Chem. 2002;373:266–276. doi: 10.1007/s00216-002-1347-2. [DOI] [PubMed] [Google Scholar]
- Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- Brieba LG, Sousa R. T7 Promoter release mediated by DNA scrunching. EMBO J. 2001;20:6826–6835. doi: 10.1093/emboj/20.23.6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentesanas PE, Chin-Bow ST, Sousa R, Mcallister WT. Characterization of Bacteriophage T7 RNA Polymerase Elongation Complexes. J Mol Biol. 2000;302:1049–1062. doi: 10.1006/jmbi.2000.4114. [DOI] [PubMed] [Google Scholar]
- Place C, Oddos J, Buc H, McAllister WT, Buckle M. Studies of contacts between T7 RNA polymerase and its promoter reveals features in common with multisubunit RNA polymerases. Biochemistry. 1999;38:4948–4857. doi: 10.1021/bi982689e. [DOI] [PubMed] [Google Scholar]
- Brieba LG, Sousa R. The T7 RNA polymerase intercalating hairpin is important for promoter opening during initiation but not for RNA displacement or transcription bubble stability during elongation. Biochemistry. 2001;40:3882–3890. doi: 10.1021/bi002716c. [DOI] [PubMed] [Google Scholar]
- Han H, Dervan PB. Visualization of RNA tertiary structure by RNA-EDTA-Fe(II) autocleavage: Analysis of tRNAPhe with uridine-EDTA-Fe(II) at position 47. Proc Natl Acad Sci USA. 1994;91:4955–4959. doi: 10.1073/pnas.91.11.4955. [DOI] [PMC free article] [PubMed] [Google Scholar]