

Abstract
The distribution of fitness effects of new mutations is a fundamental parameter in genetics. Here we present a new method by which the distribution can be estimated. The method is fairly robust to changes in population size and admixture, and it can be corrected for any residual effects if a model of the demography is available. We apply the method to extensively sampled single-nucleotide polymorphism data from humans and estimate the distribution of fitness effects for amino acid changing mutations. We show that a gamma distribution with a shape parameter of 0.23 provides a good fit to the data and we estimate that >50% of mutations are likely to have mild effects, such that they reduce fitness by between one one-thousandth and one-tenth. We also infer that <15% of new mutations are likely to have strongly deleterious effects. We estimate that on average a nonsynonymous mutation reduces fitness by a few percent and that the average strength of selection acting against a nonsynonymous polymorphism is ∼9 × 10−5. We argue that the relaxation of natural selection due to modern medicine and reduced variance in family size is not likely to lead to a rapid decline in genetic quality, but that it will be very difficult to locate most of the genes involved in complex genetic diseases.
IT has been estimated that each of us receives more than one harmful amino acid mutation each generation (Eyre-Walker and Keightley 1999). But how harmful are these mutations on average, and what proportion of mutations are weakly, mildly, and strongly deleterious? In short, what is the distribution of fitness effects of new mutations? This question is central to understanding several topics in human biology, including the genetic basis of disease and the likely consequences of relaxing natural selection through modern medicine and better living standards (Muller 1950; Crow 1997; Lynch et al. 1999). Furthermore, the distribution of fitness effects is central to our understanding of many other problems in genetics and evolution, including the maintenance of genetic variation (Charlesworth et al. 1993), the long-term survival of small populations (Lande 1994; Lynch et al. 1995), and the basis of the molecular clock (Ohta 1977).
Although the distribution of fitness effects is an important parameter in genetics and evolutionary biology, relatively little is known with certainty about its form. Mutagenesis and mutation-accumulation experiments suggest that the distribution of fitness effects is highly leptokurtic, such that most mutations appear to have effects of <1% (Keightley 1994, 1996; Davies et al. 1999; Vassilieva et al. 2000; Estes et al. 2004). This has been broadly corroborated by studies of DNA sequence evolution, although the precise form of the distribution inferred by different studies varies considerably. Piganeau and Eyre-Walker (2003) and Loewe et al. (2006) found that a gamma distribution, with a shape parameter of less than one, was consistent with nonsynonymous data from animal mitochondria and Drosophila, respectively, whereas Nielsen and Yang (2003) and Sawyer et al. (2002) showed that a normal distribution was consistent with similar data.
These studies were based on different methods, each with its own advantages and disadvantages. However, they all share two limitations. First, they are based on relatively little information. Generally these methods use either divergence data or divergence data in association with a single statistic summarizing polymorphism data, which limits the power of these analyses. Second, the use of divergence data introduces the problem of adaptive substitutions, which may influence estimates of the distribution of fitness effects (although note that this should not be a problem for the method of Loewe et al. 2006, which estimates the proportion of adaptive substitutions). Furthermore, estimates based on a combination of divergence and polymorphism data may be affected by differences in effective population sizes associated with the polymorphism and divergence data, respectively.
Despite these potential limitations, previous work has progressed toward a more precise quantification of the distribution of fitness effects in humans. Fay et al. (2001) used human single-nucleotide polymorphism data to infer that ∼20% of amino acid changing mutations were neutral in humans, with a further ∼20% of the remaining deleterious mutations being sufficiently weakly selected to contribute to polymorphism. Eyre-Walker et al. (2002; see also Yampolsky et al. 2005) used an estimate of the effective population size to estimate that >70% of mutations in humans are deleterious with strengths of selection >10−4. The distribution of fitness effects for nonsynonymous mutations that are strongly deleterious is much harder to estimate in humans because these mutations will only very rarely be seen. On the basis of data on mutations responsible for Mendelian disease, Yampolsky et al. (2005) have suggested that ∼25% of mutations have effects of >1%.
The aim of this work is twofold: first to develop a method by which the distribution of fitness effects can be inferred, overcoming some of the limitations of previous methods by using polymorphism data alone and incorporating information from the distribution of allele frequencies, and second to give a more complete estimate of the distribution of fitness effects of nonsynonymous mutations in humans.
MATERIALS AND METHODS
Method:
Under a standard population genetic model it is possible to write down expressions for the number of single-nucleotide polymorphisms (SNPs) we expect to observe in j out of n alleles at both selected (e.g., nonsynonymous) sites Pn(j) and nonselected (e.g., intron) sites Pi(j):
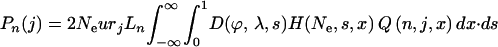 |
(1) |
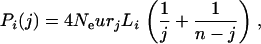 |
(2) |
where
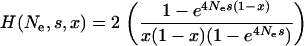 |
(3) |
and
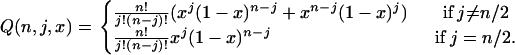 |
(4) |
u is the nucleotide mutation rate, Ne is the effective population size, and Ln and Li are the numbers of nonsynonymous and intron sites, respectively. H(Ne, s, x) is the time that a new semidominant deleterious mutation of heterozygous selection strength s spends between x and x + dx (Wright 1938) and Q(n, j, x) is the probability of observing a mutation at frequency x in the total population in j of n sequences. We do not attempt to infer the direction of mutations, so a SNP segregating at a frequency of x is equivalent to a SNP at a frequency of 1 − x. We do not orientate SNPs for a number of reasons: (i) the method works well without orientating them, (ii) the method is more general since an outgroup is not needed, and (iii) even with a close outgroup, some SNPs are likely to be misorientated. D(ϕ, λ, s) is the distribution of fitness effects that we assume here to be a gamma distribution,
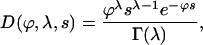 |
(5) |
where λ is the shape parameter and ϕ is a parameter that is related to the mean of the distribution, γ = 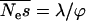 . Goodness-of-fit tests suggest that the gamma distribution is a satisfactory fit to the data (see results). We assume that all selected mutations are deleterious, although selection can be sufficiently weak that they are effectively neutral.
. Goodness-of-fit tests suggest that the gamma distribution is a satisfactory fit to the data (see results). We assume that all selected mutations are deleterious, although selection can be sufficiently weak that they are effectively neutral.
The rj parameters take into account some of the effects of demographic change. We allow each frequency category to have its own effective mutation rate urj, where rj is the mutation rate of the jth frequency category relative to the mutation rate for singletons. For example, under population size expansion the allele frequency distribution of neutral mutations is skewed toward rare alleles so the rj values are less than one. In essence we are assuming that demography affects the allele frequency distribution of both neutral and selected mutations to a similar extent. In reality this is not the case: under population size expansion, for example, the allele frequency distribution of selected mutations is likely to be skewed more dramatically than that of neutral mutations. However, simulations (see below) suggest that the approximation works well and we introduce a method by which any residual bias can be corrected if the demography of the population being studied is known.
If we assume free recombination between sites, we can write down the likelihood of observing the data for a single gene, given the parameters of the model, because the numbers of SNPs in each frequency category are Poisson distributed,
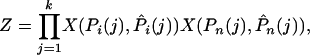 |
(6) |
where
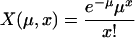 |
and k is the number of frequency categories. The likelihood for multiple genes is obtained by multiplying the likelihoods of individual genes.
Statistical analysis:
Since the model for multiple genes is parameter rich, we estimated the parameters of our model using a Monte Carlo Markov chain running the Metropolis–Hastings algorithm; this is Bayesian inference with a uniform prior. We allowed each gene to have its own Ne and u values with the effective mutation rate parameters, rj, and parameters of the gamma distribution, λ and γ, shared between genes. In effect we are assuming that demography affects all loci to a similar extent and we are estimating the overall distribution of fitness effects across loci.
All parameters, except the mutation rates, were given uniform priors with large bounds. The mutation rates were constrained to vary by twofold around the genomic average. This is in line with recent estimates of the variation in the mutation rate that is observed in the human genome (Webster et al. 2004). We needed to place bounds on the mutation rate because our data include genes that had some intron SNPs but no nonsynonymous SNPs. The pattern in such genes can be explained by any combination of Ne and u so long as 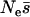 is very large; this means that Ne can become infinitely large and u infinitely small. Each chain was run for a burn in of 1,000,000 steps before being sampled for a further 50,000,000 steps. Convergence and mixing were checked graphically.
is very large; this means that Ne can become infinitely large and u infinitely small. Each chain was run for a burn in of 1,000,000 steps before being sampled for a further 50,000,000 steps. Convergence and mixing were checked graphically.
Data:
The data were downloaded from the Environmental Genome Project (EGP) website (http://egp.gs.Washington.edu) (Livingston et al. 2004). These comprise 320 autosomal genes resequenced in 90 individuals. Due to technical problems, on average only 170 of the 180 alleles were successfully resolved. However, all sites were subsequently treated as being from a sample of 170 alleles—i.e., the allele frequency was multiplied by 170 to yield an estimated number of alleles containing the SNP. Calculations (not shown) suggest that this is a good approximation, and one that is necessary to make the method computationally tractable. Although it is possible to calculate the likelihood of the data for every frequency category individually, this is very time consuming. We therefore chose to group frequency classes according to the following scheme: we considered singletons by themselves and then grouped SNPs that were present in 2–3, 4–7, 8–15, 16–31, and 32–85 alleles.
The number of nonsynonymous sites was calculated assuming a transition:transversion ratio of 3:1: i.e., Ln = L0 + 2L2/5 + L3/3, where Lx is the number of x-fold degenerate sites. CpG sites experience a 10-fold higher mutation rate than other sites (Sved and Bird 1990). We therefore calculated effective numbers of intron and nonsynonymous sites by multiplying CpG sites by 10: i.e., Ln = Ln(non-CpG) + 10 · Ln(CpG). The analysis was also run excluding CpG sites with similar results [β = 0.21 (0.14–0.27), γ = 487 (145, 1949)].
Simulations:
Simulations were performed to test the behavior of the method. Forward simulations had to be used because there is currently no other way in which to simulate population size changes with selection. The simulation was performed using a pseudo-sampling variance procedure (Kimura 1979) in which random genetic drift was simulated by generating numbers from a binomial distribution. A haploid population subject to mutation, selection, and genetic drift was simulated according to three demographic models. In the first model we follow the scheme set out by Adams and Hudson (2004): the population was allowed to equilibrate for 4N0 generations before being reduced to a size of fintN0 and then allowed to grow exponentially to a size of frecN0 over the course of 2tN0 generations. In each simulation 500,000 selected and neutral sites were independently simulated—i.e., free recombination was assumed. The mutation rate was selected such that 2frecN0u = 0.005, which prevents violation of the infinite-sites assumption, and the parameters of the gamma distribution were set at λ = 0.23, γ = 425, the values of the gamma distribution estimated from the data. We simulated data with a variety of different demographic parameter values. In the second model we simulated a model of population admixture. The population was allowed to equilibrate for 4N0 generations before being split into two equal sized populations of size N0/2, which were then allowed to evolve independently for 2tN0 generations before being remixed for the generation in which sampling takes place. Finally, we simulated a population according to the demographic model of Williamson et al. (2005) since they estimated demographic parameters for the data that we have used here. In this model the population was allowed to equilibrate for 4N0 generations before being expanded instantly to a size of frecN0, where it remained for another 2tN0 generations until sampled (please note that we use a different nomenclature from that of Williamson et al.).
RESULTS
Description:
The method we have developed is conceptually simple. Using standard population genetic theory it is possible to write down expressions for the number of polymorphisms we expect to observe segregating at a particular frequency in a sample of DNA sequences. We can write these expressions both for neutral mutations and for mutations that are subject to selection and in which the strength of selection is drawn from some distribution. Here we assume that the distribution of fitness effects can be described by a gamma distribution. This is a flexible monotonic distribution that can take a variety of shapes. It is governed by two parameters, a shape parameter (λ) and the mean of the distribution (γ). The method allows the mutation rate and effective population size to differ between loci and it can also accommodate demographic change (e.g., population size expansion and contraction).
The method requires that there are two types of site: a set of sites at which all mutations are neutral and sites at which some of the mutations are subject to selection. We assume here that mutations in introns are neutral and estimate the distribution of fitness effects for mutations that change an amino acid. Although some parts of introns do appear to be subject to selection in some organisms (Shabalina and Kondrashov 1999; Bergman and Kreitman 2001; Keightley and Gaffney 2003; Andolfatto 2005; Haddrill et al. 2005), there is little evidence of this in humans (Keightley et al. 2005).
We have applied our method to 320 genes sequenced in humans as part of the Environmental Genome Project (Livingston et al. 2004). These genes are thought to be involved in our interaction with our environment, so they are not a random selection of genes, but they represent by far the largest data set of human genes for which there are carefully sampled SNPs. The depth of sampling is particularly useful, since mutations of quite strong effect have some chance of being sampled, and hence we have more information about the distribution of fitness effects for a broader spectrum of selection coefficients. In total there are 965 nonsynonymous and 30,065 intron SNPs in the data. A summary of the data is shown in Figure 1. In line with previous results, nonsynonymous SNPs tend to segregate at lower frequencies than intron SNPs (Cargill et al. 1999), which is consistent with some of them being deleterious. Intron SNPs are also skewed toward rare variants relative to the expectation for neutral mutations in an equilibrium population; this pattern is consistent with selection, population size expansion, or admixture. Since there is little evidence that intron sites, other than those involved in splicing control, are subject to selection in humans (Keightley et al. 2005) we assume that this skew is due to population size expansion and/or admixture.
Figure 1.

The allele frequency distribution of nonsynonymous (solid bars) and intron (shaded bars) SNPs relative to the values expected for neutral mutations in an equilibrium population (open bars). Alleles have been grouped into the classes used in the analysis. Singletons were treated by themselves and then SNPs that were present in 2–3, 4–7, 8–15, 16–31, and 32–85 alleles were grouped together.
Parameter estimation:
Using our method we estimate the shape parameter of the gamma distribution to be 0.23 with 95% credibility intervals of 0.19–0.27 and the mean of the distribution to be 425 (225, 766) (Figure 2). Note that the mean of the distribution is not necessarily the same as the mean strength of selection, although it is very close in this case. This is because mutations cannot be more than lethal, so s cannot be >1. To calculate the mean strength of selection it is necessary to know the effective population size and then to condense all the probability density above s = 1 at 1. If we assume that Ne = 10,000 in humans (Jorde et al. 1997) then 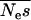 = 425 and
= 425 and 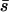 = 4.3%.
= 4.3%.
Figure 2.

The distribution of fitness effects of deleterious mutations represented as either (a) a continuous or (b) a discrete function. The dashed lines in a and the solid lines in b represent the 95% credibility intervals. (a) A transformation of the gamma distribution to a log-scale. Note also the difference in the minimum values for a and b.
Goodness of fit:
To assess whether the gamma distribution fits the data satisfactorily, we performed a goodness-of-fit test by summing the data across genes and finding the maximum-likelihood estimates of the parameters of the model. The maximum-likelihood values were found by simulated annealing (Kirkpatrick et al. 1983). Such maximum-likelihood analysis is not practical on the unsummed data since the model has too many parameters. The maximum-likelihood estimates (λ = 0.24, γ = 333) are similar to the Bayesian estimates for the summed [λ = 0.23 (0.17, 0.28), γ = 392 (180, 1149)] and the unsummed data (see above). The model yields a good fit to the data (χ2 = 4.82, d.f. = 3, P = 0.19).
Simulations:
Past demographic changes and admixture present a potential problem for any method that seeks to infer selection from the allele frequency distribution. Demographic effects can mimic the action of natural selection: for example, population size expansion will skew the allele frequency distribution toward rare alleles, which is also the pattern expected if purifying selection were acting on slightly deleterious mutations. This could be a problem in the current analysis since human populations have had a complex demographic history, with African populations showing evidence of expansion, and European populations showing evidence of a bottleneck followed by expansion (Adams and Hudson 2004). Furthermore, population mixture can also skew the allele frequency and the EGP data are sampled from the current American population, which is a mix of many populations.
Ideally we would like to simultaneously estimate the demography of our population and the distribution of fitness effects. Although progress is being made in this direction (Williamson et al. 2005), current models are quite simple and it is not clear how easy it will be to extend the methods of Williamson et al. from estimating a single selection coefficient, as they have done, to estimating a distribution of effects. Instead, we have chosen to test the robustness of our method by simulating data under a number of demographic models that involve population size expansion, bottlenecks, or admixture. We have also investigated our method under a demographic model estimated from other human data by Adams and Hudson (2004) and from this data set by Williamson et al. (2005).
Our method generally estimates the shape parameter with little bias under all demographic models (Table 1). However, the mean of the distribution is overestimated when there has been a sharp increase in population size; this overestimation can be very large if the increase in population size has been dramatic. The reason for this is as follows. In an expanding population, deleterious mutations experience higher effective population sizes than neutral mutations because they tend to be younger on average (Otto and Whitlock 1997). As a consequence, there are fewer deleterious mutations segregating than one would expect given the apparent effective population size estimated from neutral variation, and the mean strength of selection acting on deleterious mutations thus appears to be higher. In contrast to population size expansion, bottlenecks and admixture have relatively little effect on the parameter estimates.
TABLE 1.
The effect of demography on the parameter estimates
| fint | frec | t | λ | γ | |
|---|---|---|---|---|---|
| Equilibrium | |||||
| 1 | 1 | 0 | 0.24 (0.22, 0.25) | 308 (216, 440) | |
| Expansion | |||||
| 1 | 2 | 0.25 | 0.21 (0.18, 0.25) | 1,114 (420, 2,687) | |
| 4 | 0.20 (0.17, 0.24) | 1,890 (738, 4,780) | |||
| 8 | 0.20 (0.19, 0.22) | 2,436 (1,766, 3,436) | |||
| 16 | 0.16 (0.12, 0.22) | 36,900 (2,450, 187,000) | |||
| 1 | 2 | 0.5 | 0.20 (0.17, 0.22) | 1,570 (783, 3,190) | |
| 4 | 0.21 (0.18, 0.21) | 2,180 (1,770, 3,130) | |||
| 8 | 0.16 (0.12, 0.20) | 27,000 (2,930, 177,000) | |||
| 16 | 0.15 (0.12, 0.19) | 64,200 (6,130, 218,000) | |||
| 1 | 2 | 1 | 0.22 (0.20, 0.25) | 637 (376, 1,070) | |
| 4 | 0.20 (0.17, 0.22) | 2,750 (1,090, 6,760) | |||
| 8 | 0.19 (0.15, 0.23) | 8,630 (1,910, 30,700) | |||
| 16 | 0.19 (0.12, 0.27) | 26,200 (1,090, 158,000) | |||
| Bottleneck | |||||
| 0.01 | 1 | 0.01 | 0.24 (0.20, 0.29) | 156 (67, 347) | |
| 0.1 | 0.19 (0.15, 0.24) | 540 (197, 1,450) | |||
| 1 | 0.18 (0.16, 0.21) | 628 (292, 1,380) | |||
| 0.1 | 1 | 0.01 | 0.20 (0.17, 0.21) | 474 (228, 995) | |
| 0.1 | 0.22 (0.19, 0.25) | 267 (152, 469) | |||
| 1 | 0.19 (0.16, 0.21) | 643 (304, 1,351) | |||
| Admixture | |||||
| 0.125 | 0.19 (0.16, 0.21) | 981 (480, 1,968) | |||
| 0.25 | 0.19 (0.16, 0.21) | 1,190 (576, 2,620) | |||
| 0.50 | 0.20 (0.18, 0.23) | 569 (307, 971) | |||
| 1.0 | 0.22 (0.20, 0.24) | 421 (261, 675) | |||
| Adams and Hudson | |||||
| African–Hausa | 1 | 3.1 | 6.1 | 0.22 (0.20, 0.25) | 453 (370, 899) |
| African–American | 1 | 1.9 | 0.27 | 0.21 (0.19, 0.23) | 841 (523, 1,231) |
| European | 0.19 | 2.0 | 0.035 | 0.22 (0.20, 0.25) | 356 (221, 2,191) |
| Williamson et al. | |||||
| 1 | 6.25 | 0.028 | 0.19 (0.17, 0.21) | 1,600 (968, 2,765) | |
Data were simulated under a variety of different models assuming the shape parameter of the distribution was 0.23 and the mean was 425.
Simulations conducted under demographic models with parameters estimated from other human data suggest that the average strength of selection has possibly been overestimated by a few fold (Table 1). If we take the demographic model of Williamson et al. (2005) as the most appropriate, since this was estimated from the EGP data, then it seems that the mean strength of selection has been overestimated by approximately fourfold and the shape parameter slightly underestimated.
Correcting for demography:
If we have a good demographic model then it should be possible to remove any biases completely from our estimation using standard bias correction methods; if we find that the mean strength of selection is generally overestimated under the demographic model, by say threefold, then we might guess that the true value of the mean is threefold lower than we estimate from the data. We formalize this strategy as follows. First, estimate the parameters of the distribution of fitness effects using our method; let these estimates be γ0 and λ0. Second, estimate the demographic model using the neutral data. Third, simulate data under the demographic model using γ0 and λ0 and then reestimate the parameters of the distribution; let these estimates be γe1 and λe1. Now let our corrected estimates of γ and λ be
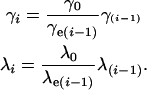 |
(7) |
We repeat the process until γi = γi−1 and λi = λi−1. As an example consider the demographic model of Williamson et al. (2005), which was estimated using the neutral variation in the EGP data set. When we simulated data under this model using γ0 = 425 and λ0 = 0.23 and estimated the parameters of the gamma distribution we obtained γe1 = 1600 and λe1 = 0.19 (Table 1). Substituting these values into Equations 7, our corrected estimates of γ and λ are γ1 = 425·425/1600 = 113 and λ1 = 0.23 · 0.23/0.19 = 0.28. If we then simulate data using these parameter estimates for the gamma distribution under the Williamson et al. model and reestimate the parameters of the gamma distribution we get γe2 = 342 (250, 515) and λe2 = 0.22 (0.20, 0.23). These values are similar to γ0 and β0, demonstrating that we have almost entirely corrected our estimates for the demographic model; i.e., we have found the values of γ and λ, which when simulated under the demographic model yield estimated values of γ and λ that are similar to those we estimated from the EGP data.
Unfortunately, the model of Williamson et al. (2005) does not fit the frequency distribution of either neutral or selected SNPs very well (goodness-of-fit tests yield χ2-values of 113 and 13 for neutral and selected distributions, respectively, P < 0.0001 and P = 0.0234) so we cannot currently conclude that the true values of λ and γ are 0.28 and 113, respectively. To correct our estimates properly we need a better demographic model. However, this analysis suggests that the shape parameter is likely to be fairly accurate and the mean strength of selection overestimated by a few fold.
DISCUSSION
We have used human SNP data to estimate the distribution of fitness effects of mutations that change an amino acid in humans. Assuming that the distribution can be described by a gamma distribution we estimate the shape of this distribution to be 0.23. The distribution is well estimated with small credibility intervals for each class of mutations (Figure 2) and a goodness-of-fit test shows that the model provides an adequate description of the data. We infer that the average strength of selection acting against a nonsynonymous mutation is 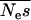 = 425 or
= 425 or 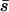 = 4.3% if we assume an effective population size of 10,000 individuals (Jorde et al. 1997). Under this distribution we infer that 19% of mutations are effectively neutral (i.e., have Nes < 1) and that 14% of mutations are slightly deleterious (1 < Nes < 10), such that they segregate in the population at moderate frequencies, but never become fixed. The remainder of the mutations are strongly deleterious such that they contribute little to polymorphism or divergence. We infer that 23, 31, and 13% have effects of Nes = 10–100, 100–1000, and 1000–10,000, respectively. If we use the values of λ and γ corrected using the demographic model of Williamson et al. (2005) the proportions are fairly similar: 20% (Nes < 1), 19%, 32%, 28%, and 1% (1000 < Nes < 10,000), the principle difference being the lack of amino acid mutations with very strongly deleterious effects.
= 4.3% if we assume an effective population size of 10,000 individuals (Jorde et al. 1997). Under this distribution we infer that 19% of mutations are effectively neutral (i.e., have Nes < 1) and that 14% of mutations are slightly deleterious (1 < Nes < 10), such that they segregate in the population at moderate frequencies, but never become fixed. The remainder of the mutations are strongly deleterious such that they contribute little to polymorphism or divergence. We infer that 23, 31, and 13% have effects of Nes = 10–100, 100–1000, and 1000–10,000, respectively. If we use the values of λ and γ corrected using the demographic model of Williamson et al. (2005) the proportions are fairly similar: 20% (Nes < 1), 19%, 32%, 28%, and 1% (1000 < Nes < 10,000), the principle difference being the lack of amino acid mutations with very strongly deleterious effects.
However, it should be noted that we have no direct information about the distribution of fitness effects of mutations with fitness effects of Nes > 100 or Nes < 1. This is because mutations with effects >100 have a very small probability (<5% that of a neutral mutation) of being detected in a sample of 170 chromosomes, and mutations with selection strengths of <1 are all effectively neutral. Our estimate of the distribution beyond these limits is therefore a projection based on the assumption that the distribution is well described by a gamma distribution. To investigate whether this projection is reasonable we reran the analysis, allowing a proportion of mutations, δ, to be strongly deleterious; this can be achieved by multiplying Equation 1 by (1 − δ), with δ being another parameter in the model that is estimated. This means that the gamma distribution is no longer constrained to allocate some of its density to strongly deleterious mutations and could therefore take a very different shape and have a different mean if needed; in this analysis the gamma distribution is estimated from the SNPs, not from the nonpolymorphic sites. Under this model the parameter estimates are λ = 0.23 (0.18, 0.29), γ = 240 (47, 584), and δ = 0.10 (0.29, 0.01), which yields a distribution that is very similar to the one estimated without the δ-parameter; the distributions look almost identical for Nes < 100 and have a very similar proportion of mutations >Nes = 100 (0.40 and 0.47 for models with and without δ, respectively). This suggests that the gamma distribution is a reasonable approximation, since the gamma distribution inferred from the polymorphism data alone is similar to that inferred with all the data (i.e., including sites that have no polymorphism) and predicts a similar number of strongly deleterious mutations.
Previous results:
The distribution of fitness effects estimated here is quite similar to that estimated for human mitochondrial data using a different method (λ = 0.39, γ = 700) (Piganeau and Eyre-Walker 2003). It is also quite similar to the distribution estimated by Yampolsky et al. (2005), using a variety of different approaches. If we assume that Ne = 10,000 in humans (Jorde et al. 1997) we infer from our estimate of the distribution that ∼11% of mutations have an effect of <10−5; 8%, 10−5–10−4; 37%, 10−4–10−2; and 44%, >10−2. The numbers given by Yampolsky et al. (2005) are 12, 14, 49, and 25%, respectively. However, we may have overestimated the mean strength of selection; if the true distribution is actually λ = 0.28 and γ = 113, as we estimate using the demographic model of Williamson et al. (2005), then the proportions are 10, 10, 51, and 29%, which agree very closely with those of Yampolsky et al. (2005).
However, the results are not in such good agreement with those of Eyre-Walker et al. (2002), who inferred that 16% of mutations had effects <10−6; 15%, 2 × 10−5–2 × 10−6; and 69%, >2 × 10−5; the corresponding numbers from our analysis are 7, 7, and 86%. The discrepancy may be due to adaptive evolution, which will tend to increase the apparent proportion of neutral mutations, when divergence data are used to infer the distribution, and to the fact that we may have overestimated the mean strength of selection.
The distribution we have estimated for humans is also consistent with the results of mutation-accumulation and mutagenesis experiments in other organisms (Keightley 1994, 1996; Davies et al. 1999; Vassilieva et al. 2000; Estes et al. 2004). These have suggested that most mutations have small effects and that the mean strength of selection on mutations is between a few percent and a few tens of percent (Lynch et al. 1999; Charlesworth et al. 2004; Fry 2004). However, it is important to appreciate that many of the estimates from the mutation-accumulation and mutagenesis experiments were derived under the assumption that all mutations have the same effect; this means that the mean strength of selection is overestimated. Furthermore, these experiments generally measure the effects of all types of mutation occurring in all parts of the genome, whereas we have estimated the mean strength of selection against point mutations that alter an amino acid. And finally, it should be emphasized that our estimate of the mean strength of selection depends heavily on the shape of the part of the distribution for which we have no direct information (i.e., for Nes > 100) and that it might have been overestimated.
Mean strength of selection acting on polymorphisms:
Although our estimate of the mean strength of selection must be treated with caution, we can estimate the mean strength of selection acting upon segregating polymorphisms with much better accuracy since this is the part of the distribution for which we have direct information. To be precise we estimate the average strength of selection acting upon a randomly sampled nonsynonymous polymorphism. This quantity can be calculated as
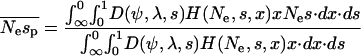 |
(8) |
For the EGP data we estimate 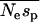 = 0.85, where sp is the strength of selection acting against nonsynonymous polymorphisms. This is in contrast to the estimate obtained by Williamson et al. (2005) of 4.45 from the same data. This could be due to any one of several differences between the methods. First, the method of Williamson et al. (2005) does not estimate the mean strength of selection. Rather, it estimates a quantity that might be regarded as the “effective” selection pressure acting against the nonsynonymous mutations; their method assumes that all nonsynonymous polymorphisms are equally deleterious and then estimates the strength of selection that would give the frequency distribution observed. Second, the discrepancy could be due to the way in which the two methods handle demography. The method of Williamson et al. is unbiased under the demographic model they implement (an instantaneous increase in population size). In contrast, our method tends to overestimate the mean strength of selection. However, this is not likely to explain the discrepancy. Even if we assume that we have overestimated the mean strength of selection by fourfold, the mean strength acting upon polymorphisms is much the same,
= 0.85, where sp is the strength of selection acting against nonsynonymous polymorphisms. This is in contrast to the estimate obtained by Williamson et al. (2005) of 4.45 from the same data. This could be due to any one of several differences between the methods. First, the method of Williamson et al. (2005) does not estimate the mean strength of selection. Rather, it estimates a quantity that might be regarded as the “effective” selection pressure acting against the nonsynonymous mutations; their method assumes that all nonsynonymous polymorphisms are equally deleterious and then estimates the strength of selection that would give the frequency distribution observed. Second, the discrepancy could be due to the way in which the two methods handle demography. The method of Williamson et al. is unbiased under the demographic model they implement (an instantaneous increase in population size). In contrast, our method tends to overestimate the mean strength of selection. However, this is not likely to explain the discrepancy. Even if we assume that we have overestimated the mean strength of selection by fourfold, the mean strength acting upon polymorphisms is much the same, 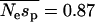 .
.
It is also of interest to estimate the average strength of selection acting against deleterious mutations that are eventually removed by natural selection, i.e., the average strength of selection acting upon polymorphisms with Nes > 1 (Loewe et al. 2006). We estimate this to be Nes = 7.61 using Equation 8 and 8.44 using the harmonic mean (Loewe et al. 2006). These are similar to the values obtained in Drosophila (Loewe et al. 2006). This may seem odd given that Drosophila has a much bigger effective population size than humans. However, analyses show that the mean strength of selection acting against segregating mutations depends on the shape of the distribution but not strongly on the mean (our unpublished results).
Assumptions:
The method assumes that there is no dominance, epistasis, or advantageous mutation and that there is free recombination. The first and second of these assumptions are unlikely to be important. In effect we have estimated the distribution of heterozygous effects of mutations across the various genetic backgrounds that the mutations experience. Our method is likely to be seriously biased only if a large proportion of deleterious mutations are completely recessive or if mutations of very small effect tend to be recessive or dominant. Advantageous mutations subject to directional selection are also unlikely to be a problem since they contribute relatively little to polymorphism if selection is relatively strong (i.e., Nes > 25) (Smith and Eyre-Walker 2002), and although they may skew the allele frequency distribution of linked variants, this pattern persists only for a short period of time—∼0.1Ne generations (Kim and Stephan 2002). However, advantageous mutations subject to balancing selection may be a serious problem since they will skew the allele frequency distribution toward common alleles in a way in which our model may not be able to cope. To investigate this further, we repeated our analysis without genes that had high levels of nonsynonymous polymorphism; the results remained qualitatively unchanged [e.g., ignoring genes with ≥10 nonsynonymous polymorphisms; λ = 0.22 (0.18, 0.27), γ = 962 (398, 1963)]. We have also assumed that there is free recombination between SNPs. There are two consequences if this assumption is violated. First, we will have underestimated the credibility intervals on our parameter estimates because we will not have taken into account the variance associated with the coalescence between alleles. Second, we will have ignored possible Hill–Robertson interference between selected mutations (McVean and Charlesworth 2000). This latter effect is unlikely to be serious since levels of nonsynonymous diversity are very low in humans (Cargill et al. 1999) and most genes are separated by substantial amounts of intergenic DNA and hence have a moderate amount of recombination between them.
Molecular clock:
The distribution of fitness effects we have estimated has a number of implications. First, it suggests that the molecular clock will not be very robust to changes in effective population size. Ohta (1977) showed that if all mutations were deleterious and the distribution of fitness effects was exponential then the rate of evolution, f, was expected to be proportional to the reciprocal of the effective population size. Similarly Kimura (1979) showed that if the distribution was gamma distributed with a shape parameter of 0.5, the rate of evolution was expected to be proportional to 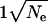 . Since a gamma distribution with a shape parameter of 1 is an exponential distribution, this suggests the generalization
. Since a gamma distribution with a shape parameter of 1 is an exponential distribution, this suggests the generalization 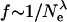 , where λ is the shape parameter of the gamma distribution (Chao and Carr 1993) (a result we will prove elsewhere). We thus expect moderate changes in the rate of evolution in response to increases or decreases in effective population size. For example, if the population size increases 10-fold we would expect the rate of evolution to decline by ∼40% unless there is an increase in the rate of adaptive evolution.
, where λ is the shape parameter of the gamma distribution (Chao and Carr 1993) (a result we will prove elsewhere). We thus expect moderate changes in the rate of evolution in response to increases or decreases in effective population size. For example, if the population size increases 10-fold we would expect the rate of evolution to decline by ∼40% unless there is an increase in the rate of adaptive evolution.
Our estimate of the distribution appears to be fairly consistent with the ratio of nonsynonymous to synonymous substitution rates in primates and rodents. Let us assume that synonymous mutations are neutral and that the distribution of fitness effects for nonsynonymous mutations is gamma. Although there is some evidence of selection on synonymous codon use in both murids and hominids (Keightley and Gaffney 2003; Urrutia and Hurst 2003; Chamary and Hurst 2004; Chimpanzee Sequencing and Analysis Consortium 2005), the level of selection seems to be small and quite similar in the two lineages; for example, the rate of synonymous substitution appears to be ∼70% of the intron substitution rate in both groups (Keightley and Gaffney 2003; Chimpanzee Sequencing and Analysis Consortium 2005). Further, let us assume that a small proportion of nonsynonymous mutations are advantageous and that these cause a proportion α of the nonsynonymous substitutions to be adaptive, the others being neutral or slightly deleterious. Under these conditions the rates of synonymous (dS) and nonsynonymous (dN) substitution are expected to be
 |
(9) |
where u is the nucleotide mutation rate and k is a constant. Hence we expect dN/dS in primates divided by dN/dS in rodent to be
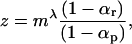 |
(10) |
where m is the ratio of the rodent and primate effective population sizes and αr and αp are the proportions of nonsynonymous substitutions that are adaptive in rodents and primates, respectively. If we assume these proportions are similar then 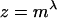 . The effective population sizes of humans and chimpanzees are in the range of 10,000–30,000 (Eyre-Walker et al. 2002), while our only estimate for a rodent, the house mouse, is in the range of 450,000–810,000 (Keightley et al. 2005). We would therefore expect dN/dS to be approximately twofold higher in primates than in rodents. The ratio of the nonsynonymous and synonymous substitution rates is 0.31 in human–chimpanzee and 0.16 in mouse–rat (Eyre-Walker et al. 2002); the ratio of these numbers is 1.9.
. The effective population sizes of humans and chimpanzees are in the range of 10,000–30,000 (Eyre-Walker et al. 2002), while our only estimate for a rodent, the house mouse, is in the range of 450,000–810,000 (Keightley et al. 2005). We would therefore expect dN/dS to be approximately twofold higher in primates than in rodents. The ratio of the nonsynonymous and synonymous substitution rates is 0.31 in human–chimpanzee and 0.16 in mouse–rat (Eyre-Walker et al. 2002); the ratio of these numbers is 1.9.
Decline in fitness:
For many years geneticists have pondered the potential consequences that modern medicine might be having upon our genetic quality (Muller 1950; Crow 1997; Lynch et al. 1999)—medicine relaxes natural selection, which allows potentially harmful mutations to accumulate. This may pose a risk to our population if selection is reimposed sometime in the future. Estimates of the rate at which amino acid mutations occur and their average effect allow us to estimate whether this is likely to be a problem. We estimate, using the numbers in Eyre-Walker and Keightley (1999), but a revised estimate for the number of genes (25,000), that we receive on average 1.8 amino acid mutations per generation. Our estimate for the average effect of mutations is 4.3%, so the decline in genetic quality per generation is predicted to be 1.8 × 4.3 = 7.7%. This estimate must be treated with caution since the mean strength of selection depends largely on mutations whose distribution we have no direct estimate of. However, there are also several reasons to believe that the rate of decline in our genetic quality is likely to be <7.7% per generation. First, natural selection cannot be completely relaxed, and it is far from relaxed for most of the world's population. Second, our estimate of the mean effect of mutations is likely to be overestimated by two- or threefold due to population size expansion. Third, although we have not included mutations that lie outside genes or mutations that are not single-nucleotide changes, these are unlikely to contribute much to the decline in genetic quality. While a fair amount of DNA outside genes is subject to natural selection (Dermitzakis and Clark 2001; Dermitzakis et al. 2003; Keightley and Gaffney 2003; Keightley et al. 2005), the selection upon these sequences is often quite weak (Keightley et al. 2005), and indels are relatively infrequent in mammals (Ophir and Graur 1997). So it seems that, at worst, human populations will suffer a decline in genetic quality of a few percent, or less, per generation.
Variance in fitness:
However, our estimate of the distribution of fitness effects suggests that, while a decline in genetic quality may not be a problem in humans, locating the genes involved in genetic disease may be. Let us assume that the distribution of effects of mutations affecting some trait, say predisposition to heart disease, is similar in shape to the distribution of fitness effects and that the number of mutations that potentially affect the trait is very large. Then the variance in a trait contributed by alleles with effects v segregating at a frequency x is
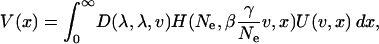 |
(11) |
where 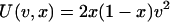 and β is a parameter that measures the association between the trait and fitness: when β = 1 the trait is fitness and when β = 0 the trait is neutral with respect to fitness. Many human diseases may not be strongly associated with fitness because they affect people later in life when they are past their natural reproductive age (Wright et al. 2003), although older individuals do help raise their grand-offspring. U(v, x) is the variance in the trait contributed by a mutation of effect v segregating at frequency x. The trait is arbitrarily scaled such that the mean effect of a mutation on the trait is 1. If we use our estimates of λ and γ it is evident that unless the trait and the alleles that affect it are completely neutral, the majority of the variance in the trait is contributed by alleles segregating at very low frequency (Figure 3). This is in agreement with a recent study of alleles associated with low levels of HDL cholesterol, in which most of the putatively harmful alleles were at very low frequency (Cohen et al. 2004). This may also explain why it has proven difficult to locate genes for many complex human genetic diseases and why many of the results cannot be replicated (Cardon and Bell 2001). The fact that some alleles of fairly large effect do segregate at moderate frequency (Lohmueller et al. 2003) suggests that a few alleles associated with disease may be completely neutral with respect to fitness or have been subject to positive selection.
and β is a parameter that measures the association between the trait and fitness: when β = 1 the trait is fitness and when β = 0 the trait is neutral with respect to fitness. Many human diseases may not be strongly associated with fitness because they affect people later in life when they are past their natural reproductive age (Wright et al. 2003), although older individuals do help raise their grand-offspring. U(v, x) is the variance in the trait contributed by a mutation of effect v segregating at frequency x. The trait is arbitrarily scaled such that the mean effect of a mutation on the trait is 1. If we use our estimates of λ and γ it is evident that unless the trait and the alleles that affect it are completely neutral, the majority of the variance in the trait is contributed by alleles segregating at very low frequency (Figure 3). This is in agreement with a recent study of alleles associated with low levels of HDL cholesterol, in which most of the putatively harmful alleles were at very low frequency (Cohen et al. 2004). This may also explain why it has proven difficult to locate genes for many complex human genetic diseases and why many of the results cannot be replicated (Cardon and Bell 2001). The fact that some alleles of fairly large effect do segregate at moderate frequency (Lohmueller et al. 2003) suggests that a few alleles associated with disease may be completely neutral with respect to fitness or have been subject to positive selection.
Figure 3.

The variance in a quantitative trait as a function of allele frequency and association of the trait with fitness. The distribution of trait effects is assumed to be a gamma distribution with a shape parameter of 0.23. The curves going from left to right show decreasing association with fitness: β = 1, 0.1, 0.01, and 0.001.
Conclusions:
The distribution of fitness effects is central to the understanding of many problems in genetics and evolution. Here we have attempted to provide a detailed description of this distribution, by fitting a population genetic model to extensively and deeply sampled single-nucleotide polymorphism data in humans. Although there are limitations to this method, particularly for inferring the distribution of fitness effects of strongly selected mutations, we estimate that the vast majority of amino-acid-changing mutations in humans have mild effects of between 1/1000 and 1/10. The estimated mean strength of selection against nonsynonymous mutations is a few percent, which suggests that declines in fitness due to modern medicine in humans are unlikely to be a problem. However, the distribution does suggest that it will be difficult to locate the majority of mutations involved in genetic disease unless the disease is completely unassociated with fitness or some of the mutations have been subject to positive selection.
Software to run this analysis is available from A.E.W.
Acknowledgments
We thank John Welch for statistical help; David Waxman for mathematical help; Scott Williamson for help with simulations; Peter Keightley, Sally Otto, Nina Stoletzki, Ken Weiss, and several anonymous referees for comments on the manuscript; and the Biotechnology and Biological Sciences Research Council and Royal Society for support.
This article is dedicated to the memory of Nick Smith, with whom A.E.W. started this work.
References
- Adams, A. M., and R. R. Hudson, 2004. Maximum-likelihood estimation of demographic parameters using the frequency spectrum of unlinked single nucleotide polymorphisms. Genetics 168: 1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437: 1149–1152. [DOI] [PubMed] [Google Scholar]
- Bergman, C. M., and M. Kreitman, 2001. Analysis of conserved noncoding DNA in Drosophila reveals similar constraints in intergenic and intronic sequences. Genome Res 11: 1335–1345. [DOI] [PubMed] [Google Scholar]
- Cardon, L. R., and J. I. Bell, 2001. Association study designs for complex diseases. Nat. Rev. Genet. 2: 91–99. [DOI] [PubMed] [Google Scholar]
- Cargill, M., D. Altshuler, J. Ireland, P. Sklar, K. Ardlie et al., 1999. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nature Genetics 22: 231–238. [DOI] [PubMed] [Google Scholar]
- Chamary, J. V., and L. D. Hurst, 2004. Similar rates but different modes of sequence evolution in introns and at exonic silent sites in rodents: evidence for selectively driven codon usage. Mol. Biol. Evol. 21: 1014–1023. [DOI] [PubMed] [Google Scholar]
- Chao, L., and D. E. Carr, 1993. The molecular clock and the relationship between population size and generation time. Evolution 47: 688–690. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., M. T. Morgan and D. Charlesworth, 1993. The effect of deleterious mutationson neutral molecular variation. Genetics 134: 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., H. Borthwick, C. Bartolome and P. Pignatelli, 2004. Estimates of the genomic rate of detrimental alleles in Drosophila melanogaster. Genetics 167: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium, 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437: 69–87. [DOI] [PubMed] [Google Scholar]
- Cohen, J. C., R. S. Kiss, A. Pertsemlidis, Y. L. Marcel, R. McPherson et al., 2004. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 305: 869–872. [DOI] [PubMed] [Google Scholar]
- Crow, J. F., 1997. The high spontaneous mutation rate: Is it health risk? Proc. Natl. Acad. Sci. USA 94: 8380–8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, E. K., A. D. Peters and P. D. Keightley, 1999. High frequency of cryptic deleterious mutations in Caenorhabditis elegans. Science 285: 1748–1751. [DOI] [PubMed] [Google Scholar]
- Dermitzakis, E. T., and A. G. Clark, 2001. Differential selection after duplication in mammalian developmental genes. Mol. Biol. Evol. 18: 557–562. [DOI] [PubMed] [Google Scholar]
- Dermitzakis, E. T., A. Reymond, N. Scamuffa, C. Ucla, E. Kirkness et al., 2003. Evolutionary discrimination of mammalian conserved non-genic sequences (CNGs). Science 302: 1033–1035. [DOI] [PubMed] [Google Scholar]
- Estes, S., P. C. Phillips, D. R. Denver, W. K. Thomas and M. Lynch, 2004. Mutation accumulation in populations of varying size: the distribution of mutational effects for fitness correlates in Caenorhabditis elegans. Genetics 166: 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker, A., and P. D. Keightley, 1999. High genomic deleterious mutation rates in hominids. Nature 397: 344–347. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A., P. D. Keightley, N. G. C. Smith and D. Gaffney, 2002. Quantifying the slightly deleterious model of molecular evolution. Mol. Biol. Evol. 19: 2142–2149. [DOI] [PubMed] [Google Scholar]
- Fay, J., G. J. Wycoff and C.-I Wu, 2001. Positive and negative selection on the human genome. Genetics 158: 1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry, J. D., 2004. On the rate and linearity of viability declines in Drosophila mutation-accumulation experiments: genomic mutation rates and synergistic epistasis revisited. Genetics 166: 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill, P. R., B. Charlesworth, D. L. Halligan and P. Andolfatto, 2005. Patterns of intron sequence evolution in Drosophila are dependent upon length and GC content. Genome Biol. 6: R67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde, L. B., M. Bamshad and A. R. Rogers, 1997. Using mitochondrial and nuclear DNA markers to reconstruct human evolution. BioEssays 20: 126–136. [DOI] [PubMed] [Google Scholar]
- Keightley, P. D., 1994. The distribution of mutation effects on viability in Drosophila melanogaster. Genetics 138: 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., 1996. Nature of deleterious mutation load in Drosophila. Genetics 144: 1993–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., and D. J. Gaffney, 2003. Functional constraints and frequency of deleterious mutations in noncoding DNA of rodents. Proc. Natl. Acad. Sci. USA 100: 13402–13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., M. J. Lercher and A. Eyre-Walker, 2005. Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biol. 3: e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160: 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1979. Model of effectively neutral mutations in which selective constraint is incorporated. Proc. Natl. Acad. Sci. USA 76: 3440–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, S., C. D. Gelatt and M. P. Vecchi, 1983. Optimization by simulated annealing. Science 220: 671–680. [DOI] [PubMed] [Google Scholar]
- Lande, R., 1994. The risk of population extinction from new deleterious mutations. Evolution 48: 1460–1469. [DOI] [PubMed] [Google Scholar]
- Livingston, R. J., A. von Niederhausern, A. G. Jegga, D. C. Crawford, C. S. Carlson et al., 2004. Pattern of sequence variation across 213 environmental response genes. Genome Res. 14: 1821–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe, L., B. Charlesworth, C. Bartolome and V. Noel, 2006. Estimating selection on nonsynonymous mutations. Genetics 172: 1079–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller, K. E., C. L. Pearce, M. Pike, E. S. Lander and J. N. Hirschhorn, 2003. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet. 33: 177–182. [DOI] [PubMed] [Google Scholar]
- Lynch, M., J. Blanchard, D. Houle, T. Kibota, S. Schultz et al., 1999. Spontaneous deleterious mutation. Evolution 53: 645–663. [DOI] [PubMed] [Google Scholar]
- Lynch, M., J. Conery and R. Burger, 1995. Mutation accumulation and the extinction of small populations. Am. Nat. 146: 489–518. [Google Scholar]
- McVean, G. A., and B. Charlesworth, 2000. The effects of Hill-Robertson interference between weakly selected mutations on patterns of molecular evolution and variation. Genetics 155: 929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, H. J., 1950. Our load of mutations. Am. J. Hum. Genet. 2: 111–176. [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., and Z. Yang, 2003. Estimating the distribution of selection coefficients from phylogenetic data with applications to mitochondrial and viral DNA. Mol. Biol. Evol. 20: 1231–1239. [DOI] [PubMed] [Google Scholar]
- Ohta, T., 1977. Extension of the neutral mutation drift hypothesis, pp. 148–167 in Molecular Evolution and Polymorphism, edited by M. Kimura. National Institute of Genetics, Mishima, Japan.
- Ophir, R., and D. Graur, 1997. Patterns and rates of indel evolution in processed pseudogenes from humans and murids. Gene 31: 191–202. [DOI] [PubMed] [Google Scholar]
- Otto, S. P., and M. C. Whitlock, 1997. The probability of fixation in populations of changing size. Genetics 146: 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piganeau, G., and A. Eyre-Walker, 2003. Estimating the distribution of fitness effects from DNA sequence data: implications for the molecular clock. Proc. Natl. Acad. Sci. USA 100: 10335–10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer, S., R. J. Kulathinal, C. D. Bustamante and D. L. Hartl, 2003. Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection. J. Mol. Evol. 57: 5154–5164. [DOI] [PubMed] [Google Scholar]
- Shabalina, S. A., and A. S. Kondrashov, 1999. Pattern of selective constraint in C. elegans and C. briggsae genomes. Genet. Res. 74: 23–30. [DOI] [PubMed] [Google Scholar]
- Smith, N. G. C., and A. Eyre-Walker, 2002. Adaptive protein evolution in Drosophila. Nature 415: 1022–1024. [DOI] [PubMed] [Google Scholar]
- Sved, J., and A. P. Bird, 1990. The expected equilibrium of the CpG dinucleotide in vertebrate genomes under a mutation model. Proc. Natl. Acad. Sci. USA 87: 4692–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urrutia, A. O., and L. D. Hurst, 2003. The signature of selection mediated by expression on human genes. Genome Res. 13: 2260–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilieva, L., A. M. Hook and M. Lynch, 2000. The fitness effects of spontaneous mutations in Caenorhabditis elegans. Evolution 54: 1234–1246. [DOI] [PubMed] [Google Scholar]
- Webster, M. T., N. G. C. Smith, M. J. Lercher and H. Ellegren, 2004. Gene expression, synteny and local similarity in human noncoding mutation rates. Mol. Biol. Evol. 21: 1820–1830. [DOI] [PubMed] [Google Scholar]
- Williamson, S. H., R. Hernandez, A. Fledel-Alon, L. Zhu, R. Nielsen et al., 2005. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc. Natl. Acad. Sci. USA 102: 7882–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, A., B. Charlesworth, I. Rudan, A. Carothers and H. Campbell, 2003. A polygenic basis for late-onset disease. Trends Genet. 19: 97–106. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1938. The distribution of gene frequencies under irreversible mutation. Proc. Natl. Acad. Sci. USA 24: 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yampolsky, L. Y., F. A. Kondrashov and A. S. Kondrashov, 2005. Distribution of the strength of selection against amino acid replacements in human proteins. Hum. Mol. Genet. 14: 3191–3201. [DOI] [PubMed] [Google Scholar]