

The title compound, bis(tetrahydrofuran)(5,10,15,20-tetra-p-tolylporphyrinato) iron(II), [Fe(C48H36N4)(C4H8O)2] or [Fe(TTP)(THF)2] (TTP = dianion of meso-tetratolylporphyrin), is a six-coordinate high-spin iron(II) tetratolylporphyrin with two tetrahydrofuran molecules as axial ligands. The FeII atom is at an inversion center. The average Fe—N distance is 2.067 (7) Å and the Fe—O distance is 2.3208 (8) Å.
Comment
Reed et al. 1980 reported the structure of the first six-coordinate high-spin iron(II) porphyrinate, [Fe(TPP)(THF)2]. Subsequently, Lecomte et al. 1986 performed an electron-density study of the compound. In this paper, we report the molecular structure of another six-coordinate high-spin iron(II) porphyrinate, the title compound, (I), [Fe(TTP)(THF)2].
The molecular structure of (I), with the atom-numbering scheme, is shown in Fig. 1. The FeII atom is located at a crystallographic inversion center. As a result, the FeII atom is centered in the porphyrin plane. The independent Fe—N bond distances are 2.0619 (9) and 2.0713 (9) Å, which are similar to those in [Fe(TPP)(THF)2] (2.054 and 2.060 Å; Reed et al., 1980). These values are substantially longer than the value of 1.996 (6) Å in the low-spin six-coordinate iron(II) porphyrinate [Fe(TPP)(THT)2] (THT is tetrahydrothiophene; Mashiko et al., 1979).
Figure 1.
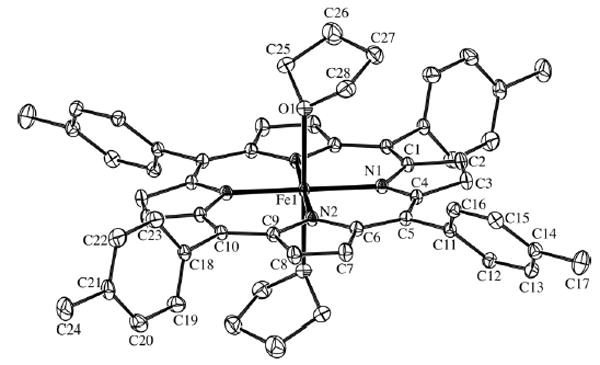
The molecular structure of (I), showing 50% probability displacement ellipsoids. H atoms have been omitted. Unlabeled atoms are related by the symmetry operation −x, 1 − y, −z.
The axial Fe—O bond distance [2.3208 (8) Å] is similar to the value of 2.351 (3) Å in [Fe(TPP)(THF)2] (Reed et al., 1980). This bond distance indicates that the THF ligand is a weak-field ligand. The Fe—O vector is tipped by 1.3 (1)° from the normal to the mean plane through the plane of the four N atoms. This is smaller than the 4.3° tip seen in [Fe(TPP)(THF)2] (Reed et al., 1980). This deviation also breaks the D4h symmetry of the FeN4O2 coordination. The THF ligand has an envelope configuration with approximate Cs symmetry. The O atom is displaced from the mean plane of the four C atoms by 0.56 (3) Å.
Experimental
All experimental manipulations were performed under a purified nitrogen atmosphere using standard Schlenk techniques. [Fe(TTP)]2O (35 mg) was reduced in tetrahydrofuran (10 ml) in the presence of excess EtSH overnight. The solution was transfered into glass tubes and layered with hexane. A week later, high-quality red crystals of (I) formed.
| Crystal data | |
| [Fe(C48H36N4)(C4H8O)2] | Dx = 1.312 Mg m−3 |
| Mr = 868.87 | Mo Kα radiation |
| Monoclinic, P21/n | Cell parameters from 7064 reflections |
| a = 13.6820 (2) Å | |
| b = 9.7320 (1) Å | θ = 2.5–31.5° |
| c = 17.1014 (2) Å | μ = 0.39 mm−1 |
| β = 104.964 (1)° | T = 100 (2) K |
| V = 2199.89 (5) Å3 | Needle, red |
| Z = 2 | 0.49 × 0.15 × 0.09 mm |
| Data collection | |
| Bruker SMARTAPEX2 CCD area-detector diffractometer | 7311 independent reflections 6283 reflections with I > 2σ(I) |
| ϕ and ω scans | Rint = 0.028 |
| Absorption correction: multi-scan | θmax = 31.5° |
| (SADABS; Sheldrick, 2004) | h = −20 → 17 |
| Tmin = 0.83, Tmax = 0.97 | k = −11 → 14 |
| 31 446 measured reflections | l = −22 → 25 |
| Refinement | |
| Refinement on F2 | w = 1/[σ2(Fo2) + (0.0487P)2 + 1.0408P] |
| R[F2 > 2σ(F2)] = 0.038 | |
| wR(F2) = 0.102 | where P = (Fo2 + 2Fc2)/3 |
| S = 1.04 | (Δ/σ)max = 0.001 |
| 7311 reflections | Δρ max = 0.53 e Å−3 |
| 288 parameters | Δρ min = −0.31 e Å−3 |
| H-atom parameters constrained | |
H atoms were positioned geometrically and treated as riding, with C—H distances in the range 0.95–0.99 Å and with Uiso(H) = 1.2Ueq(C).
Data collection: APEX2 (Bruker Nonius, 2004); cell refinement: APEX2 and SAINT (Bruker Nonius, 2004); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: PLATON (Spek, 2003); software used to prepare material for publication: SHELXL97.
Table 1.
Selected geometric parameters (Å, °).
| Fe1—N1 | 2.0619 (9) | Fe1—O1 | 2.3208 (8) |
| Fe1—N2 | 2.0713 (9) | ||
| N1—Fe1—N2 | 89.57 (4) | N2—Fe1—O1 | 88.96 (3) |
| N1—Fe1—O1 | 89.34 (3) |
Acknowledgments
The authors thank the US National Institutes of Health for support of this research under grant No. GM-38401.
Footnotes
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
References
- Bruker Nonius (2004). APEX2 and SAINT Bruker Nonius AXS, Madison, Wisconsin, USA.
- Lecomte C, Blessing RH, Coppens P, Tabard A. J Am Chem Soc. 1986;108:6942–6950. [Google Scholar]
- Mashiko T, Marchon JC, Musser DT, Reed CA, Kastner ME, Scheidt WR. J Am Chem Soc. 1979;101:3653–3655. [Google Scholar]
- Reed CA, Mashiko T, Scheidt WR, Spartalian K, Lang G. J Am Chem Soc. 1980;102:2302–2306. [Google Scholar]
- Sheldrick, G. M. (1997). SHELXS97 and SHELXL97 University of Göttingen, Germany.
- Sheldrick, G. M. (2004). SADABS Version 2.10. University of Göttingen, Germany.
- Spek AL. J Appl Cryst. 2003;36:7–13. [Google Scholar]