

Abstract
Cisplatin (CDDP), an anticancer drug, induces remarkable toxicity in the kidneys of animals and humans and it has been well documented that reactive oxygen species and the renal antioxidant system are strongly involved in acute renal damage induced by CDDP. The aim of the present study was to investigate whether or not the renal antioxidant system plays also an important role in chronic renal damage induced by repeated doses of CDDP (1 mg/kg intraperitoneally twice weekly during 10 weeks in rats). In order to elucidate it, serum creatinine and urea levels, renal glutathione and thiobarbituric acid-reactive substances (TBARS) content, as well as renal superoxide dismutase and glutathione peroxidase activities were measured in the kidney homogenates of chronically CDDP-treated rats and additionally histological studies were performed in the rat kidneys. The chronic treatment with CDDP induced a significant increase in creatinine and urea levels in serum, but the other parameters mentioned above were not significantly modified as compared to the values in nontreated rats. Taking into account these results, we conclude that chronic CDDP administration induces also severe nephrotoxicity, in contrast to CDDP acute application, without any significant modification in the activity of relevant antioxidant enzymes such as superoxide dismutase and glutathione peroxidase, renal glutathione and lipid peroxides, by which the role of the antioxidant system in chronic nephrotoxicity induced by CDDP in rats is uncertain.
INTRODUCTION
Cisplatin (cis-diamminedichloroplatinum II) (CDDP) is one of the most potent antitumor drugs [1]. High doses of CDDP produce the impairment of kidney function, which is recognized as the main side-effect and the most important dose-limiting factor [2]. The alterations induced by CDDP in the kidney functions were characterized by signs of injury, such as changes in urine volume, body weight, in glutathione status, increase of products of lipid peroxidation, and changes in creatinine clearance [3]. Histologically, the acute toxic tubular necrosis observed after CDDP administration is similar to that produced in intoxication by mercury or cadmium [4] suggesting that CDDP nephropathy may be attributable ultimately to the toxicity of the platinum molecules which induce damage in the proximal tubular cells.
CDDP is able to generate reactive oxygen species (ROS) such as superoxide anion and hydroxyl radical [5, 6, 7] and to inhibit the activity of antioxidant enzymes in renal tissue [8]. Furthermore, glutathione depletion and increase of thiobarbituric acid-reactive substances (TBARS) induced by CDDP were found and they are determinant steps in oxidative stress in the kidney tissue, which leads to nephrotoxicity [9]. However, in contrast to the former studies on acute nephrotoxicity induced by CDDP in rats, little has been done to investigate the role of ROS in the development of chronic nephrotoxicity induced in rats by this drug [10].
Taking into account that in human beings CDDP chemotherapy is carried out at doses in which the anti-tumor agent is already nephrotoxic (higher than 2 mg/kg) and that more than one cycle of drug administration is necessary to obtain effective results against solid tumors, we decided to investigate the behavior of the antioxidant enzymes superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px), the lipid peroxides and the glutathione (GSH) content in kidney homogenates from rats submitted to CDDP-induced chronic nephrotoxicity.
MATERIALS AND METHODS
Chemicals
Serum creatinine and urea were measured spectrophotometrically with assay kits purchased from the Carlos J Finlay, Biological Products Enterprise Havana, Cuba. All reagents used in determinations of GSH, SOD, GSH-Px, thiobarbituric acid-reactive substances (TBARS), and CDDP were purchased from Sigma Chemicals (St Louis, Mo). Other reagents of analytical grade were obtained from normal commercial sources.
Animals and treatments
Male Sprague-Dawley rats (200–250g) were used in the experiment. The animals were purchased from the National Center for Laboratory Animal Production (CENPALAB, Havana, Cuba), housed in an environmentally (t = 25°C) and air humidity (60%) controlled room with a 12-hour light-dark cycle, and kept on a standard laboratory diet and drinking water ad libitum. The experiments were conducted in accordance with the ethical guidelines for investigations in laboratory animals and were approved by the Ethical Committee for Animal Experimentation of the National Center for Scientific Research (CNIC).
Two groups of eight rats were used in the experiment: one control group without treatment, and one group receiving 1 mg/kg of CDDP twice weekly for 10 weeks as it was reported by Choie et al [4]. CDDP was injected IP and was dissolved in physiologic saline (1 mg/mL) within one hour before use. One week after ending the treatment period, blood was collected from the posterior cava vein in each animal, under ether anesthesia, to determine serum levels of blood urea and creatinine using assay kits. The kidneys were removed and used, one of them for biochemical assays and the other one for histopathological examination.
Biochemical assays
Kidney homogenates were obtained using a tissue homogenator Ultraturrax T-25 Polytron at 4°C. The homogenates (1:10 w/v) were prepared by using a 100 mM KCl buffer (pH 7) containing EDTA 0.3 mM (buffer 1), for GSH, TBARS, GSH-Px, and SOD determinations. All homogenates were centrifuged at 600g for 60 minutes at 4°C and the supernatants were used for biochemical assays.
Determination of GSH content
GSH was determined spectrophotometrically by a slightly modified version of the method of Beutler et al [11]. One mL of the kidney homogenate, as described before, was mixed with 1.5 mL of 5% metaphosphoric acid and centrifuged at 3000g for 10 minutes at room temperature. Five hundred microliters of this acidic supernatant was mixed with 2 mL of 0.2 M phosphate buffer and 0.25 mL of 0.04% 5,5′-dithiobis(-2-nitrobenzoic acid). Absorbance of the yellow solution was measured at 412 nm within 10 minutes. A molar extinction coefficient (13.6 Mcm−1) that describes the formation of the thiolate anion by the reaction of sulfhydryl groups with 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) at 412 nm was used to quantify GSH.
Determination of SOD activity
The SOD activity was determined by a modified version of the method of Minami and Yoshikawa [13]. Fifty microliters of the kidney homogenate was mixed with 450 μL of cold deionized water, 125 μL of chloroform, and 250 μL of ethanol. The mixture was centrifuged at 8000g for 2 minutes at 4°C. Five hundred microliters of the extract was added to the reaction mixture containing 500 μL of 72.4 mM tris-cacodylate buffer with 3.5 mM diethylene pentaacetic acid (pH 8.2), 100 μL of 16% TritonX-100, and 250 μL of 0.9 mM nitroblue tetrazolium (NBT). The reaction mixture was incubated for 5 minutes at 37°C before adding 10 μL of 9 mM pyrogallol (dissolved in 10 mM HCL). Then, it was incubated for exactly 5 minutes at 37°C. The reaction was stopped with the addition of 300 μL of 2 M formic buffer (pH 3.5) containing 16% TritonX-100. The absorbance was measured at 540 nm in the spectrophotometer. One unit of SOD enzymatic activity is equal to the amount of enzyme that diminishes the initial absorbance of nitroblue tetrazolium by 50%.
Dertermination of GSH-Px activity
GSH-Px was measured using a modified version of the method of Faraji et al [14]. All reaction mixtures were dissolved in 20 mM sodium phosphate buffer containing 6 mM EDTA (pH 7.0). The reaction mixture consisted of 98.8 μL of phosphate buffer, 700 μL of 2.86 mM GSH, 100 μL of 1 mM sodium azide, 100 μL of 1 mM NADPH, and 4.2 μL of GSH reductase (0.5 units). Then, 10 μL of the tissue homogenate supernatant was added to the reaction mixture and incubated at room temperature for 10–15 minutes. Afterward, 10 μL of 30 mM t-butyl hydroperoxide (dissolved in bidistilled water) was added to the reaction mixture and measured at 340 nm for 7 minutes in the spectrophotometer. A molar extinction coefficient of 6.22 × 103 M cm−1 was used to determine the activity of GSH-Px. The enzyme activity is expressed as international units of enzymatic activity/mg of protein. International units are expressed as μmol of hydroperoxides transformed per min/mL of enzyme.
Lipid peroxidation assay
This assay is used to determine thiobarbituric acid-reactive substances (TBARS) level as described by Ohkawa et al [12]. Two hundred microliters of tissue homogenate supernatant was added to 100 μL of sodium dodecyl sulfate (SDS), 750 μL of 20% acetic acid (pH 3.5), 750 μL of 0.6% thiobarbituric acid, and 300 μL of distilled water and were incubated at 95°C for 60 minutes. The samples were allowed to cool at room temperature. Then 2.5 mL of butanol: pyridine (15:1) and 500 μL of distilled water were added, vortexed, and centrifuged at 2000g for 15 minutes. The absorbance of 3 mL of the colored layer was measured at 532 nm spectrophotometrically using 1,1,3,3-tetraethoxypropane as a standard.
Protein assay
Protein concentrations were determined by the method of Lowry [15] using bovine serum albumin as a standard.
Histopathological examination
The left kidneys were quickly removed and fixed for 2 hours in a solution containing 150 mL ethanol, 60 mL formaldehyde, 15 mL acetic acid, and 1g picric acid. Thereafter the samples were included in phosphate-buffered formaldehyde until they were embedded in paraffin, sectioned at 3 μm, stained with hematoxylin and eosin (H/E), and evaluated by light microscopy.
Statistical analysis
Statistical analysis was performed using analysis of variance (ANOVA). Differences between treatments were determined by the Mann-Whitney test. The results were expressed as the mean ± SD of eight values in each group and a statistical probability of P < .05 was considered to be significant.
RESULTS
Table 1 shows the changes in serum creatinine, blood urea nitrogen, and body weight after chronic treatment with CDDP during 10 weeks. In CDDP-administered rats, serum creatinine and urea concentrations were increased significantly above those of control rats. Mean urea level was about 5.9-fold higher in the CDDP (P < .001) group compared to the control group, and creatinine was increased about 2.4-fold (P < .001). Body weight of animals treated with CDDP was slightly lower than that of controls, but this decrease was not statistically significant.
Table 1.
Nephrotoxicity after chronic treatment with CDDP. Rats were injected IP with CDDP (1 mg/kg) twice weekly for 10 weeks and sacrificed one week after the last dose. Values are means ± SD. N = 8. ∗ denotes that P < .05.
| Group | Body weight (g) | Creatinine (μM) | Urea (mM) |
| Mean ± SD | Mean ± SD | Mean ± SD | |
| Control | 474.5 ± 93.9 | 113.4 ± 4.9 | 6.6 ± 0.64 |
| CDDP (1 mg/kg) | 417.2 ± 37.8 | 281.2 ± 45.4∗ | 39.1 ± 19.9∗ |
Table 2 shows antioxidant status of rat kidney after chronic treatment with CDDP. SOD and GSH values were not significantly modified with respect to those of control rats. TBARS, which indicate the extent of lipid peroxidation, also remained unchanged in the kidney of animals chronically treated with CDDP. The GSH-Px activity was little decreased in the CDDP group, but this decrease was not statistically significant.
Table 2.
Antioxidant status of rat kidney after chronic treatment with CDDP. Each value represents mean ± SD of eight animals.
| TBARS | SOD | GSH | GSH-Px | |
| Group | (nmol/mg prot) | (U/μg prot) | (nmol/mg prot) | (UI/mg prot) |
| Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | |
| Control | 0.3 ± 0.08 | 7.4 ± 0.99 | 8.8 ± 1.68 | 12.2 ± 2.61 |
| Cisplatin (1 mg/kg) | 0.3 ± 0.08 | 8.7 ± 1.70 | 8.9 ± 1.50 | 9.8 ± 2.02 |
The toxic effect of chronic CDDP administration was confirmed by the detection of morphologic alterations in kidney slices of treated animals. Figure 1 shows the normal histological feature of a kidney belonging to a nontreated control rat.
Figure 1.

Normal histological structure of rat kidney. HE 250X.
The kidneys of rats chronically treated with CDDP were enlarged and the surface was markedly granular. Histologically, the most characteristic feature was massive tubular dilation in the corticomedullary region and also in the cortical area accompanied with flattening and necrosis of tubular epithelial cells (Figure 2). Also, mononuclear cell infiltration and interstitial fibrosis between the dilated tubules were observed (Figure 3).
Figure 2.
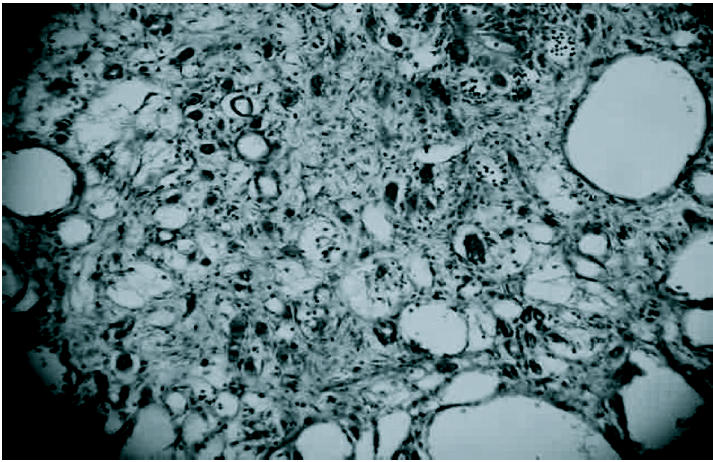
Massive tubular dilation accompanied with flattening and necrosis of tubular epithelial cells in kidney of rat chronically treated with CDDP. HE 250X.
Figure 3.
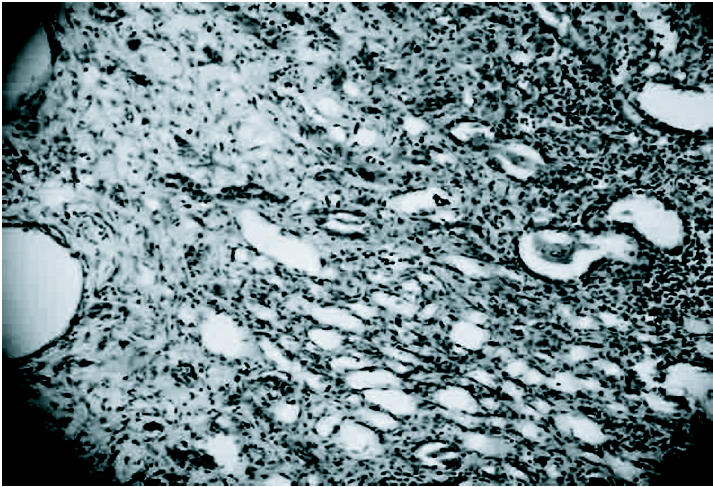
Mononuclear cell infiltration and interstitial fibrosis between the dilated tubules in kidney of rat chronically treated with CDDP. HE 250X.
DISCUSSION
Using an experimental model of CDDP-induced chronic nephrotoxicity in rats, we evaluated the renal activity of the key antioxidant enzymes (SOD, GSH-Px), the glutathione level and TBARS content of tissue as an index of lipid peroxidation.
Our results demonstrated that repeated treatment with CDDP (1 mg/kg, intraperitoneally, twice weekly) for 10 weeks induced biochemical and histological signs of renal injury (Table 1, Figures 2 and 3) without any significant reduction in the activity of SOD and GSH-Px. Neither GSH nor lipid peroxides levels were significantly modified in this chronic model of renal failure (Table 2).
However, several studies demonstrated that CDDP-induced acute nephrotoxicity is mediated by depletion of renal GSH and by impaired activity of SOD and GSH-Px as well as an increase in renal lipid peroxidation [16, 17, 18, 19, 20]. In relation to renal GSH levels, whereas some reports show GSH depletion in acute CDDP nephrotoxicity, others show increased or normal GSH levels under the same experimental conditions. According to Meyer and Madias [2] these observations are not necessarily contradictory; they may represent different stages of the toxic response to CDDP.
On the other hand, a possible explanation for the absence of changes in most of the antioxidant/oxidant parameters measured in the rat kidney may be an adaptation to the continuous stress induced by treatment with low doses of CDDP.
Trachtman et al [10] examined whether there is increased production of ROS during chronic renal failure induced in rats subjected to 3/4 nephrectomy three weeks after surgery. This study revealed no detectable renal production of ROS in rats with chronic renal failure. Thus, the role of lipid peroxidation and its position in the chain of events that leads to CDDP nephrotoxicity still remains controversial. However, Kruidering et al [21] have suggested that although free-radical generation is not the direct cause of cisplatin-induced renal injury, it may contribute to CDDP nephrotoxicity, since the in vivo administration of antioxidants to animals can attenuate or inhibit the renal damage.
Also induction of heme oxygenase-1 (HO-1) is thought to serve as an immediate protective response against the nephrotoxicity of CDDP. Heme oxygenase (HO) catalyzes the first and rate-limiting step in the degradation of heme to biliverdin and bilirubin, respectively. Several studies have ascribed the protective effect of HO activity against oxidative-stress-related toxicity to increased levels of bilirubin [22]. Its HO-1 isoenzyme is a widely distributed heat-shock protein and is induced by a variety of stimuli, including UV irradiation, heavy metals, inflammation, and pro-oxidant states [23]. The assumption that HO may serve as a safeguard of cellular redox levels concurs with the reports describing the first case of human hereditary HO-1 deficiency showing the important role of HO-1 in protecting the kidney against chronic low-level oxidative insult [24]. Moreover, studies using transgenic mice deficient in HO-1 (-/-) showed that absence of HO-1 resulted in more severe renal failure and greater renal injury as compared to wild-type mice treated with CDDP. Furthermore, increased HO activity was shown to protect against the nephrotoxicity of CDDP in rats while inhibition of HO exacerbated its toxic effect [25, 26]. Currently research is in progress in our laboratory in order to elucidate the modulation of this inducible enzyme in the course of chronic renal nephrotoxicity induced by CDDP.
Taking into account the results of this study, we can conclude that chronic CDDP administration produces severe nephrotoxicity in rats without any significant modification in the activity of endogenous antioxidant enzymes, renal GSH and lipid peroxides. Further studies are needed to clarify whether the antioxidant enzymes within the rat kidneys can be modulated in CDDP-induced chronic renal failure and, if so, how it will be relevant in the early course of chronic renal failure.
References
- 1.Lebwohl D, Canetta R. Clinical development of platinum complexes in cancer therapy: an historical perspective and an update. Eur J Cancer. 1998;34(10):1522–1534. doi: 10.1016/s0959-8049(98)00224-x. [DOI] [PubMed] [Google Scholar]
- 2.Meyer KB, Madias NE. Cisplatin nephrotoxicity. Miner Electrolyte Metab. 1994;20(4):201–213. [PubMed] [Google Scholar]
- 3.Kersten L, Bräunlich H, Keppler BK, Gliesing C, Wendelin M, Westphal J. Comparative nephrotoxicity of some antitumour-active platinum and ruthenium complexes in rats. J Appl Toxicol. 1998;18(2):93–101. doi: 10.1002/(sici)1099-1263(199803/04)18:2<93::aid-jat472>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 4.Choie DD, Longnecker DS, del Campo AA. Acute and chronic cisplatin nephropathy in rats. Lab Invest. 1981;44(5):397–402. [PubMed] [Google Scholar]
- 5.Masuda H, Tanaka T, Takahama UT. Cisplatin generates superoxide anion by interaction with DNA in a cell-free system. Biochem Biophys Res Commun. 1994;203(2):1175–1180. doi: 10.1006/bbrc.1994.2306. [DOI] [PubMed] [Google Scholar]
- 6.Baliga R, Zhang Z, Baliga M, Ueda N, Shah S. In vitro and in vivo evidence suggesting a role for iron in cisplatin-induced nephrotoxicity. Kidney Int. 1998;53(2):394–401. doi: 10.1046/j.1523-1755.1998.00767.x. [DOI] [PubMed] [Google Scholar]
- 7.Matsushima H, Yonemura K, Ohishi K, Hishida A. The role of oxygen free radicals in cisplatin-induced acute renal failure in rats. J Lab Clin Med. 1998;131(6):518–526. doi: 10.1016/s0022-2143(98)90060-9. [DOI] [PubMed] [Google Scholar]
- 8.Appenroth D, Fröb S, Kersten L, Splinter EK, Winnefeld K. Protective effects of vitamin E and C on cisplatin nephrotoxicity in developing rats. Arch Toxicol. 1997;71(11):677–683. doi: 10.1007/s002040050444. [DOI] [PubMed] [Google Scholar]
- 9.Zhang JG, Lindup WE. Role of mitochondria in cisplatin-induced oxidative damage exhibited by rat renal cortical slices. Biochem Pharmacol. 1993;45(11):2215–2222. [PubMed] [Google Scholar]
- 10.Trachtman H, Wilson D, Rao PS. The role of oxygen free radicals in the development of chronic renal failure. Life Sci. 1992;50(24):1877–1883. doi: 10.1016/0024-3205(92)90548-4. [DOI] [PubMed] [Google Scholar]
- 11.Beutler F, Duron O, Mikus B. Improved method for the determination of blood glutathione. J Lab Clin Med. 1963;16(6):882–888. [PubMed] [Google Scholar]
- 12.Ohkawa H, Orishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95(2):351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 13.Minami M, Yoshikawa H. A simplified assay method of superoxide dismutase activity for clinical use. Clin Chim Acta. 1979;92(3):337–342. doi: 10.1016/0009-8981(79)90211-0. [DOI] [PubMed] [Google Scholar]
- 14.Faraji B, Kang HK, Valentine JL. Methods compared for determining glutathione peroxidase activity in blood. Clin Chem. 1987;33(4):539–543. [PubMed] [Google Scholar]
- 15.Lowry OH, Rosebrough NJ, Far AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 16.Somani SM, Husain K, Whitworth C, Trammel GL, Malafa M, Rybak LP. Dose-dependent protection by lipoic acid against cisplatin-induced nephrotoxicity in rats: antioxidant defense system. Pharmacol Toxicol. 2000;86(5):234–241. doi: 10.1034/j.1600-0773.2000.d01-41.x. [DOI] [PubMed] [Google Scholar]
- 17.Saad SY, Al-Rikabi AC. Protection effects of taurine supplementation against cisplatin-induced nephrotoxicity in rats. Chemotherapy. 2002;48(1):42–48. doi: 10.1159/000048587. [DOI] [PubMed] [Google Scholar]
- 18.Davis CA, Nick HS, Agarwal A. Manganese superoxide dismutase attenuates cisplatin-induced renal injury: importance of superoxide. J Am Soc Nephrol. 2001;12(12):2683–2690. doi: 10.1681/ASN.V12122683. [DOI] [PubMed] [Google Scholar]
- 19.Borrego A, Zamora ZB, González R, et al. Protection by ozone preconditioning is mediated by the antioxidant system in cisplatin-induced nephrotoxicity in rats. Mediators Inflamm. 2004;13(1):13–19. doi: 10.1080/09629350410001664806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez R, Borrego A, Zamora ZB, et al. Reversion by ozone treatment of acute nephrotoxicity induced by cisplatin in rats. Mediators Inflamm. 2004;13(5-6):307–312. doi: 10.1155/S0962935104000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kruidering M, Van de Water B, De Heer E, Mulder GJ, Nagelkerke JF. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J Pharmacol Exp Ther. 1997;280(2):638–649. [PubMed] [Google Scholar]
- 22.Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 gene ablation and expression. J Am Soc Nephrol. 2000;11(5):965–973. doi: 10.1681/ASN.V115965. [DOI] [PubMed] [Google Scholar]
- 23.Dennery PA. Regulation and role of heme oxygenase in oxidative injury. Curr Top Cell Regul. 2000;36:181–199. doi: 10.1016/s0070-2137(01)80008-x. [DOI] [PubMed] [Google Scholar]
- 24.Ohta K, Yachie A, Fujimoto K, et al. Tubular injury as a cardinal pathologic feature in human heme oxygenase-1 deficiency. Am J Kidney Dis. 2000;35(5):863–870. doi: 10.1016/s0272-6386(00)70256-3. [DOI] [PubMed] [Google Scholar]
- 25.Agarwal A, Balla J, Alam J, Croatt AJ, Nath KA. Induction of heme oxygenase in toxic renal injury: a protective role in cisplatin nephrotoxicity in the rat. Kidney Int. 1995;48(4):1298–1307. doi: 10.1038/ki.1995.414. [DOI] [PubMed] [Google Scholar]
- 26.Schaaf GJ, Maas RF, De Groene EM, Fink-Gremmels J. Management of oxidative stress by heme oxygenase-1 in cisplatin-induced toxicity in renal tubular cells. Free Radic Res. 2002;36(8):835–843. doi: 10.1080/1071576021000005267. [DOI] [PubMed] [Google Scholar]