

Abstract
When polymorphism and divergence data are available for multiple loci, extended forms of the McDonald–Kreitman test can be used to estimate the average proportion of the amino acid divergence due to adaptive evolution—a statistic denoted 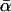 . But such tests are subject to many biases. Most serious is the possibility that high estimates of
. But such tests are subject to many biases. Most serious is the possibility that high estimates of  reflect demographic changes rather than adaptive substitution. Testing for between-locus variation in α is one possible way of distinguishing between demography and selection. However, such tests have yielded contradictory results, and their efficacy is unclear. Estimates of
reflect demographic changes rather than adaptive substitution. Testing for between-locus variation in α is one possible way of distinguishing between demography and selection. However, such tests have yielded contradictory results, and their efficacy is unclear. Estimates of 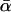 from the same model organisms have also varied widely. This study clarifies the reasons for these discrepancies, identifying several method-specific biases in widely used estimators and assessing the power of the methods. As part of this process, a new maximum-likelihood estimator is introduced. This estimator is applied to a newly compiled data set of 115 genes from Drosophila simulans, each with each orthologs from D. melanogaster and D. yakuba. In this way, it is estimated that
from the same model organisms have also varied widely. This study clarifies the reasons for these discrepancies, identifying several method-specific biases in widely used estimators and assessing the power of the methods. As part of this process, a new maximum-likelihood estimator is introduced. This estimator is applied to a newly compiled data set of 115 genes from Drosophila simulans, each with each orthologs from D. melanogaster and D. yakuba. In this way, it is estimated that 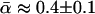 , a value that does not vary substantially between different loci or over different periods of divergence. The implications of these results are discussed.
, a value that does not vary substantially between different loci or over different periods of divergence. The implications of these results are discussed.
THE McDonald–Kreitman test (McDonald and Kreitman 1991; Kreitman and Akashi 1995) is an important technique for quantifying the contribution of positive Darwinian selection to molecular evolution. The test compares levels of polymorphism within a species to measures of divergence between species and relies on the assumption that a certain class of mutations can be treated as effectively neutral, a priori. Following McDonald and Kreitman, most studies have focused on protein-coding sequences and used synonymous mutations as their assumed-neutral referent. As such, the tests compare levels of synonymous polymorphism (Ps) and divergence (Ds) with their nonsynonymous (amino acid changing) equivalents (Pn and Dn). The focus of many studies has been to estimate the proportion of the nonsynonymous divergence, Dn, that was due to adaptive evolution, a statistic that is denoted α.
A serious problem with these tests is that levels of polymorphism are typically low in most population samples at most loci, especially if rare variants are excluded, and this means that single-locus estimates of α can be unreliable. To solve this problem, many methods of combining data from multiple loci have been introduced (Fay et al. 2001; Bustamante et al. 2002; Smith and Eyre-Walker 2002; Sawyer et al. 2003; Bierne and Eyre-Walker 2004). Such methods can be used to estimate  , the average value of α across the sampled loci.
, the average value of α across the sampled loci.
However, it is now clear that different variants of the test have given different results when applied to data from the same model organism. Consider, for example, published results using polymorphism data from Drosophila simulans. Smith and Eyre-Walker (2002) introduced a heuristic estimator of 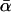 that they applied to a data set of 35 loci. Measuring divergence from D. yakuba, they estimated that
that they applied to a data set of 35 loci. Measuring divergence from D. yakuba, they estimated that 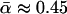 (i.e., that ∼45% of the divergence between D. simulans and D. yakuba was driven by positive selection). In contrast, Fay et al. (2002) used their own earlier estimator (Fay et al. 2001) on the 23-locus data set of Begun (2001), with divergence measured from the common ancestor with D. melanogaster; they obtained an estimate of
(i.e., that ∼45% of the divergence between D. simulans and D. yakuba was driven by positive selection). In contrast, Fay et al. (2002) used their own earlier estimator (Fay et al. 2001) on the 23-locus data set of Begun (2001), with divergence measured from the common ancestor with D. melanogaster; they obtained an estimate of  . An even higher estimate was obtained by Sawyer et al. (2003), whose distinctive version of the test is set within a firm probabilistic framework (Sawyer and Hartl 1992; Bustamante et al. 2001). Using a set of 56 D. simulans loci, measuring divergence from D. melanogaster, they estimated that ∼94% of the nonsynonymous divergence was adaptively driven. Finally, Bierne and Eyre-Walker (2004) introduced a maximum-likelihood estimator, which they applied to several data sets. Their largest data set, of 75 D. simulans loci, yielded estimates close to that of Smith and Eyre-Walker (2002) when divergence was measured from D. melanogaster, but this increased to ∼
. An even higher estimate was obtained by Sawyer et al. (2003), whose distinctive version of the test is set within a firm probabilistic framework (Sawyer and Hartl 1992; Bustamante et al. 2001). Using a set of 56 D. simulans loci, measuring divergence from D. melanogaster, they estimated that ∼94% of the nonsynonymous divergence was adaptively driven. Finally, Bierne and Eyre-Walker (2004) introduced a maximum-likelihood estimator, which they applied to several data sets. Their largest data set, of 75 D. simulans loci, yielded estimates close to that of Smith and Eyre-Walker (2002) when divergence was measured from D. melanogaster, but this increased to ∼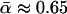 when divergence was measured along the simulans lineage alone.
when divergence was measured along the simulans lineage alone.
Just as different studies have yielded different estimates of 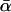 , there has also been disagreement about whether α varies significantly between loci. The existence of such between-locus variation is of great importance because of a criticism that can be leveled at the McDonald–Kreitman approach, namely that high estimates of
, there has also been disagreement about whether α varies significantly between loci. The existence of such between-locus variation is of great importance because of a criticism that can be leveled at the McDonald–Kreitman approach, namely that high estimates of  reflect not adaptive evolution, but rather changes in selective constraint over the history of the lineage (McDonald and Kreitman 1991; Eyre-Walker 2002; Fay et al. 2002). If average levels of selective constraint have increased, then contemporary polymorphism will reflect a different level of constraint compared to what prevailed over the period of divergence, and this biases upward estimates of
reflect not adaptive evolution, but rather changes in selective constraint over the history of the lineage (McDonald and Kreitman 1991; Eyre-Walker 2002; Fay et al. 2002). If average levels of selective constraint have increased, then contemporary polymorphism will reflect a different level of constraint compared to what prevailed over the period of divergence, and this biases upward estimates of 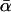 . While this may appear to be an ad hoc explanation for the high estimates that have appeared in the literature, it is far from implausible that an increase in selective constraint has occurred in many model organisms. This is because changes in effective population size can alter the proportion of mutations that are effectively neutral, with
. While this may appear to be an ad hoc explanation for the high estimates that have appeared in the literature, it is far from implausible that an increase in selective constraint has occurred in many model organisms. This is because changes in effective population size can alter the proportion of mutations that are effectively neutral, with 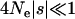 , and so demographic processes may have a major influence on selective constraint.
, and so demographic processes may have a major influence on selective constraint.
To discriminate between adaptive evolution and changes in Ne, Fay et al. (2002) argued that the former would be apparent at only a small subset of loci, while the latter would affect all loci. As such, they suggested, between-locus variation in α might indicate that adaptive evolution was truly the cause of high estimates of  . This argument is open to criticism, because the quantitative effects of, say, a population size increase might differ dramatically between loci, if the loci in question generate very different spectrums of selection coefficients (e.g., Gillespie 1991; Bierne and Eyre-Walker 2004). There is also the theoretical possibility that all of the sampled loci have undergone adaptive substitution at similar rates. Nevertheless, it is intuitively plausible that the effects of a demographic change would be more uniform across the genome than would the response to a novel selective pressure. As such, the presence or absence of substantial between-locus variation in α is relevant for the trustworthiness, or otherwise, of the high estimates of
. This argument is open to criticism, because the quantitative effects of, say, a population size increase might differ dramatically between loci, if the loci in question generate very different spectrums of selection coefficients (e.g., Gillespie 1991; Bierne and Eyre-Walker 2004). There is also the theoretical possibility that all of the sampled loci have undergone adaptive substitution at similar rates. Nevertheless, it is intuitively plausible that the effects of a demographic change would be more uniform across the genome than would the response to a novel selective pressure. As such, the presence or absence of substantial between-locus variation in α is relevant for the trustworthiness, or otherwise, of the high estimates of  .
.
Unfortunately, because of the high error variance associated with single-locus estimates, testing for significant between-locus variation is not trivial. Fay et al. (2002) addressed the problem by combining divergence measures from individual loci, with polymorphism values summed across all loci. Their results from 45 polymorphic loci from D. melanogaster were strikingly diverse, and so they concluded that high levels of adaptive substitution had occurred. However, the significance of the variation observed could not be determined quantitatively. Bierne and Eyre-Walker (2004) tackled the same problem in a different and more formal manner. Specifically, their maximum-likelihood approach, combined with model selection methods, allowed them to compare the fit of models where α varied across loci with models where α took a single fixed value. Intriguingly, and contrary to the findings of Fay et al. (2002), their data sets showed no evidence of significant between-locus variation. This result is particularly surprising in the light of the differing estimates of 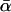 obtained by the studies mentioned above, each of which had many genes in common with the data set of Bierne and Eyre-Walker (2004).
obtained by the studies mentioned above, each of which had many genes in common with the data set of Bierne and Eyre-Walker (2004).
This study returns to the estimation of α-values in D. simulans and has two related goals. The first is to shed light on the different published estimates of 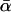 . In particular, we wish to determine whether such differences are due to the different assumptions or approximations employed by the methods or whether they reflect true differences in the data. The second goal is to explore the power of any method to test for significant between-locus variation in α. Particular attention is paid to the possibility that Bierne and Eyre-Walker's (2004) detection of a constant α across genes was an artifact, not of Drosophila demography, but of their estimation procedure—a possibility that is strong for a number of rather technical reasons, explained in what follows.
. In particular, we wish to determine whether such differences are due to the different assumptions or approximations employed by the methods or whether they reflect true differences in the data. The second goal is to explore the power of any method to test for significant between-locus variation in α. Particular attention is paid to the possibility that Bierne and Eyre-Walker's (2004) detection of a constant α across genes was an artifact, not of Drosophila demography, but of their estimation procedure—a possibility that is strong for a number of rather technical reasons, explained in what follows.
To achieve these goals, a refined and extended version of Bierne and Eyre-Walker's (2004) maximum-likelihood estimator is first introduced. This estimator and others are then used on a greatly enlarged data set of D. simulans genes, each with orthologs from both D. melanogaster and D. yakuba. Finally, numerical simulations are used to test the accuracy and power of the methods.
MATERIALS AND METHODS
Data:
Sequences:
Partial sequences of 122 protein-coding genes with multiple alleles from D. simulans were assembled. For each, an attempt was made to locate orthologous sequences from both D. melanogaster and D. yakuba. When no D. yakuba gene was annotated on GenBank, the yakuba genome Release 1.0 draft assembly (http://genome.ucsc.edu/cgi-bin/hgGateway?db=droYak1) was searched, verifying orthology by reciprocal blast. For seven loci (all male accessory gland proteins or Acp's), no convincing yakuba ortholog was found, leaving a total of 115 genes. For these genes, between 2 and 70 simulans alleles (median 8) were available (for full details see supplemental material 1 at http://www.genetics.org/supplemental/). This data set includes the 75 genes analyzed by Bierne and Eyre-Walker (2004), which in turn include the 35 genes analyzed by Smith and Eyre-Walker (2002). All alignments were produced by hand and are available on request.
Divergence and polymorphism estimates:
Estimates of the number of synonymous and nonsynonymous sites and of Dn and Ds were obtained from codeml, part of the PAML software package (Goldman and Yang 1994; Yang and Nielsen 1998). Unrooted trees of all three species were analyzed in each case. To avoid falsely counting a sampled polymorphism as a fixed difference, the D. simulans sequence sent to codeml was a composite of multiple alleles where necessary. During the estimation, the dN/dS ratio was allowed to take a different value along each of the three branches, and the 3 × 4 model of base composition was used. Noninteger estimates of Dn and Ds were rounded to the nearest integer for use in the likelihood estimator. Estimates of Pn and Ps were obtained from software written in conjunction with Jane Charlesworth. When three or more codons were segregating at a site, the most parsimonious path linking them was found, averaging across possible mutational orders, but excluding multiple hits. Ambiguous codons were not included in the count.
Methods:
Maximum-likelihood estimator:
The maximum-likelihood (ML) estimator is closely based on the method of Bierne and Eyre-Walker (2004), with modifications drawn from the theoretical work of Sawyer and Hartl (1992), which forms the basis of the methods of Bustamante et al. (2002) and Sawyer et al. (2003). These methods all rely on the assumption that sites evolve independently. Consideration of this, and other simplifying assumptions, is found in the discussion. Under the assumption of independence, each of the four quantities measured (Ps, Pn, Ds, and Dn) is approximately Poisson distributed, which leads to a likelihood function of the form
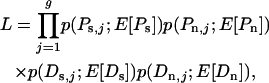 |
(1) |
where g is the number of loci analyzed, E[.] denotes an expectation, and 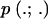 is the Poisson distribution:
is the Poisson distribution: 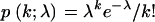 (Bustamante et al. 2002; Sawyer et al. 2003; Bierne and Eyre-Walker 2004).
(Bustamante et al. 2002; Sawyer et al. 2003; Bierne and Eyre-Walker 2004).
To complete the model, we require the expected values of each of the four quantities. These can be found from standard population genetics theory (Sawyer and Hartl 1992). Consider first a single site where mutations occur at rate μ and are subject to a common strength of selection, s. Scaling both quantities by the haploid effective population size, Ne, we define  and
and  . The level of polymorphism expected in a random sample of n alleles can now be written as
. The level of polymorphism expected in a random sample of n alleles can now be written as
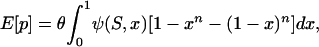 |
(2) |
where
 |
In this expression, 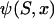 is a diffusion approximation for the expected time the mutation spends at frequency x (e.g., Ewens 1979; Sawyer and Hartl 1992); and the term in brackets is the probability that a sample of n alleles contains both mutant and wild type.
is a diffusion approximation for the expected time the mutation spends at frequency x (e.g., Ewens 1979; Sawyer and Hartl 1992); and the term in brackets is the probability that a sample of n alleles contains both mutant and wild type.
The expected level of divergence per generation is the product of the expected number of mutants appearing, 2Nμ, and their probability of reaching fixation. However, given a limited sample of alleles, the divergence measured may be inflated by falsely counting segregating polymorphisms as fixed differences (e.g., Sawyer and Hartl 1992). Taking this into account leads to the expression
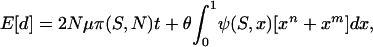 |
(3) |
where
 |
Here, 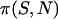 is the approximate fixation probability (Kimura 1957), t is the total length of the divergence in generations, and xn is the probability that all n alleles carry the polymorphic mutant. The coefficient m varies depending on the way divergence is measured. If the total divergence between a species pair is required, m is the number of alleles sampled from the comparison species (m = 1 for the present work). When the divergence along a single lineage is required (i.e., the lineage leading to D. simulans from its common ancestor with D. melanogaster), we set m = 2 because divergence estimates will be inflated only if both melanogaster and yakuba sequences carry a polymorphic mutation.
is the approximate fixation probability (Kimura 1957), t is the total length of the divergence in generations, and xn is the probability that all n alleles carry the polymorphic mutant. The coefficient m varies depending on the way divergence is measured. If the total divergence between a species pair is required, m is the number of alleles sampled from the comparison species (m = 1 for the present work). When the divergence along a single lineage is required (i.e., the lineage leading to D. simulans from its common ancestor with D. melanogaster), we set m = 2 because divergence estimates will be inflated only if both melanogaster and yakuba sequences carry a polymorphic mutation.
In this work, it is assumed that all synonymous mutations are effectively neutral, and so 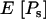 and
and 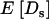 follow from taking the limit
follow from taking the limit 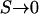 in Equations 2 and 3 and then multiplying each by the number of synonymous sites. To determine the equivalent expressions for nonsynonymous mutations we must model natural selection, and here there is a choice of approach. The first possibility is to specify a distribution of selection coefficients applying to all mutations. This approach faces difficulties fitting areas of the distribution where S is not small. This is because strongly beneficial mutations are unlikely to contribute greatly to polymorphism (e.g., Ewens 1979), and strongly deleterious mutations contribute little to either polymorphism or divergence. A second approach is to treat different categories of mutations separately, for example, by estimating the size of a class of severely deleterious mutations. A potential problem with this approach is its failure to deal adequately with mutations that do not fall unambiguously into any of the specified categories, notably mildly deleterious mutations. Here, a hybrid method is implemented; this method attempts to combine the strengths of the two approaches and includes each as a special case.
in Equations 2 and 3 and then multiplying each by the number of synonymous sites. To determine the equivalent expressions for nonsynonymous mutations we must model natural selection, and here there is a choice of approach. The first possibility is to specify a distribution of selection coefficients applying to all mutations. This approach faces difficulties fitting areas of the distribution where S is not small. This is because strongly beneficial mutations are unlikely to contribute greatly to polymorphism (e.g., Ewens 1979), and strongly deleterious mutations contribute little to either polymorphism or divergence. A second approach is to treat different categories of mutations separately, for example, by estimating the size of a class of severely deleterious mutations. A potential problem with this approach is its failure to deal adequately with mutations that do not fall unambiguously into any of the specified categories, notably mildly deleterious mutations. Here, a hybrid method is implemented; this method attempts to combine the strengths of the two approaches and includes each as a special case.
To model strongly deleterious mutations, we define f ( ) as a measure of selective constraint. In particular, it is assumed that a proportion 1 − f of nonsynonymous mutations are sufficiently deleterious so as to contribute negligible amounts to either divergence or polymorphism. For the remaining, weakly selected mutations, the scaled selection coefficient, S, is treated as a random variable drawn from a partially reflected exponential distribution,
) as a measure of selective constraint. In particular, it is assumed that a proportion 1 − f of nonsynonymous mutations are sufficiently deleterious so as to contribute negligible amounts to either divergence or polymorphism. For the remaining, weakly selected mutations, the scaled selection coefficient, S, is treated as a random variable drawn from a partially reflected exponential distribution,
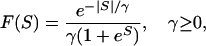 |
(4) |
where  , the distribution's sole parameter, is a natural measure of selection strength. Equation 4 can be derived from a mechanical model of molecular evolution, in which most mutations are deleterious, but where mildly deleterious mutations that reach fixation create the opportunity for beneficial mutations of equivalent strength—thereby undoing the damage caused by the substitution (Bulmer 1991; Piganeau and Eyre-Walker 2003). This is more plausible than the assumption that all such mutations are deleterious at equilibrium (Gillespie 1995).
, the distribution's sole parameter, is a natural measure of selection strength. Equation 4 can be derived from a mechanical model of molecular evolution, in which most mutations are deleterious, but where mildly deleterious mutations that reach fixation create the opportunity for beneficial mutations of equivalent strength—thereby undoing the damage caused by the substitution (Bulmer 1991; Piganeau and Eyre-Walker 2003). This is more plausible than the assumption that all such mutations are deleterious at equilibrium (Gillespie 1995).
Together, the parameters f and γ model deleterious mutations and weakly selected beneficial mutations resulting from mildly deleterious substitutions. To model true adaptive evolution, we use the parameter α ( ), defined as the proportion of the nonsynonymous divergence driven by positive natural selection of reasonable strength (Smith and Eyre-Walker 2002). This entails the assumption that such positively selected substitutions contribute little to sampled polymorphism.
), defined as the proportion of the nonsynonymous divergence driven by positive natural selection of reasonable strength (Smith and Eyre-Walker 2002). This entails the assumption that such positively selected substitutions contribute little to sampled polymorphism.
Using Equations 2–4, the quantities  ,
,  ,
,  , and
, and  can now be expressed in terms of the parameters θ, f, γ, and α and the scaled divergence time
can now be expressed in terms of the parameters θ, f, γ, and α and the scaled divergence time 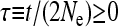 . Using Ls and Ln to denote the numbers of synonymous and nonsynonymous sites, respectively, we have
. Using Ls and Ln to denote the numbers of synonymous and nonsynonymous sites, respectively, we have
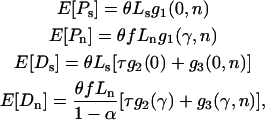 |
(5) |
where the integrals have been expressed via three functions 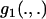 ,
, 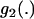 , and
, and 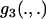 , which are defined in appendix a.
, which are defined in appendix a.
The different parameterizations:
The likelihood model specified by Equations 1 and 5 takes a maximum of 5g free parameters (where g is the number of loci analyzed) but this is far too many values to estimate from the 4g principal observations. The number of free parameters can be drastically reduced by assuming that certain parameters are common to all genes or by assigning them predetermined fixed values. These are the approaches taken for the four parameters θ, f, γ, and τ. Table 1 describes 13 different likelihood models in which one or more of these parameters are assumed to be common to all genes. Also included are models for which f = 1 at all loci (forcing selective constraint to be modeled solely though variation in the parameter γ) and models for which  (meaning that weakly selected mutations are not explicitly modeled). Note that θ, γ, and τ each depend on Ne, and so when any of these parameters is assumed to be common to all genes, corrections must be made to the likelihood function for X-linked genes. Here, we follow Sawyer et al. (2003) and assume that the Ne of X-linked genes is three-fourths that of autosomal loci. Because this assumption is questionable, separate analyses are also carried out for autosomal loci alone.
(meaning that weakly selected mutations are not explicitly modeled). Note that θ, γ, and τ each depend on Ne, and so when any of these parameters is assumed to be common to all genes, corrections must be made to the likelihood function for X-linked genes. Here, we follow Sawyer et al. (2003) and assume that the Ne of X-linked genes is three-fourths that of autosomal loci. Because this assumption is questionable, separate analyses are also carried out for autosomal loci alone.
TABLE 1.
Likelihood models
| Model | K1 | θ | τ | f | γ |
|---|---|---|---|---|---|
| 1 | 2 | θ | τ | 1 | 0 |
| 2 | 3 | θ | τ | f | 0 |
| 3 | 3 | θ | τ | 1 | γ |
| 4 | 4 | θ | τ | f | γ |
| 5 | g + 2 | θ | τ | fj | 0 |
| 6 | g + 2 | θ | τ | 1 | γj |
| 7 | g + 3 | θ | τ | fj | γ |
| 8 | g + 3 | θ | τ | f | γj |
| 9 | 2g + 2 | θ | τ | fj | γj |
| 10 | 3g | θj | τj | fj | 0 |
| 11 | 3g | θj | τj | 1 | γj |
| 12 | 3g + 1 | θj | τj | fj | γ |
| 13 | 3g + 1 | θj | τj | f | γj |
| Model | K2 | Description |
|---|---|---|
| i | 0 | αj = 0 |
| ii | 1 | αj = α |
| iii | 2 | αj ∼ pdfbeta(a, b) |
| iv | 3 | αj ∼ pdfdelta(α0, α1, q) |
The parameterizations of the different likelihood models are shown. The types of parameter are θ = 4Neμ (where μ is the nucleotide mutation rate), 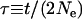 (where t is the divergence time in generations), f (the measure of selective constraint),
(where t is the divergence time in generations), f (the measure of selective constraint),  (where
(where  is the mean absolute selection coefficient) and α (the proportion of Dn attributed to adaptive evolution). A subscript j indicates that a parameter is assigned individually to each locus, while the absence of this subscript indicates a shared parameter, assumed common to all loci. For models of classes iii and iv, α is treated as a random variable drawn from a distribution whose parameters (a and b or α0, α1, and q) are estimated. The total number of parameters in a given model is K1 + K2, and g is the number of genes.
is the mean absolute selection coefficient) and α (the proportion of Dn attributed to adaptive evolution). A subscript j indicates that a parameter is assigned individually to each locus, while the absence of this subscript indicates a shared parameter, assumed common to all loci. For models of classes iii and iv, α is treated as a random variable drawn from a distribution whose parameters (a and b or α0, α1, and q) are estimated. The total number of parameters in a given model is K1 + K2, and g is the number of genes.
The parameter α, which quantifies adaptive evolution, is of special importance and here a different approach is taken. Models are included for which α is fixed at zero for all loci and for which α is a free parameter, common to all loci. Together, this allows us to test the null hypothesis of no adaptive evolution and to estimate 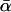 , the “average” rate across the loci. Confidence intervals for
, the “average” rate across the loci. Confidence intervals for 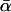 can also be obtained from the curvature of the likelihood surface (e.g., Bierne and Eyre-Walker 2004). To test for between-locus variation in α, the simplest approach—assigning gene-specific values—usually results in overparameterization. Bierne and Eyre-Walker (2004) solved this problem by treating each α-value as a random variable drawn from a given probability density: pdf(α). In this case, the nonsynonymous divergence term in Equation 1 is replaced by the integral:
can also be obtained from the curvature of the likelihood surface (e.g., Bierne and Eyre-Walker 2004). To test for between-locus variation in α, the simplest approach—assigning gene-specific values—usually results in overparameterization. Bierne and Eyre-Walker (2004) solved this problem by treating each α-value as a random variable drawn from a given probability density: pdf(α). In this case, the nonsynonymous divergence term in Equation 1 is replaced by the integral:
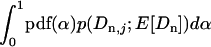 |
(6) |
(noting, from Equations 5, that 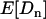 is a function of α). In this way, rather than estimate α for each locus, we need estimate only the parameters needed to specify pdf(α). For the form of pdf(α), Bierne and Eyre-Walker (2004) chose the two-parameter beta distribution,
is a function of α). In this way, rather than estimate α for each locus, we need estimate only the parameters needed to specify pdf(α). For the form of pdf(α), Bierne and Eyre-Walker (2004) chose the two-parameter beta distribution,
 |
(7) |
where B(a, b) is the normalizing beta function (Abramowitz and Stegun 1965). Bierne and Eyre-Walker used an approximation for the integral of Equations 6 and 7, but as appendix a suggests, this can be quite inaccurate, so here, an exact version is implemented.
Although the beta distribution of Equation 7 has some desirable properties, it also has some limitations. In particular, the distribution can be bimodal only if the peaks are located at α = 0 and α = 1. However, alternative scenarios are biologically plausible; for example, some loci might undergo limited adaptive evolution, and others undergo none. In addition, the beta distribution converges to the fixed-α model only as the parameters become infinite, in which case the integral Equation 6 becomes difficult to calculate. For these reasons, in addition to the beta distribution, we also implement a second pdf(α). This distribution consists solely of two weighted spikes of probability, such that α is assumed to take the value α = α0 with probability q and the value α = α1 otherwise. Formally, this distribution can be written as
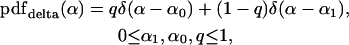 |
(8) |
where δ(.) is Dirac's delta function, which vanishes if its argument is nonzero (Abramowitz and Stegun 1965).
In total, then, the parameter α is treated in four different ways: (i) fixed at zero for all genes, (ii) fixed at an arbitrary value common to all genes, (iii) beta distributed, and (iv) two-spike distributed. These different approaches add between 0 and 3 free parameters to the model (Table 1). Combined with the different combinations of the other parameters, a grand total of 13 × 4 = 52 likelihood models can be specified. These are referred to by notation set out in Table 1. So, for example, 4iv refers to a model where each of θ, τ, f, and γ is set as a universal parameter common to all genes, while α is assumed to be drawn from the two-spiked distribution; this model has a total of K1 + K2 = 7 free parameters.
For each model, the maximum-likelihood estimates (MLEs) were obtained via a simulated annealing algorithm, written in C (source code available on request; details in appendix a).
Model selection criteria:
Although many of the models described in Table 1 are nested, this typically involves setting parameters at extremes of their ranges. In addition, the models contain very different numbers of free parameters. For these reasons, a method of model selection must be chosen with care (e.g., Posada and Crandall 2001; Kuha 2004). Bierne and Eyre-Walker (2004) used the Akaike information criterion (AIC), which selects the model minimizing the quantity
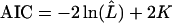 |
(9) |
(Akaike 1974), where 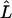 is the maximized likelihood (i.e., Equation 1 with all parameters at their MLEs) and K is the number of free parameters in the model (K = K1 + K2 in the notation of Table 1). The AIC can perform well in situations where model complexity grows with sample size—as it does for models with gene-specific parameters. However, for these same models, the ratio of datapoints to parameters will always be small (<3), and in such situations the AIC can perform poorly (e.g., Shibata 1976; Hurvich and Tsai 1989). As a result, we also use the “second-order AIC”:
is the maximized likelihood (i.e., Equation 1 with all parameters at their MLEs) and K is the number of free parameters in the model (K = K1 + K2 in the notation of Table 1). The AIC can perform well in situations where model complexity grows with sample size—as it does for models with gene-specific parameters. However, for these same models, the ratio of datapoints to parameters will always be small (<3), and in such situations the AIC can perform poorly (e.g., Shibata 1976; Hurvich and Tsai 1989). As a result, we also use the “second-order AIC”:
 |
(10) |
(Hurvich and Tsai 1989). In Equation 10, the quantity 4g is the effective sample size, reflecting the fact that four principal measurements have been taken from each gene.
Other possible objections to the AIC also apply to the AICc. For example, the constants appearing in both (9) and (10) result from a more-or-less arbitrary choice of discrepancy metric (Linhart and Zucchini 1986; Kass and Raftery 1995), and neither one takes parameter uncertainty into account. For these reasons, the Bayesian information criterion (BIC) is also used:
 |
(11) |
Equation 11 is closely related to the standard Bayes factor (Kass and Raftery 1995), but does not rely on detailed specification of prior probabilities for the parameters. The strategy of using the AIC and BIC together follows the general recommendations of Kuha (2004).
RESULTS
Results are reported here for three sets of Ds and Dn measurements: these are the total divergence between D. yakuba and D. simulans, the total divergence between D. melanogaster and simulans, and the divergence along the simulans lineage alone. Table 2 contains details of the selected models and ML parameter estimates for each data set, while the full results for all likelihood models are given as supplemental information 2 (http://www.genetics.org/supplemental/).
TABLE 2.
Maximum-likelihood results
| Criterion | Model chosen | 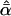 |
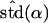 |
Shared-parameter MLEs | |
|---|---|---|---|---|---|
| yakuba–simulans | AIC/BIC | 10ii | 0.405 | 0 |
 = 0.405 = 0.405 |
| AICc | 5iv | 0.428 | 0.351 |
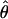 = 0.03, = 0.03,  = 9.06 = 9.06 |
|
 = 0.00, = 0.00,  = 0.78 = 0.78 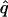 = 0.45 = 0.45 | |||||
| melanogaster–simulans | AIC | 10iv | 0.405 | 0.101 |
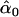 = 0.39, = 0.39, 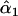 = 0.98, = 0.98,  = 0.97 = 0.97 |
| BIC | 10ii | 0.436 | 0 |
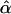 = 0.436 = 0.436 |
|
| AICc | 5iv | 0.404 | 0.375 |
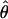 = 0.02, = 0.02, 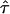 = 2.46 = 2.46 |
|
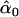 = 0.04, = 0.04,  = 0.79, = 0.79,  = 0.51 = 0.51 | |||||
| D. simulans lineage | AIC | 10iv | 0.436 | 0.097 |
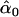 = 0.42, = 0.42,  = 0.98, = 0.98,  = 0.97 = 0.97 |
| BIC/AICc | 5iv | 0.430 | 0.324 |
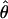 = 0.03, = 0.03,  = 0.63 = 0.63 |
|
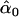 = 0.19, = 0.19, 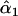 = 0.87 = 0.87  = 0.65 = 0.65 |
Likelihood models supported by the different model selection criteria are shown. Three sets of results are presented, with the divergence to D. simulans measured from D. yakuba, D. melanogaster, and the common ancestor of melanogaster and simulans. For the chosen models, the maximum-likelihood estimates of the across-locus mean and standard deviation of α are shown. These quantities, denoted 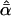 and
and 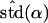 , were determined from the MLE parameters of pdf(α). Also shown are the estimated values of parameters common to all loci.
, were determined from the MLE parameters of pdf(α). Also shown are the estimated values of parameters common to all loci.
Table 2 shows that the results over the different periods of divergence, and for the different model selection criteria, are consistent in most respects. Most importantly, all selected models estimate 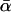 at between 40 and 45%. These estimates are significantly different from zero; indeed, models of type ii (with α as a free parameter) receive substantially more support than models of type i (with α set at zero) under all criteria and for all models incorporating selective constraint. Similarly, γ = 0 for all selected models, and so none point to the segregation of mildly deleterious mutations. Shared parameter estimates of θ and τ are also reasonably consistent, with θ staying roughly constant, and τ changing in the direction expected.
at between 40 and 45%. These estimates are significantly different from zero; indeed, models of type ii (with α as a free parameter) receive substantially more support than models of type i (with α set at zero) under all criteria and for all models incorporating selective constraint. Similarly, γ = 0 for all selected models, and so none point to the segregation of mildly deleterious mutations. Shared parameter estimates of θ and τ are also reasonably consistent, with θ staying roughly constant, and τ changing in the direction expected.
While these MLEs show a broad consistency, it is clear from Table 2 that different model selection criteria provide very different answers to the question of whether α varies significantly between loci. For all three sets of divergence, the AICc selects models in which θ and τ are shared between genes, but α is drawn from a two-spiked distribution with a high variance. In contrast, the AIC and BIC select one of two models: 10ii in which α takes a single common value of ∼40% or 10iv in which α follows a two-spiked model, but with the great majority of the probability density (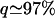 ) concentrated around a spike at ∼40%. Both of these models are of class 10, with locus-specific values of θ and τ, and this indicates between-locus variation in Ne. Furthermore, this result is not due to the presence of X-linked loci in the data set. This follows from the fact that the AIC and the BIC continue to select these parameter-rich models when X-linked loci are excluded; estimates of
) concentrated around a spike at ∼40%. Both of these models are of class 10, with locus-specific values of θ and τ, and this indicates between-locus variation in Ne. Furthermore, this result is not due to the presence of X-linked loci in the data set. This follows from the fact that the AIC and the BIC continue to select these parameter-rich models when X-linked loci are excluded; estimates of 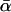 also remain stable (for full details see supplemental information 2 at http://www.genetics.org/supplemental/). Examining the results of all models together, it is clear that the detection of between-locus variation in α and the overall size of the model are closely linked. Specifically, variable-α models improve the likelihood readily for the small models 1–8 (all of which have K < 2g), while for the parameter-rich models 9–13 both the beta and two-spiked distributions tend to converge to single spikes of zero width. To determine whether between-locus variation in α really is present, then, we must determine which of the model selection criteria is most reliable.
also remain stable (for full details see supplemental information 2 at http://www.genetics.org/supplemental/). Examining the results of all models together, it is clear that the detection of between-locus variation in α and the overall size of the model are closely linked. Specifically, variable-α models improve the likelihood readily for the small models 1–8 (all of which have K < 2g), while for the parameter-rich models 9–13 both the beta and two-spiked distributions tend to converge to single spikes of zero width. To determine whether between-locus variation in α really is present, then, we must determine which of the model selection criteria is most reliable.
SIMULATIONS
To test the performance of the model selection criteria and the power of the method to detect between-locus variation, the ML estimator was tested on simulated data. The simulated data sets were designed to closely resemble the real data, with divergence measured from D. melanogaster. Simulated data sets were generated to conform to the assumptions of likelihood models 5i–iv and 10i–iv and included various levels of between-locus variation in α. In total, data sets of 25 different kinds were generated, and for each set of assumptions, 100 simulated data sets were generated, making 2500 data sets in total. For each data set, likelihood scores and MLEs were obtained for each of 12 different models, 2i–iv, 5i–iv, and 10i–iv. As a result, the true model, used to simulate the data, was always fitted. Full details of the simulation procedures and a summary of the results are given in appendix b.
Simulation results:
A surprising result of the simulations was that the BIC, and particularly the AICc, performed poorly; both criteria consistently selected models that were too small (i.e., contained many fewer parameters than the true model). Importantly, this led to the spurious detection of between-locus variation in α, when it was not present. In contrast to the other criteria, the standard AIC performed very well. It selected the true model in the majority of cases and the true class of model (i.e., 5 or 10) in almost every case. Performance in detecting between-locus variation in α was more mixed for two reasons. First, the two-spiked models (type iv) detected variation more readily than did the beta-distribution models (type iii), regardless of the true distribution of the α-values; this suggests that our power to detect the precise form of the between-locus variation is limited. Second, the rate of detection of between-locus variation declined rapidly as the magnitude of the variation also declined; in other words, extreme variation was almost always detected, but a constant-α model was regularly and falsely selected when variation was limited.
Considering the real data (Table 2), these observations imply that the anomalous results from the AICc can be safely disregarded. The results therefore strengthen the suggestion that extreme between-locus variation in α is not present in D. simulans. Nevertheless, limited variation cannot be ruled out and is, indeed, indicated by the selection of model 10iv under some conditions.
COMPARISON OF ESTIMATORS
While the results in Table 2 are consistent with each other, they disagree with some published estimates of  in D. simulans. To try to clarify the reasons for this, Table 3 contains a variety of estimates of
in D. simulans. To try to clarify the reasons for this, Table 3 contains a variety of estimates of 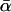 , complete with confidence intervals, each obtained from the current data set. Included in Table 3 are estimates from three of the ML models described in Table 1. The remaining three estimates use the methods of Bierne and Eyre-Walker (2004) (BEW), Smith and Eyre-Walker (2002) (SEW), and Fay et al. (2001) (FWW). To understand Table 3, the differences in assumptions between these various estimators must first be understood.
, complete with confidence intervals, each obtained from the current data set. Included in Table 3 are estimates from three of the ML models described in Table 1. The remaining three estimates use the methods of Bierne and Eyre-Walker (2004) (BEW), Smith and Eyre-Walker (2002) (SEW), and Fay et al. (2001) (FWW). To understand Table 3, the differences in assumptions between these various estimators must first be understood.
TABLE 3.
Comparison of estimators of 
| Method | yakuba–simulans | melanogaster–simulans | D. simulans lineage |
|---|---|---|---|
| ML 10ii | 0.405 (0.324, 0.476) | 0.436 (0.347, 0.514) | 0.485 (0.384, 0.565) |
| BEW (ML 10ii′) | 0.405 (0.328, 0.475) | 0.449 (0.362, 0.525) | 0.573 (0.485, 0.648) |
| SEW | 0.408 (0.264, 0.527) | 0.441 (0.305, 0.563) | 0.522 (0.374, 0.644) |
| ML 2ii | 0.492 (0.431, 0.548) | 0.517 (0.453, 0.575) | 0.583 (0.516, 0.643) |
| FWW | 0.491 (0.348, 0.608) | 0.519 (0.384, 0.627) | 0.589 (0.429, 0.706) |
| ML 11ii | 0.839 (0.810, 0.864) | 0.796 (0.761, 0.827) | 0.849 (0.815, 0.876) |
Various estimates of  are shown, with confidence intervals to those estimates in parentheses. Rows labelled ML 10ii, 2ii, and 11ii present results from the ML models defined in Table 1. BEW labels results from a variant of model 10ii lacking the correction to the expected divergence and so replicating the selected model 2d of Bierne and Eyre-Walker (2004). For these estimators, confidence intervals are defined as the α-values that reduce the maximized log-likelihood by 2 units. SEW and FWW denote the heuristic estimators of, respectively, Smith and Eyre-Walker (2002) and Fay et al. (2001), and for these estimators, 95% confidence intervals were obtained from 10,000 bootstrap resamplings of the genes.
are shown, with confidence intervals to those estimates in parentheses. Rows labelled ML 10ii, 2ii, and 11ii present results from the ML models defined in Table 1. BEW labels results from a variant of model 10ii lacking the correction to the expected divergence and so replicating the selected model 2d of Bierne and Eyre-Walker (2004). For these estimators, confidence intervals are defined as the α-values that reduce the maximized log-likelihood by 2 units. SEW and FWW denote the heuristic estimators of, respectively, Smith and Eyre-Walker (2002) and Fay et al. (2001), and for these estimators, 95% confidence intervals were obtained from 10,000 bootstrap resamplings of the genes.
Description of other estimators:
Closest to the present estimator is the method of Bierne and Eyre-Walker (2004). Most of the changes introduced here are simply additions to that work. Specifically, Bierne and Eyre-Walker's study did not include models with a continuous distribution of selection coefficients (Equation 4) or the two-spiked distribution of α (Equation 8). However, there is also one important difference that affects all of the ML models: Bierne and Eyre-Walker did not model sampling explicitly, and so the functions  ,
,  , and
, and 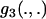 did not appear in their equivalents of Equations 5. If the parameters θ and τ are locus specific, and mildly deleterious mutations are not modeled (i.e., if
did not appear in their equivalents of Equations 5. If the parameters θ and τ are locus specific, and mildly deleterious mutations are not modeled (i.e., if 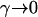 ), then two of these functions,
), then two of these functions, 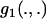 and
and  , become irrelevant, because they may be absorbed into the definition of other parameters. The same thing applies to the third function,
, become irrelevant, because they may be absorbed into the definition of other parameters. The same thing applies to the third function,  , but only if ML estimates of
, but only if ML estimates of 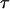 are allowed to become negative. If, as with the current approach, these scaled divergence times are constrained to be greater than zero, then the inclusion or neglect of
are allowed to become negative. If, as with the current approach, these scaled divergence times are constrained to be greater than zero, then the inclusion or neglect of 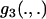 can alter results. Such an outcome is expected only over very short divergences, however, as
can alter results. Such an outcome is expected only over very short divergences, however, as  results from the second term in Equation 3, which is the correction to the expected divergence from segregating polymorphism. The estimators of Fay et al. (2001) and Smith and Eyre-Walker (2002) are similar to that of Bierne and Eyre-Walker (2004), in that they too neglect the correction to the expected divergence and also do not allow for a continuous distribution of selection coefficients. However, these methods are not based on formal likelihood equations and are subject to intrinsic biases. The most serious bias affects the estimator of Fay et al. (2001), which is equivalent to a single-locus test using values summed across all loci. This estimator can produce artifactually inflated estimates if the sampled loci show a negative correlation between
results from the second term in Equation 3, which is the correction to the expected divergence from segregating polymorphism. The estimators of Fay et al. (2001) and Smith and Eyre-Walker (2002) are similar to that of Bierne and Eyre-Walker (2004), in that they too neglect the correction to the expected divergence and also do not allow for a continuous distribution of selection coefficients. However, these methods are not based on formal likelihood equations and are subject to intrinsic biases. The most serious bias affects the estimator of Fay et al. (2001), which is equivalent to a single-locus test using values summed across all loci. This estimator can produce artifactually inflated estimates if the sampled loci show a negative correlation between 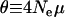 , their expected level of neutral polymorphism, and f, the proportion of nonsynonymous mutations that evolve neutrally. Such a correlation is not implausible and is expected to occur if effective population size varies between loci, and a fraction of mutations are “nearly neutral,” with
, their expected level of neutral polymorphism, and f, the proportion of nonsynonymous mutations that evolve neutrally. Such a correlation is not implausible and is expected to occur if effective population size varies between loci, and a fraction of mutations are “nearly neutral,” with  and
and 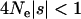 (e.g., Ohta 1992; Smith and Eyre-Walker 2002). The estimator of Smith and Eyre-Walker (2002) was designed explicitly to correct for such a correlation, but it too can be biased, particularly if expected levels of neutral polymorphism are low at any of the loci. Finally, both of the heuristic estimators are expected to yield inflated estimates of
(e.g., Ohta 1992; Smith and Eyre-Walker 2002). The estimator of Smith and Eyre-Walker (2002) was designed explicitly to correct for such a correlation, but it too can be biased, particularly if expected levels of neutral polymorphism are low at any of the loci. Finally, both of the heuristic estimators are expected to yield inflated estimates of 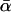 if there is a large amount of between-locus variation in α. These biases are explained in detail in appendix c, where the heuristic estimators are derived from the likelihood equations.
if there is a large amount of between-locus variation in α. These biases are explained in detail in appendix c, where the heuristic estimators are derived from the likelihood equations.
A method that does not appear in Table 3 is the estimator of Sawyer et al. (2003). This estimator resembles the current approach in that it combines the complete likelihood function of Sawyer and Hartl (1992)—including the corrections to the divergence estimates—with a continuous distribution of selection coefficients. But rather than Equation 4, Sawyer et al. (2003) used a normal distribution, with mean and variance estimated from the data. The flexibility of the normal distribution is appropriate, because these authors used the distribution to model adaptive evolution, rather than using the parameter α as here. A second important difference is that Sawyer et al. combined their likelihood function with prior distributions to carry out a hierarchical Bayesian analysis. As such, they did not use model selection to choose between various parameterizations, but instead obtained estimates that were smoothed over a range of parameter values (e.g., Holder and Lewis 2003). To achieve convergence of their estimator, Sawyer et al. (2003) restricted themselves to a situation where all genes had a common value of the parameter f, having excluded genes that yielded anomalous values in initial runs. The resulting model is thus qualitatively similar to ML model 11ii from the present work, and this is included in Table 3.
Results from other estimators:
How, then, do the results from the different estimators compare? Several of the estimates shown differ markedly from the ∼40% estimate obtained with the present method. Most notable are the very high ∼80% estimates obtained from ML model 11ii. Examining the results from all models, it is clear that estimates of this magnitude were obtained whenever selective constraint was modeled via a continuous distribution of selection coefficients and so included a large class of mildly deleterious mutations. This was the case for all models where the parameter γ was free to vary, but the flexibility of f was restricted (see Table 1 and supplemental information 2 at http://www.genetics.org/supplemental/). Much lower estimates were obtained whenever it was assumed that mutants were either effectively neutral or strongly deleterious (i.e., whenever γ→0 was assumed). This helps to explain the extremely high estimates of ∼94% obtained by Sawyer et al. (2003). For while there are many differences between their work and the current approach, their modeling of natural selection via a continuous distribution of selection coefficients must be an important factor in their estimate. In general, it is clear that this assumption can lead to liberal estimates. To demonstrate this, model 11ii was fitted to the 500 simulated data sets generated under the assumptions of models 5i and 10i (appendix b). In every single case, the presence of significant levels of adaptive evolution was indicated (a type I error rate of 100%) and 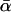 , which was zero in reality, was estimated at ∼60%.
, which was zero in reality, was estimated at ∼60%.
While the results from model 11ii are the most extreme that appear in Table 3, anomalously high estimates of ∼50% were also obtained from the FWW estimator and ML model 2ii. These inflated estimates may indicate the existence of a negative correlation between θ and f, since this is expected to bias the FWW estimator and cannot be accommodated by the four-parameter ML 2ii (appendix c). To test this possibility, MLEs for locus-specific θj and fj were examined. It was found that 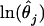 and
and 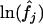 are indeed weakly but significantly negatively correlated (for example, using MLEs from Model 10ii, melanogaster–simulans divergence, and excluding outliers with values <10−9, it is found that ρ = −0.21, p = 0.03).
are indeed weakly but significantly negatively correlated (for example, using MLEs from Model 10ii, melanogaster–simulans divergence, and excluding outliers with values <10−9, it is found that ρ = −0.21, p = 0.03).
Testing the heuristic estimators on the simulated data sets further confirmed their biases (see appendix b for full results). As expected, the FWW estimator performed particularly poorly on data sets where locus-specific f- and θ-values were negatively correlated. Indeed, as with model 11ii, a type I error rate of 100% was obtained for those data sets where this correlation was extreme. Both heuristic estimators were also shown to yield inflated estimates of  when true α-values were highly variable among loci. In contrast, the ML methods performed well in these cases, with low levels of type I error and accurate estimation of
when true α-values were highly variable among loci. In contrast, the ML methods performed well in these cases, with low levels of type I error and accurate estimation of  . The agreement between the SEW estimator and the ML method on the real data therefore provides further evidence for the lack, in these data, of extreme between-locus variation in α.
. The agreement between the SEW estimator and the ML method on the real data therefore provides further evidence for the lack, in these data, of extreme between-locus variation in α.
The final anomaly apparent in Table 3 appears only in the fourth column, where the divergence is measured along the D. simulans lineage alone. In this case, the BEW and SEW estimators, which agreed well with the present method over larger divergences, are now greatly inflated. The most telling difference is the increase of the BEW estimate to almost 60%. Given the very close similarity of the methods, this difference must stem from the earlier estimator's noninclusion of the correction to the expected divergence for undetected segregating polymorphism—i.e., its lack of the function  in Equations 5. The consistency of these estimators over longer divergences shows that this sampling correction becomes important only when segregating polymorphisms constitute a nonnegligible fraction of the inferred divergence, a situation that will occur only when the true divergence is small. This is likely to be part of the explanation for the very high estimates of
in Equations 5. The consistency of these estimators over longer divergences shows that this sampling correction becomes important only when segregating polymorphisms constitute a nonnegligible fraction of the inferred divergence, a situation that will occur only when the true divergence is small. This is likely to be part of the explanation for the very high estimates of 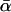 along the simulans lineage obtained by Fay et al. (2002) and Bierne and Eyre-Walker (2004).
along the simulans lineage obtained by Fay et al. (2002) and Bierne and Eyre-Walker (2004).
DISCUSSION
The McDonald–Kreitman test and its variants are among the most important methods we have for quantifying the rate of adaptive substitution. But all such tests are subject to a number of serious biases. Some sources of bias apply only to particular implementations of the test and stem from the assumptions and approximations made by different authors. In particular, this study has identified three ways in which estimates of  may be artificially inflated. These are (1) the incorrect assumption that mildly deleterious mutations are segregating, (2) the use of inadequately parameterized or heuristic estimators subject to biases, and (3) the failure to correct for the fact that divergence estimates may be inflated by segregating polymorphism. Of these potential problems, 1 and some instances of 2 have been noted in the literature (e.g., Smith and Eyre-Walker 2002; Eyre-Walker 2002; Sawyer et al. 2003; Bierne and Eyre-Walker 2004). Problem 3 has received less attention, but it reflects a wider difficulty with estimating short divergences (Ho and Larson 2006) and may explain other anomalous results from McDonald–Kreitman tests along the D. simulans lineage (e.g., Kern et al. 2004). The maximum-likelihood estimator introduced here deals with all three sources of potential error. Furthermore, the estimator can clarify the effects of varying assumptions and, when combined with model selection procedures, can discriminate between different classes of model. This was evident, for example, in the rejection of likelihood models such as 2ii and 11ii that yielded anomalous estimates (Tables 2 and 3).
may be artificially inflated. These are (1) the incorrect assumption that mildly deleterious mutations are segregating, (2) the use of inadequately parameterized or heuristic estimators subject to biases, and (3) the failure to correct for the fact that divergence estimates may be inflated by segregating polymorphism. Of these potential problems, 1 and some instances of 2 have been noted in the literature (e.g., Smith and Eyre-Walker 2002; Eyre-Walker 2002; Sawyer et al. 2003; Bierne and Eyre-Walker 2004). Problem 3 has received less attention, but it reflects a wider difficulty with estimating short divergences (Ho and Larson 2006) and may explain other anomalous results from McDonald–Kreitman tests along the D. simulans lineage (e.g., Kern et al. 2004). The maximum-likelihood estimator introduced here deals with all three sources of potential error. Furthermore, the estimator can clarify the effects of varying assumptions and, when combined with model selection procedures, can discriminate between different classes of model. This was evident, for example, in the rejection of likelihood models such as 2ii and 11ii that yielded anomalous estimates (Tables 2 and 3).
However, it must be acknowledged that model selection is a process of seeking the least inadequate model from a predefined set, all of which may be grossly inadequate as a representation of reality. Indeed, the second set of biases that afflict the McDonald–Kreitman test is due to unrealistic assumptions shared by all of the methods. Some of these assumptions, although undoubtedly false, are unlikely to create spurious evidence of substantial adaptive evolution. For example, the methods here all assume that synonymous mutations are selectively neutral, an assumption contradicted by clear evidence of selection for codon usage in D. simulans (Akashi and Schaeffer 1997; Begun 2001; McVean and Vieira 2001). However, the resulting bias appears to be deteriorating in both simulans (Begun 2001) and melanogaster (Akashi 1996), and theoretical and empirical works both suggest that the influence of such selection on the present results will be limited (Charlesworth 1994; Eyre-Walker 2002; Bierne and Eyre-Walker 2004). Similarly, the equations used here have relied on the assumption that sites evolve independently, and this assumption will also be violated, both by epistasis (Kondrashov et al. 2002) and especially by linkage (Begun 2001, 2002). Again, however, this may not greatly compromise the McDonald–Kreitman approach (Sawyer et al. 2003; Bierne and Eyre-Walker 2004; Williamson et al. 2005). This is because, in many cases, the principal effect of linked selection is a localized reduction in effective population size (e.g., Charlesworth 1994; Charlesworth et al. 1995; Gillespie 2001), and, as such, it may be captured adequately by models in which Ne is allowed vary over the genome.
By common consent, the Achilles heel of the McDonald–Kreitman approach is another assumption—the rough constancy of selective constraint. This is because the assumption is unlikely to hold unless the demographic histories of the species involved have also remained fairly stable (McDonald and Kreitman 1991; Fay et al. 2001; Eyre-Walker 2002; Williamson et al. 2005).
To explore the possibility that a demographic event is responsible for the high estimates of 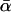 , this study adopted the strategy of Fay et al. (2002): assuming that adaptive evolution will manifest itself sporadically across the genome and then testing for between-locus variation in α. However, rather than the heuristic approach of Fay et al. (2002), which suggested the presence of substantial variation in D. melanogaster, we focused on the formal approach of Bierne and Eyre-Walker (2004), with which they detected no significant variation anywhere in the melanogaster subgroup. This study addressed the possibility that these conflicting results, like the different estimates of
, this study adopted the strategy of Fay et al. (2002): assuming that adaptive evolution will manifest itself sporadically across the genome and then testing for between-locus variation in α. However, rather than the heuristic approach of Fay et al. (2002), which suggested the presence of substantial variation in D. melanogaster, we focused on the formal approach of Bierne and Eyre-Walker (2004), with which they detected no significant variation anywhere in the melanogaster subgroup. This study addressed the possibility that these conflicting results, like the different estimates of 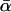 , were due to methodological artifacts. In particular, a series of extensions and refinements were introduced to determine whether Bierne and Eyre-Walker's failure to detect significant between-locus variation was a false negative. Possible sources of type II error investigated here were (i) inappropriate choice of model selection criterion, (ii) approximations made to the likelihood function, (iii) the choice of a beta distribution to model the between-locus variation, (iv) the approximate implementation of this distribution, (v) the limited number of loci involved, or (vi) a simple lack of power.
, were due to methodological artifacts. In particular, a series of extensions and refinements were introduced to determine whether Bierne and Eyre-Walker's failure to detect significant between-locus variation was a false negative. Possible sources of type II error investigated here were (i) inappropriate choice of model selection criterion, (ii) approximations made to the likelihood function, (iii) the choice of a beta distribution to model the between-locus variation, (iv) the approximate implementation of this distribution, (v) the limited number of loci involved, or (vi) a simple lack of power.
While results were to some extent equivocal, they do suggest that the presence of very high levels of between-locus variation in α really can be rejected. For example, the new two-spiked model did detect the presence of between-locus variation under some conditions, but this variation was very limited and under other conditions was absent altogether (see the AIC results in Table 2). Similarly, simulations suggested that the method has low power to detect limited between-locus variation, but that this power increases greatly with the extent of the variation (appendix b).
Other results also argue against the presence of high levels of between-locus variation in D. simulans. For example, the present estimate of 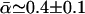 is very close to that of Smith and Eyre-Walker (2002) for the yakuba–simulans divergence, despite a more than trebling of the number of loci analyzed and the upward bias of the earlier estimator when α is highly variable (appendix c). Furthermore, previous estimates that have differed greatly from 40% have been attributed, at least in part, to methodological biases.
is very close to that of Smith and Eyre-Walker (2002) for the yakuba–simulans divergence, despite a more than trebling of the number of loci analyzed and the upward bias of the earlier estimator when α is highly variable (appendix c). Furthermore, previous estimates that have differed greatly from 40% have been attributed, at least in part, to methodological biases.
It is possible, of course, that adaptive substitution has constituted a roughly constant proportion of substitutions at most of the loci sampled, especially when the far from random sample of loci is taken into account. Nonetheless, it is more intuitively plausible that the lack of substantial between-locus variation in α is due to a demographic artifact.
Furthermore, another aspect of the results presented here allows us to make strong inferences about the kind of demographic event that could have given rise to the high 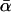 -estimates: this is the remarkable constancy of those estimates over the three different periods of divergence (Table 2). This finding allows us to locate any demographic event firmly within the D. simulans lineage (since that is the only period of divergence shared by all three conditions). It also allows us to exclude the possibility that an extended bottleneck in D. simulans inflated the estimate (since the fraction of the divergence due to any bottleneck would decline as the period of divergence increased). As such, the most plausible form of any demographic artifact would be an increase in Ne late in the history of the D. simulans lineage—and this, of course, is entirely consistent with the relatively recent spread of D. simulans out of Africa (Lachaise et al. 1988). Such an interpretation, however, is far from conclusive. Some recent studies have indicated that the effective population size of D. simulans has remained fairly stable (Li et al. 1999; Takahata and Satta 2002). Furthermore, the out-of-Africa expansion could not have created artifactual evidence of adaptive evolution if, as evidence suggests, non-African populations have a lower Ne than do African populations (Andolfatto 2001; Eyre-Walker 2002; Schöfl and Schlötterer 2004). That said, the causes of the reduced diversity in non-African populations have been disputed (Hamblin and Veuille 1999; Begun and Whitley 2000; Wall et al. 2002; Gravot et al. 2004).
-estimates: this is the remarkable constancy of those estimates over the three different periods of divergence (Table 2). This finding allows us to locate any demographic event firmly within the D. simulans lineage (since that is the only period of divergence shared by all three conditions). It also allows us to exclude the possibility that an extended bottleneck in D. simulans inflated the estimate (since the fraction of the divergence due to any bottleneck would decline as the period of divergence increased). As such, the most plausible form of any demographic artifact would be an increase in Ne late in the history of the D. simulans lineage—and this, of course, is entirely consistent with the relatively recent spread of D. simulans out of Africa (Lachaise et al. 1988). Such an interpretation, however, is far from conclusive. Some recent studies have indicated that the effective population size of D. simulans has remained fairly stable (Li et al. 1999; Takahata and Satta 2002). Furthermore, the out-of-Africa expansion could not have created artifactual evidence of adaptive evolution if, as evidence suggests, non-African populations have a lower Ne than do African populations (Andolfatto 2001; Eyre-Walker 2002; Schöfl and Schlötterer 2004). That said, the causes of the reduced diversity in non-African populations have been disputed (Hamblin and Veuille 1999; Begun and Whitley 2000; Wall et al. 2002; Gravot et al. 2004).
To resolve these issues, future extensions of the McDonald–Kreitman approach will have to make use of additional sources of information. One possibility, already being explored, is to exploit the frequency spectrum of mutations, although here a lack of robustness to the simplifying assumptions may be a problem (Bustamante et al. 2001; Williamson et al. 2005; Zhu and Bustamante 2005). A second possibility that deserves further attention is to extend the element of cross-species comparison in a more formal manner (Fay et al. 2002; Bierne and Eyre-Walker 2004).
Acknowledgments
I first thank Adam Eyre-Walker who has provided a great deal of help and encouragement throughout this project. In addition, all of the following people generously provided expert help and/or computer code: Nicolas Bierne, Mark Broom, Jane Charlesworth, Emmanuel Ladoukakis, Ted Phelps, David Waxman, and Meg Woolfit. Peter Andolfatto, Lindell Bromham, Rob Lanfear, Jess Thomas, David Begun, and anonymous reviewers also helped to improve the manuscript.
APPENDIX A
The functions 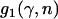 ,
, 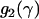 , and
, and 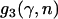 of Equations 5 are defined, and an exact form of the integral involving the beta distribution (Equations 6 and 7) is described. From Equations 2–5 it quickly follows that
of Equations 5 are defined, and an exact form of the integral involving the beta distribution (Equations 6 and 7) is described. From Equations 2–5 it quickly follows that
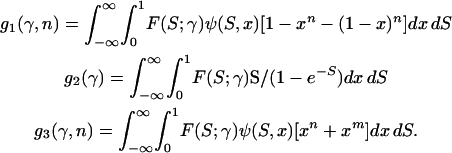 |
(A1) |
These simplify greatly in the limit γ→0:
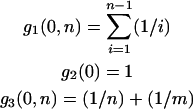 |
(A2) |
(e.g., Sawyer and Hartl 1992). In the general case, the expressions of Equation A1 can be calculated directly by numerical integration, but the computational load can be greatly reduced by writing the integrals over S in terms of special functions, for which well-developed numerical recipes are available (full details of these derivations will appear elsewhere). To calculate these functions, the ML estimation software made extensive use of the GNU scientific library of mathematical functions in C (Galassi et al. 2004).
Special functions from the GNU scientific library were also used to calculate the likelihood of the nonsynonymous divergence, when α was treated as a beta-distributed random variable (Equations 6 and 7). If we define 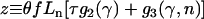 , then the integral required is
, then the integral required is
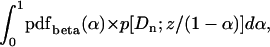 |
(A3) |
where we have used the beta distribution, Equation 7, and the Poisson distribution, Equation 1. To calculate Equation A3 Bierne and Eyre-Walker (2004) used an approximate integration technique. This approximation, which is explained by Yang (1994), is
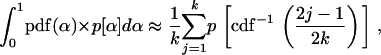 |
(A4) |
where k is some positive integer (k = 10 in the published results of Bierne and Eyre-Walker 2004), and cdf−1(α) is the inverse of the cumulative distribution of α:  . Numerical evaluation of Equations A3 and A4 shows that Equation A4, while a good approximation in most relevant parameter regimes, can be very inaccurate in some cases (results not shown). Such inaccuracy means that a constant-α model might be falsely rejected or receive false support. With this in mind, the present work calculated Equation A3 exactly. To do this, note that Equation A3 can be written as the product of a constant factor,
. Numerical evaluation of Equations A3 and A4 shows that Equation A4, while a good approximation in most relevant parameter regimes, can be very inaccurate in some cases (results not shown). Such inaccuracy means that a constant-α model might be falsely rejected or receive false support. With this in mind, the present work calculated Equation A3 exactly. To do this, note that Equation A3 can be written as the product of a constant factor, 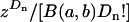 , and an integral. This integral can be written in terms of special functions as
, and an integral. This integral can be written in terms of special functions as
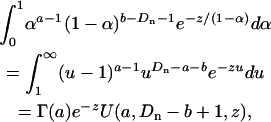 |
(A5) |
where Γ is Euler's gamma function and U is Kummer's U-function, also known as Tricomi's psi, or a confluent hypergeometric function of the second kind (Abramowitz and Stegun 1965). GNU scientific library routines for calculating Kummer's U were found to be accurate in most parameter regimes, but for the ML estimator, the routines were altered to give acceptable results throughout the relevant range.
APPENDIX B
Details of the methods used to generate the simulated data sets are described, and the results of the simulations are summarized.
Simulated data sets were identical to the real data in terms of gene number, gene lengths, and number of alleles sampled. In each case, the four principal observations, Dn, Ds, Pn, and Ps, were independent random integers drawn from Poisson distributions. The expected values of these distributions varied under different conditions. These conditions were chosen partly to reflect the real data and selected models and partly to test the behavior of the estimators under specific extreme conditions.
When data sets were simulated under the assumptions of likelihood model 10, the expected values of the four Poisson distributions were taken from the real data as
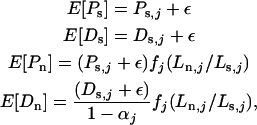 |
(B1) |
where ɛ is a small positive constant, without which zero-valued measurements would never generate nonzero simulated values. In all reported simulations, we took ɛ = 0.01. When a data set was simulated under the assumptions of likelihood model 5, the expected values were taken directly from Equations 5, with common θ- and τ-values obtained from the real data, via
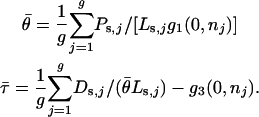 |
(B2) |
Five different methods were used to generate the true αj values. These methods, which are set out in Table B1, include the null model of no adaptive evolution, scenarios chosen to reflect the MLEs and model selection from the real data, and scenarios designed to evaluate the success of the estimators in the presence of extreme between-locus variation in the αj.
TABLE B1.
Methods for generating true αj-values for simulated data sets
| Model | Description | Equation |
|---|---|---|
| i | αj = 0 | |
| ii | αj = 0.4 | |
| iii(a) | αj ∼ pdfbeta(α; a = 1.0, b = 1.5) | 7 |
| iii(b) | αj ∼ pdfbeta(α; a = 3.0, b = 4.5) | 7 |
| iv | αj ∼ pdfdelta(α; α0 = 0.0, α1 = 0.8, q = 0.5) | 8 |
The five methods of generating the true αj-values for the simulated data sets are shown. In all cases except model i, the expected value of α is E[α] = 0.4, a value estimated from the real data. The variances of the two beta distributions are Var[α] ∼ 0.07 for model iii(a) and Var[α] ∼ 0.03 for model iii(b), making the former more than twice as variable. The variance of the two-spiked distribution is even greater: Var[α] = 0.16.
Finally three different methods were used to generate the locus-specific fj-values (see Table B2). In two of these, each f was simply a random number independently drawn from a beta distribution. The third method contrived a negative correlation between the fj and θj, similar to, but more extreme than that observed in the MLEs from the real data. (A correlation of this magnitude would be expected if the distribution of selection coefficients were exponential; see Gillespie 1991 and Ohta 1992.) Because it relies on the presence of locus-specific θj-values, this method could be used only when the true model was of type 10. By combining all of these methods in various combinations, simulated data sets of 25 types could be created, and 100 data sets were generated under each set of conditions. We note here that multiple ML estimations were obtained for each data set under each condition to ensure that the true MLEs were found.
TABLE B2.
Methods for generating true fj-values for simulated data sets
| Model | Description | |
|---|---|---|
| a | fj ∼ pdfbeta(f ; a = 0.5, b = 4.5) | |
| b | fj ∼ pdfbeta(f ; a = 2.0, b = 18.0) | |
| c |
|
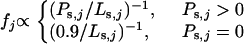
The three methods of generating the true fj-values for the simulated data sets are shown. Model c is used only when the true model is of type 10 (with locus-specific θj-values) and generates a negative correlation between θj and fj. The constant of proportionality was chosen such that  ; this value, chosen to reflect estimates from the real data, holds for all three models.
; this value, chosen to reflect estimates from the real data, holds for all three models.
Summaries of the results, which provide evidence for the assertions made in the main text, are given in Tables B3–B6. The poor performance of the BIC and the AICc is evident from Table B3. The BIC performed least well when the true model was of type 5 and the fj were highly variable (model b of Table B2); in this case, model 2iii was consistently and erroneously selected. The AICc performed least well when the true model was of type 10, in which case the AIC and BIC gave similar (and accurate) results, but the AICc consistently selected models 5iv or 2iii. Note that this pattern mirrors closely the results with the real data (Table 2), suggesting that a high-parameter model best characterizes these data.
TABLE B3.
Model selection with simulated data sets
| True model
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5i: αj = 0
|
5ii: αj = 0.40
|
5iii(a): αj ∼ pdfbeta
|
5iii(b): αj ∼ pdfbeta
|
5iv: αj ∼ pdfdelta
|
|||||||||||
| AIC | BIC | AICc | AIC | BIC | AICc | AIC | BIC | AICc | AIC | BIC | AICc | AIC | BIC | AICc | |
| 2iii | — | — | — | — | — | — | 0.08 | 0.08 | 0.08 | — | — | — | — | — | — |
| — | 0.10 | — | — | 0.67 | — | 0.10 | 0.99 | 0.10 | — | 0.81 | — | — | 0.88 | — | |
| 2iv | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| — | 0.74 | — | — | 0.21 | — | — | — | — | — | — | — | — | — | — | |
| 5i | 0.93 | 0.99 | 0.98 | — | — | — | — | — | — | — | — | — | — | — | — |
| 0.93 | 0.16 | 0.96 | — | — | — | — | — | — | — | — | — | — | — | — | |
| 5ii | 0.07 | 0.01 | 0.02 | 0.99 | 1.00 | 1.00 | 0.01 | 0.03 | 0.02 | 0.58 | 0.78 | 0.67 | — | — | — |
| 0.07 | — | 0.04 | 1.00 | 0.12 | 1.00 | 0.01 | — | 0.02 | 0.66 | 0.15 | 0.76 | — | — | — | |
| 5iii | — | — | — | — | — | — | 0.29 | 0.32 | 0.29 | 0.02 | 0.03 | 0.02 | — | — | — |
| — | — | — | — | — | — | 0.08 | 0.01 | 0.09 | — | — | 0.01 | — | — | — | |
| 5iv | — | — | — | 0.01 | — | — | 0.62 | 0.57 | 0.61 | 0.40 | 0.19 | 0.31 | 1.00 | 1.00 | 1.00 |
| — | — | — | — | — | — | 0.81 | — | 0.79 | 0.34 | 0.04 | 0.23 | 1.00 | 0.12 | 1.00 | |
| True model
| |||||||||||||||
| 10i: αj = 0
|
10ii: αj = 0.40
|
10iii(a): αj ∼ pdfbeta
|
10iii(b): αj ∼ pdfbeta
|
10iv: αj ∼ pdfdelta
|
|||||||||||
| AIC | BIC | AICc | AIC | BIC | AICc | AIC | BIC | AICc | AIC | BIC | AICc | AIC | BIC | AICc | |
| 2iii | — | — | — | — | — | — | 0.15 | 0.15 | 0.15 | 0.01 | 0.01 | 0.01 | — | — | — |
| — | — | — | — | — | — | 0.09 | 0.09 | 0.09 | — | — | — | — | — | — | |
| — | 0.69 | 1.00 | — | 0.61 | 1.00 | 0.12 | 0.80 | 0.98 | — | 0.72 | 0.99 | — | 0.51 | 0.95 | |
| 5i | — | — | 0.03 | — | — | — | — | — | — | — | — | — | — | — | — |
| — | — | 0.02 | — | — | — | — | — | — | — | — | — | — | — | — | |
| — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |
| 5ii | — | — | — | — | — | 0.01 | — | — | — | — | — | — | — | — | — |
| — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |
| — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |
| 5iii | — | — | 0.02 | — | — | — | — | — | 0.28 | — | — | 0.02 | — | — | 0.08 |
| — | — | 0.01 | — | — | — | — | — | 0.06 | — | — | — | — | — | — | |
| — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |
| 5iv | — | — | 0.95 | — | — | 0.99 | — | — | 0.57 | — | — | 0.97 | — | — | 0.92 |
| — | — | 0.97 | — | — | 1.00 | — | — | 0.85 | — | — | 1.00 | — | — | 1.00 | |
| — | — | — | — | — | — | — | — | — | — | — | 0.01 | — | — | 0.05 | |
| 10i | 1.00 | 1.00 | — | — | 0.01 | — | — | — | — | — | — | — | — | — | — |
| 0.98 | 1.00 | — | — | — | — | — | — | — | — | — | — | — | — | — | |
| 0.93 | 0.31 | — | — | — | — | — | — | — | — | — | — | — | — | — | |
| 10ii | — | — | — | 0.97 | 0.99 | — | 0.32 | 0.38 | — | 0.84 | 0.97 | — | 0.02 | 0.15 | — |
| 0.02 | — | — | 1.00 | 1.00 | — | 0.37 | 0.45 | — | 0.91 | 0.97 | — | 0.16 | 0.22 | — | |
| 0.07 | — | — | 1.00 | 0.39 | — | 0.63 | 0.19 | — | 0.99 | 0.27 | — | 0.98 | 0.49 | — | |
| 10iii | — | — | — | — | — | — | 0.08 | 0.09 | — | 0.01 | 0.01 | — | — | — | — |
| — | — | — | — | — | — | 0.02 | 0.02 | — | — | — | — | — | — | — | |
| — | — | — | — | — | — | 0.03 | — | — | — | — | — | — | — | — | |
| 10iv | — | — | — | 0.03 | — | — | 0.45 | 0.38 | — | 0.14 | 0.01 | — | 0.98 | 0.85 | — |
| — | — | — | — | — | — | 0.52 | 0.44 | — | 0.09 | 0.03 | — | 0.84 | 0.78 | — | |
| — | — | — | — | — | — | 0.22 | 0.01 | — | 0.01 | 0.01 | — | 0.02 | — | — | |
The likelihood models chosen by various model selection criteria for simulated data sets are shown. Each entry shows the proportion of 100 simulated data sets for which a given likelihood model was selected, with a dash indicating that the model was never selected. Models that were not selected under any conditions are omitted. Entries are grouped according to the class of true model used to generate the simulated data (Table B1). Each section contains entries for the two (or three) methods used to generate the true fj-values (Table B2), with results for methods a, b, and c (if appropriate) given in descending order. The sections where the selected model agrees with the true model are underlined, and, when a single model was chosen for the great majority of data sets (>75%), this is indicated in italics.
TABLE B4.
Estimates of  from simulated data sets
from simulated data sets
| True model
|
|||||
|---|---|---|---|---|---|
| 5i: αj = 0 | 5ii: αj = 0.40 | 5iii(a): αj ∼ pdfbeta | 5iii(b): αj ∼ pdfbeta | 5iv: αj ∼ pdfdelta | |
| 2ii | 0.02 (0.03) | 0.39 (0.04) | 0.62 (0.09) | 0.45 (0.05) | 0.66 (0.04) |
| 0.03 (0.04) | 0.39 (0.04) | 0.63 (0.08) | 0.46 (0.04) | 0.66 (0.04) | |
| 2iii | 0.19 (0.02) | 0.29 (0.02) | 0.32 (0.02) | 0.30 (0.02) | 0.34 (0.03) |
| 0.16 (0.03) | 0.34 (0.02) | 0.38 (0.03) | 0.35 (0.03) | 0.43 (0.04) | |
| 2iv | 0.17 (0.02) | 0.23 (0.03) | 0.28 (0.11) | 0.23 (0.05) | 0.24 (0.06) |
| 0.14 (0.03) | 0.26 (0.03) | 0.44 (0.09) | 0.28 (0.04) | 0.40 (0.07) | |
| 5ii | 0.02 (0.03) | 0.39 (0.04) | 0.67 (0.13) | 0.45 (0.05) | 0.66 (0.04) |
| 0.03 (0.03) | 0.39 (0.04) | 0.68 (0.13) | 0.45 (0.04) | 0.66 (0.03) | |
| 5iii | 0.02 (0.03) | 0.39 (0.04) | 0.42 (0.08) | 0.43 (0.04) | 0.52 (0.07) |
| 0.03 (0.03) | 0.39 (0.04) | 0.43 (0.06) | 0.44 (0.04) | 0.57 (0.08) | |
| 5iv | 0.02 (0.03) | 0.39 (0.04) | 0.43 (0.07) | 0.41 (0.05) | 0.49 (0.06) |
| 0.03 (0.03) | 0.39 (0.04) | 0.44 (0.06) | 0.42 (0.05) | 0.48 (0.06) | |
| 10ii | 0.03 (0.04) | 0.40 (0.04) | 0.56 (0.08) | 0.44 (0.05) | 0.61 (0.04) |
| 0.03 (0.04) | 0.40 (0.04) | 0.56 (0.07) | 0.45 (0.04) | 0.62 (0.04) | |
| 10iii | 0.03 (0.04) | 0.40 (0.04) | 0.41 (0.18) | 0.43 (0.06) | 0.59 (0.06) |
| 0.03 (0.04) | 0.40 (0.04) | 0.45 (0.15) | 0.44 (0.04) | 0.61 (0.06) | |
| 10iv | 0.03 (0.04) | 0.40 (0.04) | 0.42 (0.06) | 0.43 (0.05) | 0.47 (0.07) |
| 0.03 (0.04) | 0.40 (0.04) | 0.43 (0.06) | 0.44 (0.04) | 0.47 (0.06) | |
| SEW | 0.01 (0.07) [0.95] | 0.40 (0.04) [0.94] | 0.62 (0.09) [0.22] | 0.45 (0.05) [0.78] | 0.66 (0.04) [0.02] |
| 0.01 (0.08) [0.89] | 0.40 (0.04) [0.95] | 0.63 (0.08) [0.13] | 0.46 (0.04) [0.72] | 0.66 (0.03) [0.00] | |
| FWW | 0.00 (0.06) [0.93] | 0.40 (0.04) [0.94] | 0.62 (0.09) [0.24] | 0.45 (0.05) [0.82] | 0.66 (0.04) [0.00] |
| 0.01 (0.07) [0.91] | 0.40 (0.04) [0.96] | 0.63 (0.08) [0.09] | 0.46 (0.04) [0.69] | 0.66 (0.03) [0.00] | |
| 11ii | 0.63 (0.03) | ||||
| 0.67 (0.03) | |||||
| True model
| |||||
| 10i: αj = 0 | 10ii: αj = 0.40 | 10iii(a): αj ∼ pdfbeta | 10iii(b): αj ∼ pdfbeta | 10iv: αj ∼ pdfdelta | |
| 2ii | 0.06 (0.08) | 0.40 (0.08) | 0.63 (0.11) | 0.45 (0.10) | 0.65 (0.07) |
| 0.03 (0.05) | 0.40 (0.05) | 0.63 (0.01) | 0.47 (0.06) | 0.66 (0.04) | |
| 0.61 (0.03) | 0.76 (0.02) | 0.85 (0.04) | 0.78 (0.02) | 0.86 (0.02) | |
| 2iii | 0.18 (0.03) | 0.27 (0.03) | 0.30 (0.03) | 0.28 (0.03) | 0.32 (0.03) |
| 0.16 (0.02) | 0.31 (0.03) | 0.36 (0.04) | 0.33 (0.03) | 0.41 (0.04) | |
| 0.33 (0.03) | 0.49 (0.03) | 0.53 (0.03) | 0.51 (0.03) | 0.58 (0.04) | |
| 2iv | 0.15 (0.03) | 0.20 (0.04) | 0.29 (0.15) | 0.20 (0.05) | 0.23 (0.07) |
| 0.13 (0.03) | 0.23 (0.04) | 0.40 (0.12) | 0.25 (0.05) | 0.38 (0.08) | |
| 0.37 (0.06) | 0.61 (0.04) | 0.69 (0.06) | 0.63 (0.04) | 0.74 (0.05) | |
| 5ii | 0.07 (0.08) | 0.41 (0.08) | 0.71 (0.14) | 0.47 (0.10) | 0.67 (0.07) |
| 0.04 (0.05) | 0.41 (0.05) | 0.68 (0.13) | 0.47 (0.06) | 0.67 (0.04) | |
| 0.61 (0.03) | 0.76 (0.02) | 0.85 (0.05) | 0.78 (0.05) | 0.66 (0.03) | |
| 5iii | 0.09 (0.06) | 0.37 (0.05) | 0.43 (0.06) | 0.40 (0.05) | 0.51 (0.06) |
| 0.06 (0.04) | 0.38 (0.04) | 0.44 (0.05) | 0.41 (0.05) | 0.51 (0.05) | |
| 0.15 (0.07) | 0.53 (0.06) | 0.58 (0.06) | 0.56 (0.06) | 0.65 (0.06) | |
| 5iv | 0.16 (0.08) | 0.43 (0.06) | 0.46 (0.06) | 0.45 (0.06) | 0.53 (0.06) |
| 0.16 (0.08) | 0.43 (0.06) | 0.46 (0.06) | 0.45 (0.05) | 0.52 (0.05) | |
| 0.27 (0.06) | 0.52 (0.05) | 0.56 (0.06) | 0.53 (0.05) | 0.60 (0.06) | |
| 10ii | 0.01 (0.02) | 0.36 (0.05) | 0.55 (0.10) | 0.40 (0.06) | 0.56 (0.07) |
| 0.01 (0.02) | 0.35 (0.04) | 0.53 (0.08) | 0.40 (0.05) | 0.58 (0.06) | |
| 0.04 (0.05) | 0.40 (0.06) | 0.61 (0.09) | 0.46 (0.05) | 0.66 (0.03) | |
| 10iii | 0.01 (0.02) | 0.36 (0.05) | 0.29 (0.20) | 0.38 (0.09) | 0.54 (0.11) |
| 0.01 (0.02) | 0.35 (0.04) | 0.35 (0.20) | 0.40 (0.05) | 0.57 (0.08) | |
| 0.04 (0.05) | 0.39 (0.06) | 0.38 (0.24) | 0.45 (0.06) | 0.65 (0.03) | |
| 10iv | 0.01 (0.02) | 0.36 (0.05) | 0.39 (0.08) | 0.39 (0.06) | 0.48 (0.09) |
| 0.01 (0.02) | 0.36 (0.04) | 0.39 (0.06) | 0.39 (0.06) | 0.47 (0.08) | |
| 0.04 (0.05) | 0.40 (0.06) | 0.46 (0.08) | 0.45 (0.05) | 0.59 (0.08) | |
| SEW | 0.09 (0.09) [0.79] | 0.45 (0.06) [0.83] | 0.67 (0.10) [0.16] | 0.51 (0.07) [0.53] | 0.69 (0.05) [0.01] |
| 0.10 (0.07) [0.74] | 0.45 (0.05) [0.71] | 0.67 (0.08) [0.02] | 0.52 (0.05) [0.36] | 0.69 (0.04) [0.00] | |
| 0.33 (0.09) [0.29] | 0.58 (0.06) [0.43] | 0.73 (0.07) [0.03] | 0.62 (0.05) [0.27] | 0.76 (0.04) [0.00] | |
| FWW | 0.02 (0.15) [0.93] | 0.41 (0.08) [0.92] | 0.64 (0.10) [0.38] | 0.46 (0.10) [0.81] | 0.66 (0.07) [0.13] |
| 0.01 (0.08) [0.95] | 0.41 (0.05) [0.97] | 0.64 (0.08) [0.14] | 0.47 (0.06) [0.72] | 0.67 (0.04) [0.02] | |
| 0.62 (0.03) [0.00] | 0.77 (0.02) [0.00] | 0.85 (0.04) [0.00] | 0.79 (0.02) [0.00] | 0.87 (0.02) [0.00] | |
| 11ii | 0.55 (0.05) | ||||
| 0.60 (0.03) | |||||
| 0.70 (0.03) | |||||
Mean estimates of 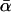 obtained from 100 data sets are shown. The standard deviations of these estimates over the data sets are also shown in parentheses. Results from the FWW and SEW estimators are included in addition to the various ML models, and for these estimators the proportion of data sets for which the true value of
obtained from 100 data sets are shown. The standard deviations of these estimates over the data sets are also shown in parentheses. Results from the FWW and SEW estimators are included in addition to the various ML models, and for these estimators the proportion of data sets for which the true value of  appeared in the 95% bootstrap confidence intervals is shown in brackets. Layout otherwise resembles Table B3. Models that were regularly selected by the AIC, whether or not they are the true model, are underlined (see Table B3). Mean estimates that differ by >0.15 from the true value of
appeared in the 95% bootstrap confidence intervals is shown in brackets. Layout otherwise resembles Table B3. Models that were regularly selected by the AIC, whether or not they are the true model, are underlined (see Table B3). Mean estimates that differ by >0.15 from the true value of  (either 0.4 or 0.0) are indicated in italics.
(either 0.4 or 0.0) are indicated in italics.
TABLE B5.
False detection of adaptive evolution in simulated data sets
| True model
|
||
|---|---|---|
| 5i | 10i | |
| 2 | 0.08 | 0.35 |
| 0.10 | 0.18 | |
| 1.00 | ||
| 5 | 0.07 | 0.36 |
| 0.07 | 0.25 | |
| 1.00 | ||
| 10 | 0.08 | 0.00 |
| 0.13 | 0.02 | |
| 0.07 | ||
| SEW | 0.01 | 0.08 |
| 0.00 | 0.12 | |
| 0.39 | ||
| FWW | 0.00 | 0.02 |
| 0.01 | 0.00 | |
| 1.00 | ||
| MHz | 0.08 | 0.05 |
| 0.11 | 0.05 | |
| 0.10 | ||
| 11 | 1.00 | 1.00 |
| 1.00 | 1.00 | |
| 1.00 | ||
Entries show the proportion of data sets for which false evidence of adaptive evolution was obtained from various estimators. In all cases, the true model was of type i (with α fixed at zero). For the ML estimators, entries show the proportion of 100 simulated data sets for which a type ii model (with α as a free parameter) was preferred to the true type i model, under the AIC model selection criterion. For the SEW and FWW estimators, entries show the proportion of data sets for which a positive estimate of  was obtained in at least 95% of the bootstrap resamplings. Also shown (MHz) is the number of false positives obtained from the Mantel-Haenszel test (Snedecor and Cochran 1980; Bartolomé et al. 2005). Layout is otherwise identical to Table B3. Conditions where the estimators faired particularly poorly (yielding false positives in excess of 15%), are indicated in italics. Note that false negatives (i.e., failures to detect selection that was present in the true model) were extremely rare under all estimators (always <2%).
was obtained in at least 95% of the bootstrap resamplings. Also shown (MHz) is the number of false positives obtained from the Mantel-Haenszel test (Snedecor and Cochran 1980; Bartolomé et al. 2005). Layout is otherwise identical to Table B3. Conditions where the estimators faired particularly poorly (yielding false positives in excess of 15%), are indicated in italics. Note that false negatives (i.e., failures to detect selection that was present in the true model) were extremely rare under all estimators (always <2%).
TABLE B6.
Evidence of between-locus variation in α from simulated data sets
| Beta distribution: true model
|
Two-spiked distribution: true model
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 5ii | 5iii(a) | 5iii(b) | 5iv | 5ii | 5iii(a) | 5iii(b) | 5iv | |
| 5 | 0.00 | 0.95 | 0.12 | 0.92 | 0.00 | 0.99 | 0.42 | 1.00 |
| 0.00 | 0.91 | 0.05 | 0.40 | 0.01 | 0.99 | 0.34 | 1.00 | |
| 10 | 0.00 | 0.26 | 0.00 | 0.00 | 0.00 | 0.68 | 0.03 | 0.96 |
| 0.00 | 0.20 | 0.00 | 0.00 | 0.00 | 0.76 | 0.05 | 0.95 | |
| Beta distribution: true model
|
Two-spiked distribution: true model
|
|||||||
| 10ii | 10iii(a) | 10iii(b) | 10iv | 10ii | 10iii(a) | 10iii(b) | 10iv | |
| 10 | 0.00 | 0.45 | 0.02 | 0.05 | 0.03 | 0.68 | 0.16 | 0.98 |
| 0.00 | 0.31 | 0.01 | 0.01 | 0.00 | 0.63 | 0.09 | 0.84 | |
| 0.00 | 0.17 | 0.00 | 0.00 | 0.00 | 0.34 | 0.01 | 0.02 | |
The proportion of 100 simulated data sets for which the AIC favored a model with variable α over a model in which α took a common value at all loci. The left half compares models of type ii (fixed α) to models of type iii (beta-distributed α), and the right half compares models of type ii to models of type iv (two-spike distributed α). Layout is otherwise identical to Table B3. Conditions when the estimation faired particularly poorly (supporting variable α when the true model had a fixed value, or vice versa), are indicated in italics. Not shown are results when models were too small (i.e., of type 2 or of type 5 when the true model was of type 10). In these cases, between-locus variation in α was detected in almost every case, whether or not it was present in the data.
Table B4 shows the generally accurate estimation of  under the ML method with the AIC. Performance is seen to decline, however, in the extreme cases—i.e., when the αj were highly variable and an extreme negative correlation was present between the fj and θj [models iii(a) and iv of Table B1 and model c of Table B2]. Also clearly evident is the poor performance of the SEW and FWW estimators if either of these conditions held and of ML model 11ii.
under the ML method with the AIC. Performance is seen to decline, however, in the extreme cases—i.e., when the αj were highly variable and an extreme negative correlation was present between the fj and θj [models iii(a) and iv of Table B1 and model c of Table B2]. Also clearly evident is the poor performance of the SEW and FWW estimators if either of these conditions held and of ML model 11ii.
Table B5 shows that false detection of selection was common if the ML model was much smaller than the true model (meaning that the models commonly selected by the BIC and AICc often gave misleading results), but that performance was good otherwise. Also indicated is the very high rate of false positives obtained under the SEW and, especially, the FWW estimators, when the fj and θj were negatively correlated (model c of Table B2). The poor performance of model 11ii is also evident.
Table B6 shows the generally poor performance of the type iii beta-distribution models in detecting between-locus variation in α, especially when the true model was parameter rich (of class 10). Also shown is the superior performance of the two-spiked distribution in detecting such variation [note that when the true model was 10iii(a), models of type iv were preferred to constant-α models more regularly than were the true type iii models]. Also clearly evident is the failure of all methods to detect between-locus variation in α when it was of limited magnitude [i.e., when the true model was of type iii(b)].
APPENDIX C
Brief derivations of the heuristic estimators of 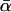 introduced by Fay et al. (2001) and Smith and Eyre-Walker (2002) are presented. These derivations clarify the biases to which these estimators are subject. The estimator of Fay et al. (2001) (FWW) is given by
introduced by Fay et al. (2001) and Smith and Eyre-Walker (2002) are presented. These derivations clarify the biases to which these estimators are subject. The estimator of Fay et al. (2001) (FWW) is given by
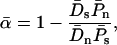 |
(C1) |
where overbars denote the average over all genes; this is, of course, equivalent to summing the values over all loci. The estimator of Smith and Eyre-Walker (SEW) is
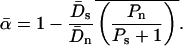 |
(C2) |
These estimators can be derived from Equations 1 and 5, on the assumptions that no mildly deleterious mutations are segregating (i.e., that γ→0) and that the correction to the estimated divergence resulting from segregating polymorphism, 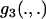 , can be neglected [this will be so on the condition that
, can be neglected [this will be so on the condition that  ].
].
Consider first the factor containing the divergence measures, which is common to both estimators. If there are no correlations between the parameters, then the expected value of this factor is
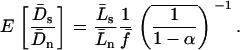 |
(C3) |
The factor containing α can be clarified using two series expansions and excluding higher-order terms; this yields
 |
(C4) |
Because  is not explicitly canceled, this suggests that both estimators will provide an upwardly biased estimate of
is not explicitly canceled, this suggests that both estimators will provide an upwardly biased estimate of  if α is highly variable between loci. The same bias can be demonstrated more rigorously from Jensen's inequality.
if α is highly variable between loci. The same bias can be demonstrated more rigorously from Jensen's inequality.
Now consider the factors, unique to each of the estimators, that contain the polymorphism measures. Using Equations C3 and C4 in Equations C1 and C2 shows that the purpose of these polymorphism factors is to cancel the quantity  from Equation C3. To understand these factors, define
from Equation C3. To understand these factors, define 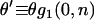 as the expected neutral polymorphism at a single site in the sample of alleles. The expression contained in the FWW estimator is then
as the expected neutral polymorphism at a single site in the sample of alleles. The expression contained in the FWW estimator is then
 |
(C5) |
Equation C5 will equal the quantity required,  , only if
, only if  and f do not covary over loci. Negative covariation, such as is expected under the nearly neutral theory of evolution (e.g., Ohta 1992; see main text) will lead to an underestimation of
and f do not covary over loci. Negative covariation, such as is expected under the nearly neutral theory of evolution (e.g., Ohta 1992; see main text) will lead to an underestimation of  and so an overestimation of
and so an overestimation of  .
.
To understand the equivalent factor for the SEW estimator, consider a Poisson deviate, X, with expected value λ. In this case,  . Using this result, we obtain
. Using this result, we obtain
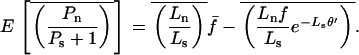 |
(C6) |
Because there is typically little variation in the Ln/Ls ratio, Equation C6 may closely approximate the required  , but only if the second term of Equation C6 is very small. This is guaranteed by the presence of the factor
, but only if the second term of Equation C6 is very small. This is guaranteed by the presence of the factor  , if all values of
, if all values of  are reasonably large. Recalling from Equations 5 that
are reasonably large. Recalling from Equations 5 that 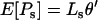 , this explains why Smith and Eyre-Walker (2002) excluded genes with low values of Ps from their analysis. This is dangerous, however, as excluding genes in this way means that Equation C6 must be replaced with a conditional expectation. This results in a complex expression and, more importantly, can upwardly bias the estimate of
, this explains why Smith and Eyre-Walker (2002) excluded genes with low values of Ps from their analysis. This is dangerous, however, as excluding genes in this way means that Equation C6 must be replaced with a conditional expectation. This results in a complex expression and, more importantly, can upwardly bias the estimate of 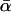 . That said, for many real data sets, this bias is unlikely to be substantial.
. That said, for many real data sets, this bias is unlikely to be substantial.
References
- Abramowitz, M., and I. Stegun, 1965. Handbook of Mathematical Functions. Dover, New York.
- Akaike, H., 1974. A new look at statistical model identification. IEEE Trans. Automat. Control 19: 716–723.
- Akashi, H., 1996. Molecular evolution between Drosophila melanogaster and D. simulans: reduced codon bias, faster rates of amino acid substitution, and large proteins in D. melanogaster. Genetics 144: 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., and S. W. Schaeffer, 1997. Natural selection and the frequency distributions of “silent” DNA polymorphism in Drosophila. Genetics 146: 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., 2001. Contrasting patterns of X–linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Mol. Biol. Evol. 18: 279–290. [DOI] [PubMed] [Google Scholar]
- Bartolomé, C., X. Maside, S. Yi, A. L. Grant and B. Charlesworth, 2005. Patterns of selection on synonymous and nonsynonymous variatns in Drosophila miranda. Genetics 169: 1495–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun, D. J., 2001. The frequency distribution of nucleotide variation in Drosophila simulans. Mol. Biol. Evol. 18: 1343–1352. [DOI] [PubMed] [Google Scholar]
- Begun, D. J., 2002. Protein variation in Drosophila simulans, and comparison of genes from centromeric versus noncentromeric regions of chromosome 3. Mol. Biol. Evol. 19: 201–203. [DOI] [PubMed] [Google Scholar]
- Begun, D. J., and P. Whitley, 2000. Reduced X-linked nucleotide polymorphism in Drosophila simulans. Proc. Natl. Acad. Sci. USA 97: 5960–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne, N., and A. Eyre-Walker, 2004. The genomic rate of adaptive amino acid substitution in Drosophila. Mol. Biol. Evol. 21: 1350–1360. [DOI] [PubMed] [Google Scholar]
- Bulmer, M., 1991. The selection-mutation-drift theory of synonymous codon usage. Genetics 129: 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante, C. D., J. Wakeley, S. A. Sawyer and D. L. Hartl, 2001. Directional selection and the site-frequency spectrum. Genetics 159: 1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante, C. D., R. Nielsen, S. A. Sawyer, K. M. Olsen, M. D. Purugganan et al., 2002. The cost of inbreeding in Arabidopsis. Nature 416: 531–534. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., 1994. The effect of background selection against deleterious mutations on weakly selected, linked variants. Genet. Res. 63: 213–227. [DOI] [PubMed] [Google Scholar]
- Charlesworth, D., B. Charlesworth and M. T. Morgan, 1995. The pattern of neutral molecular variation under the background selection model. Genetics 141: 1619–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., 1979. Mathematical Population Genetics. Springer, Berlin.
- Eyre-Walker, A., 2002. Changing effective population size and the McDonald–Kreitman test. Genetics 162: 2017–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J. C., G. J. Wycoff and C.-I. Wu, 2001. Positive and negative selection on the human genome. Genetics 158: 1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J. C., G. J. Wycoff and C.-I. Wu, 2002. Testing the neutral theory of molecular evolution with genomic data from Drosophila. Nature 415: 1024–1026. [DOI] [PubMed] [Google Scholar]
- Galassi, M., J. Davies, J. Theiler, B. Gough, G. Jungman et al., 2004. GNU Scientific Library Reference Manual: Ed. 1.6, for GSL Version 1.6. Network Theory, Bristol, UK.
- Gillespie, J. H., 1991. The Causes of Molecular Evolution (Oxford Series in Ecology and Evolution). Oxford University Press, Oxford.
- Gillespie, J. H., 1995. On Ohta's hypothesis: most amino acid substitutions are deleterious. J. Mol. Evol. 40: 64–69. [Google Scholar]
- Gillespie, J. H., 2001. Is the population size of a species relevant to its evolution? Evolution 55: 2161–2169. [DOI] [PubMed] [Google Scholar]
- Goldman, N., and Z. Yang, 1994. A codon based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11: 725–736. [DOI] [PubMed] [Google Scholar]
- Gravot, E., M. Huet and M. Veuille, 2004. Effect of breeding structure on population genetic parameters in Drosophila. Genetics 166: 779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin, M. T., and M. Veuille, 1999. Population structure among African and derived populations of Drosophila simulans: evidence for ancient subdivision and recent admixture. Genetics 153: 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, S. Y. W., and G. Larson, 2006. Molecular clocks: when times are a-changin'. Trends Ecol. Evol. 22: 79–83. [DOI] [PubMed] [Google Scholar]
- Holder, M., and P. O. Lewis, 2003. Phylogenetic estimation: traditional and Bayesian approaches. Nat. Rev. Genet. 4: 275–284. [DOI] [PubMed] [Google Scholar]
- Hurvich, C. M., and C.-L. Tsai, 1989. Regression and time series model selection in small samples. Biometrika 76: 297–307. [Google Scholar]
- Kass, R. E., and A. E. Raftery, 1995. Bayes factors. J. Am. Stat. Assoc. 90: 773–795. [Google Scholar]
- Kern, A. D., C. D. Jones and D. J. Begun, 2004. Molecular population genetics of male accessory gland proteins in the Drosophila simulans complex. Genetics 167: 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1957. Some problems of stochastic processes in genetics. Ann. Math. Stat. 28: 882–901. [Google Scholar]
- Kondrashov, A., S. Sunyaev and F. Kondrashov, 2002. Dobzhansky-Muller incompatibilities in protein evolution. Proc. Natl. Acad. Sci. USA 99: 14878–14883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman, M., and H. Akashi, 1995. Molecular evidence for natural selection. Annu. Rev. Ecol. Syst. 26: 403–422. [Google Scholar]
- Kuha, J., 2004. AIC and BIC: comparisons of assumptions and performance. Sociol. Methods Res. 33: 188–229. [Google Scholar]
- Lachaise, D., M.-L. Cariou, J. R. David, F. Lemeunier, L. Tsacas et al., 1988. Historical biogeography of the Drosophila melanogaster species subgroup. Evol. Biol. 22: 159–225. [Google Scholar]
- Li, Y. J., Y. Satta and N. Takahata, 1999. Paleo-demography of the Drosophila melanogaster subgroup: application of the maximum likelihood method. Genes Genet. Syst. 74: 117–127. [DOI] [PubMed] [Google Scholar]
- Linhart, H., and W. Zucchini, 1986. Model Selection. John Wiley & Sons, New York.
- McDonald, J. H., and M. Kreitman, 1991. Adaptive evolution at the Adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- McVean, G., and J. Vieira, 2001. Inferring parameters of mutation, selection and demography from patterns of synonymous site evolution in Drosophila. Genetics 157: 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., 1992. The nearly neutral theory of molecular evolution. Annu. Rev. Ecol. Syst. 23: 263–286. [Google Scholar]
- Piganeau, G., and A. Eyre-Walker, 2003. Estimating the distribution of fitness effects from DNA sequence data: implications for the molecular clock. Proc. Natl. Acad. Sci. USA 100: 10335–10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada, D., and K. A. Crandall, 2001. Selecting the best fit model of nucleotide substitution. Syst. Biol. 50: 580–601. [PubMed] [Google Scholar]
- Sawyer, S. A., and D. L. Hartl, 1992. Population genetics of polymorphism and divergence. Genetics 132: 1161–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer, S. A., R. J. Kulathinal, C. D. Bustamante and D. L. Hartl, 2003. Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection. J. Mol. Evol. 57: S154–S164. [DOI] [PubMed] [Google Scholar]
- Schöfl, G., and C. Schlötterer, 2004. Patterns of microsatellite variability among X chromosomes and autosomes indicate a high frequency of beneficial mutations in non-African D. simulans. Mol. Biol. Evol. 21: 1384–1390. [DOI] [PubMed] [Google Scholar]
- Shibata, R., 1976. Selection of the order of an autoregressive model by Akaike's information criterion. Biometrika 63: 117–126. [Google Scholar]
- Smith, N. G. C., and A. Eyre-Walker, 2002. Adaptive protein evolution in Drosophila. Nature 415: 1022–1024. [DOI] [PubMed] [Google Scholar]
- Snedecor, G. W., and W. G. Cochran, 1980. Statistical Methods. Iowa State University Press, Ames, IA.
- Takahata, N., and Y. Satta, 2002. Pre-speciation coalescence and the effective size of ancestral populations, pp. 52–71 in Modern Developments in Theoretical Population Genetics, the Legacy of Gustave Malecot, edited by M. Slatkin and M. Veuille. Oxford University Press, Oxford.
- Wall, J. D., P. Andolfatto and M. Przeworski, 2002. Testing models of selection and demography in Drosophila simulans. Genetics 162: 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, S. H., R. Hernandez, A. Fledel-Alon, R. Nielsen and C. D. Bustamante, 2005. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc. Natl. Acad. Sci. USA 102: 7882–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., 1994. Maximum likelihood estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39: 306–314. [DOI] [PubMed] [Google Scholar]
- Yang, Z., and R. Nielsen, 1998. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J. Mol. Evol. 46: 409–418. [DOI] [PubMed] [Google Scholar]
- Zhu, L., and C. D. Bustamante, 2005. A composite likelihood approach for detecting directional selection from DNA sequence data. Genetics 170: 1411–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]