

Abstract
Determining the dimensionality of G provides an important perspective on the genetic basis of a multivariate suite of traits. Since the introduction of Fisher's geometric model, the number of genetically independent traits underlying a set of functionally related phenotypic traits has been recognized as an important factor influencing the response to selection. Here, we show how the effective dimensionality of G can be established, using a method for the determination of the dimensionality of the effect space from a multivariate general linear model introduced by Amemiya (1985). We compare this approach with two other available methods, factor-analytic modeling and bootstrapping, using a half-sib experiment that estimated G for eight cuticular hydrocarbons of Drosophila serrata. In our example, eight pheromone traits were shown to be adequately represented by only two underlying genetic dimensions by Amemiya's approach and factor-analytic modeling of the covariance structure at the sire level. In contrast, bootstrapping identified four dimensions with significant genetic variance. A simulation study indicated that while the performance of Amemiya's method was more sensitive to power constraints, it performed as well or better than factor-analytic modeling in correctly identifying the original genetic dimensions at moderate to high levels of heritability. The bootstrap approach consistently overestimated the number of dimensions in all cases and performed less well than Amemiya's method at subspace recovery.
GENETIC variance–covariance (G) matrices conveniently summarize the genetic relationships among a suite of traits and are a central parameter in the determination of the multivariate response to selection (Lande 1979). Although individual elements of G, single-trait genetic variances along the diagonal and bivariate genetic covariances off the diagonal, are frequently the subject of hypothesis testing and biological interpretation, such a compartmentalized approach to the analysis of a set of functionally related traits is of limited value (Pease and Bull 1988; Charlesworth 1990; Blows and Hoffmann 2005). The multivariate pattern of genetic covariance represented by G can be analyzed by determining the eigenstructure of this symmetrical matrix and, subsequently, how the genetic variance is partitioned among genetically independent traits. Of particular importance is the rank of G, that is, how many genetically independent traits are represented by a set of functionally related phenotypic traits, a question that can be traced back to Fisher's (1930) geometric model (Orr 2000).
The distribution of the eigenvalues of G (the genetic variances of the genetically independent traits defined by the eigenvectors) allows an unambiguous definition of a genetic constraint. A singular G matrix (one or more of the eigenvalues are zero) suggests that an absolute genetic constraint exists (Pease and Bull 1988; Blows and Hoffmann 2005; Mezey and Houle 2005) and the effective dimensionality of G may be less than the number of traits measured. In turn, the eigenvectors of G have become important components of a number of approaches to the study of multivariate genetic constraints (Schluter 1996; Blows et al. 2004; McGuigan et al. 2005) and investigations of how selection may change the genetic variance (Blows et al. 2004; Hine et al. 2004).
Importantly, the singular nature (or otherwise) of G cannot be determined from a cursory viewing of the elements of G; G may be still be singular in the presence of genetic variance in all individual traits, for instance (Dickerson 1955; Charlesworth 1990). Determining the dimensionality of a covariance matrix, and the subsequent estimation of nonnegative definite covariance matrices, has received considerable attention in the statistical literature (Amemiya 1985; Anderson and Amemiya 1991; Calvin and Dykstra 1991; Mathew et al. 1994; Sun et al. 2003). However, the effective dimensionality of G has been addressed only on a few occasions, primarily using resampling approaches to generate confidence intervals for the eigenvalues of G (Kirkpatrick et al. 1990; Mezey and Houle 2005) and to find whether one or more eigenvalues of G were significantly different from zero (Kirkpatrick et al. 1990). In a recent departure from this approach, factor-analytic modeling has been adopted as a way of fitting genetic principal components in a framework that will also allow direct hypothesis tests of the number of genetic dimensions required to explain genetic covariation among environment and traits (Thompson et al. 2003; Kirkpatrick and Meyer 2004).
Here, we assess the utility of three methods for determining the dimensionality of G. First, we introduce how the effective dimensionality of G can be established, using a method for the determination of the dimensionality of the effect space from a multivariate general linear model (Amemiya 1985; Amemiya et al. 1990; Anderson and Amemiya 1991). We outline how the effective number of dimensions of a genetic covariance matrix obtained from either one or two random factor experimental designs commonly used in evolutionary studies can be determined. We then show how the original genetic covariance matrix can be partitioned so that a new, nonnegative definite covariance matrix can be constructed that contains only those genetic dimensions that have strong statistical support. Conveniently, this method requires calculating only the eigenvectors and eigenvalues of a symmetrical matrix without iteration, an approach that can be implemented in many commonly used software packages. Second, factor-analytic modeling in a restricted maximum-likelihood framework is utilized to demonstrate how dimensionality may be determined in many genetic experimental designs using a mixed-model approach, again resulting in a guaranteed positive semidefinite estimate of G. Finally, the bootstrap approach, recently used to establish full rank in Drosophila melanogaster wing shape by Mezey and Houle (2005), is implemented.
We compare the performance of all three methods in a simulation study conducted on 100 simulated data sets of known dimensionality, as well as applying them to a multivariate set of contact pheromones (cuticular hydrocarbons) from a standard half-sib breeding design using D. serrata. We have been investigating the effect of directional selection on the genetic variance in male sexually selected display traits of D. serrata (Blows et al. 2004; Hine et al. 2004). In this system, multiple male cuticular hydrocarbons act as contact pheromones and are under linear sexual selection. The vast majority of genetic variance in these male traits was found to be orientated in such a way that it was almost orthogonal to the direction of sexual selection in both a laboratory-adapted (Blows et al. 2004) and a field (Hine et al. 2004) population. These results suggested that sexual selection might have depleted genetic variance, resulting in an ill-conditioned G matrix that was effectively not of full rank.
MATERIALS AND METHODS
Method I: the dimensionality of the effect space from a linear model:
The dimensionality of a covariance matrix:
To begin, consider the simple case of an experimental design that could be analyzed appropriately by a one-way multivariate analysis of variance; for example, multiple traits are measured on a number of individuals from a set of families. In this case, extraction of the between- and within-mean square matrices (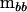 and
and 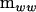 , respectively) is the first step in the estimation of the between-source (family) variance–covariance matrix (
, respectively) is the first step in the estimation of the between-source (family) variance–covariance matrix (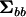 ), using
), using
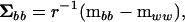 |
(1) |
where r is the coefficient of the variance components at the between-source level. The characteristic roots (λi) of 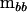 in the metric of
in the metric of 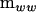 can be found by solving
can be found by solving
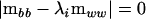 |
(2) |
(Amemiya 1985). If λi ≥ 1 for all i = 1, 2,…, p, then the covariance matrix 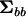 is nonnegative definite. If any λi < 1, then
is nonnegative definite. If any λi < 1, then 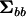 is indefinite as will often be the case with estimates of G (Hill and Thompson 1978). Solving the polynomial in λ represented by (2) requires specialized software. Instead, one can use linear algebra to find the character roots of
is indefinite as will often be the case with estimates of G (Hill and Thompson 1978). Solving the polynomial in λ represented by (2) requires specialized software. Instead, one can use linear algebra to find the character roots of 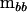 in the metric of
in the metric of 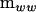 as defined by the eigenvalues (λi) of
as defined by the eigenvalues (λi) of
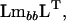 |
where L is a lower triangular matrix and defined as the transpose inverse of U (upper triangular), which in turn is the Cholesky root of 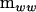 (i.e.,
(i.e., 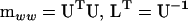 ). Note that the notation here differs from that of Amemiya (1985) to avoid confusion with lower (L) and upper (U) matrix notation. The classical Cholesky decomposition is a lower triangular matrix (
). Note that the notation here differs from that of Amemiya (1985) to avoid confusion with lower (L) and upper (U) matrix notation. The classical Cholesky decomposition is a lower triangular matrix (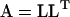 ), but many common mathematical and statistical programs (such as Matlab and SAS IML) use the upper triangular matrix convention (
), but many common mathematical and statistical programs (such as Matlab and SAS IML) use the upper triangular matrix convention (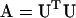 ). Be aware of which convention your program of choice uses and ensure that the inverse of the Cholesky root is arranged in lower triangular form to define L.
). Be aware of which convention your program of choice uses and ensure that the inverse of the Cholesky root is arranged in lower triangular form to define L.
When the existence of genetic constraints is of interest, the initial null hypothesis will be full rank (p) as a G matrix of full rank indicates there are no genetic constraints on the evolution of the set of traits. Let k be the number of characteristic roots such that λi ≥ 1. Amemiya et al. (1990; Anderson and Amemiya 1991) provide a way of determining how many of the k dimensions are supported statistically to determine the effective number of dimensions (m). As a first step, the null hypothesis that m ≤ k can be accepted immediately, because, at most, there is evidence for the same number of dimensions as there are λi ≥ 1. Next, we test the new null hypothesis that m ≤ k − 1. If this null hypothesis is accepted, as outlined below, we continue to test m ≤ k − 2 and so on until one in the series of null hypotheses is rejected or the null hypothesis that m ≤ 0 is accepted, indicating a lack of support for any effect.
To test the null hypothesis that m ≤ b (0 < b < k − 1), order the λi ≥ 1 from greatest to smallest (λ1 ≥ λ2 ≥ ⋯ ≥ λk ≥ 1) and to each apply the equation
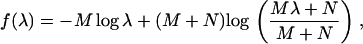 |
where M and N are the degrees of freedom at the effect (between) and error (within) levels, respectively. The test statistic for the current null hypothesis is then
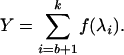 |
(3) |
To determine whether to reject the null hypothesis, the test statistic can be compared to the distribution of Y for 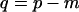 in Table 1 of Amemiya et al. (1990). Rejecting the null hypothesis m ≤ b indicates that there is evidence for b + 1 dimensions.
in Table 1 of Amemiya et al. (1990). Rejecting the null hypothesis m ≤ b indicates that there is evidence for b + 1 dimensions.
TABLE 1.
Mean squares matrices for the sire, dam, and error levels (msire, mdam, and mwithin) of the genetic example
| Z,Z-5,9-C25:2 | Z-9-C25:1 | Z-9-C26:1 | 2-Me-C26 | Z,Z-5,9-C27:2 | 2-Me-C28 | Z,Z-5,9-C29:2 | 2-Me-C30 | |
|---|---|---|---|---|---|---|---|---|
 | ||||||||
| Z,Z-5,9-C25:2 | 1.42485 | |||||||
| Z-9-C25:1 | 1.17053 | 1.72790 | ||||||
| Z-9-C26:1 | 1.04237 | 1.06351 | 1.50895 | |||||
| 2-Me-C26 | 0.97056 | 0.77925 | 0.83568 | 1.87726 | ||||
| Z,Z-5,9-C27:2 | 1.29040 | 1.16890 | 1.24746 | 1.09149 | 1.94400 | |||
| 2-Me-C28 | 1.07244 | 1.01256 | 1.00965 | 1.47972 | 1.20990 | 1.58924 | ||
| Z,Z-5,9-C29:2 | 0.96852 | 0.82302 | 0.88823 | 1.10838 | 1.25591 | 1.12072 | 1.43868 | |
| 2-Me-C30 | 1.00050 | 0.98903 | 0.95456 | 1.13624 | 1.12015 | 1.45576 | 0.99213 | 1.47852 |
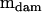 | ||||||||
| Z,Z-5,9-C25:2 | 1.22593 | |||||||
| Z-9-C25:1 | 0.87826 | 1.09057 | ||||||
| Z-9-C26:1 | 0.86307 | 0.74886 | 1.24298 | |||||
| 2-Me-C26 | 0.79774 | 0.56762 | 0.62148 | 1.11744 | ||||
| Z,Z-5,9-C27:2 | 0.88252 | 0.71058 | 0.84255 | 0.69857 | 1.15926 | |||
| 2-Me-C28 | 0.99639 | 0.79044 | 0.86157 | 0.98433 | 0.97022 | 1.25831 | ||
| Z,Z-5,9-C29:2 | 0.68691 | 0.64920 | 0.58280 | 0.49107 | 0.76609 | 0.73672 | 1.19128 | |
| 2-Me-C30 | 0.95055 | 0.76700 | 0.86477 | 0.75362 | 0.96116 | 1.19387 | 0.75942 | 1.27590 |
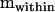 | ||||||||
| Z,Z-5,9-C25:2 | 0.61504 | |||||||
| Z-9-C25:1 | 0.50150 | 0.62708 | ||||||
| Z-9-C26:1 | 0.44486 | 0.37147 | 0.61021 | |||||
| 2-Me-C26 | 0.27630 | 0.23740 | 0.24827 | 0.55274 | ||||
| Z,Z-5,9-C27:2 | 0.36139 | 0.32113 | 0.35733 | 0.29132 | 0.51862 | |||
| 2-Me-C28 | 0.39277 | 0.36746 | 0.37803 | 0.43972 | 0.37528 | 0.56743 | ||
| Z,Z-5,9-C29:2 | 0.25354 | 0.22090 | 0.20932 | 0.18974 | 0.29587 | 0.27775 | 0.67254 | |
| 2-Me-C30 | 0.39763 | 0.36972 | 0.39217 | 0.33342 | 0.37951 | 0.55363 | 0.30152 | 0.60817 |
Constructing a nonnegative estimate of a covariance matrix:
It is possible to construct a nonnegative matrix of rank k from the eigenvectors corresponding to those λi ≥ 1 (Amemiya 1985). Let the characteristic vectors of 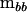 in the metric of
in the metric of 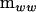 from (2) form the columns of a matrix T. Define the
from (2) form the columns of a matrix T. Define the 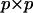 matrix P as
matrix P as
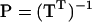 |
(Amemiya 1985). Using P we can now express the original difference 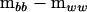 as
as
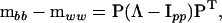 |
where 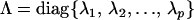 and Ipp is the
and Ipp is the 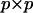 identity matrix. The characteristic roots (λi) of
identity matrix. The characteristic roots (λi) of 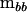 in the metric of
in the metric of 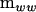 can be used to partition
can be used to partition 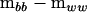 into a nonnegative definite matrix and a negative definite matrix,
into a nonnegative definite matrix and a negative definite matrix,
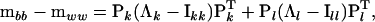 |
(4) |
where Pk is a 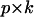 matrix containing the first k columns of P, Pl contains the remaining p − k columns of P,
matrix containing the first k columns of P, Pl contains the remaining p − k columns of P, 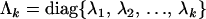 , and
, and 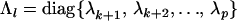 . The first term on the right-hand side of (4) then represents the nonnegative definite portion of
. The first term on the right-hand side of (4) then represents the nonnegative definite portion of 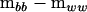 and the second term represents the negative definite portion. By dropping the second term representing the negative definite portion, the negative eigenvalues are set to zero. The remaining term is then the projection of
and the second term represents the negative definite portion. By dropping the second term representing the negative definite portion, the negative eigenvalues are set to zero. The remaining term is then the projection of 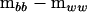 in the metric of
in the metric of 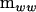 onto the set of all nonnegative definite matrices (Amemiya 1985).
onto the set of all nonnegative definite matrices (Amemiya 1985).
Again, in practice it is easier not to deal with estimating the characteristic vectors of 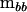 in the metric of
in the metric of 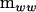 using (2). Alternatively, let Q be the matrix containing the eigenvectors of
using (2). Alternatively, let Q be the matrix containing the eigenvectors of 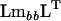 as columns (Amemiya 1985). Define the
as columns (Amemiya 1985). Define the 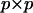 matrix P (Amemiya 1985):
matrix P (Amemiya 1985):
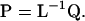 |
Having established the effective number of dimensions, we can construct a new covariance matrix of rank m that is nonnegative definite and contains only the dimensions supported by the hypothesis-testing procedure, rather than of rank k from the eigenvectors corresponding to those λi ≥ 1. The first m columns of P (forming the 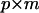 matrix Pm) are associated with the characteristic roots of
matrix Pm) are associated with the characteristic roots of 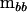 in the metric of
in the metric of 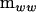 that are statistically supported. Pm is used to construct a nonnegative definite covariance matrix of rank m by
that are statistically supported. Pm is used to construct a nonnegative definite covariance matrix of rank m by
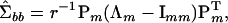 |
(5) |
where 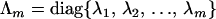 and Imm is the
and Imm is the 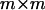 identity matrix. The first m eigenvectors of
identity matrix. The first m eigenvectors of 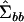 represent the dimensions of the original covariance matrix that have strong statistical support as defined by the hypothesis-testing procedure outlined above. The remaining eigenvectors will have eigenvalues equal to zero. Amemiya (1985, appendix) goes on to show that (5) is equivalent to the restricted maximum-likelihood estimator of
represent the dimensions of the original covariance matrix that have strong statistical support as defined by the hypothesis-testing procedure outlined above. The remaining eigenvectors will have eigenvalues equal to zero. Amemiya (1985, appendix) goes on to show that (5) is equivalent to the restricted maximum-likelihood estimator of 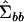 for balanced experimental designs.
for balanced experimental designs.
Method II: factor-analytic modeling:
The dimensionality of a covariance matrix:
Although Amemiya's approach allows the determination of the number of dimensions of a G matrix, it is restricted in the experimental designs that can be accommodated by the approach. Factor-analytic modeling is commonly used to determine the minimum number of factors (dimensions) required to explain the pattern of covariation among a set of variables (Krzanowski and Lai 1988). Although not explicitly the goal of their development of factor-analytic modeling for fitting genetic principal components (Kirkpatrick and Meyer 2004; Meyer and Kirkpatrick 2005), factor-analytic modeling provides an alternative way to determine the dimensionality of G matrices because likelihood-ratio tests can be made of the improvement in fit as each principal component is added (or subtracted) from the model (Kirkpatrick and Meyer 2004; Meyer and Kirkpatrick 2005). This approach displays great promise as it allows for almost any experimental design to be used, and tests of dimensionality can be conducted within a restricted maximum-likelihood framework.
Kirkpatrick and Meyer (2004) introduced a new algorithm to fit genetic principal components that we do not attempt to implement here. Rather, as suggested by these authors, alternative algorithms are available, and here we outline a readily available approach using standard statistical software (Proc Mixed in SAS). In particular, the reduced-rank model in which the specific variances are assumed to be zero can be specified in Proc Mixed for the level of interest that contains the additive genetic variance components (the sire level in a half-sib design, for example), using the factor-analytic model [FA0(m)] covariance structure. At the genetic level of interest then, a reduced-rank covariance matrix 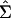 is given by
is given by
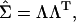 |
(6) |
where 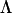 (
(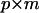 ) is a lower triangular matrix of constants that represent the factor loadings of the m latent variables. A series of nested hypothesis tests may be conducted to determine how many genetic dimensions are required to explain the observed patterns of genetic covariance. A full model is first fit (m = p), and factors are sequentially dropped until the model achieves a significantly worse fit compared to the previous model (in which it is nested), indicating that the amount of variation accounted for by the tested factor is sufficient for the factor to be retained. Since
) is a lower triangular matrix of constants that represent the factor loadings of the m latent variables. A series of nested hypothesis tests may be conducted to determine how many genetic dimensions are required to explain the observed patterns of genetic covariance. A full model is first fit (m = p), and factors are sequentially dropped until the model achieves a significantly worse fit compared to the previous model (in which it is nested), indicating that the amount of variation accounted for by the tested factor is sufficient for the factor to be retained. Since 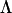 is a lower triangular matrix, one less parameter is estimated in each subsequent column. Therefore, the degrees of freedom associated with each log-likelihood-ratio test are determined by the change in the number of parameters required in
is a lower triangular matrix, one less parameter is estimated in each subsequent column. Therefore, the degrees of freedom associated with each log-likelihood-ratio test are determined by the change in the number of parameters required in 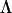 given by p − m + 1.
given by p − m + 1.
Constructing a nonnegative estimate of a covariance matrix:
Conveniently, fitting the reduced model will guarantee a nonnegative definite variance–covariance matrix with the desired number of dimensions. For example, fitting m ≤ p factors will result in a G matrix that is positive semidefinite with m dimensions, even if a number of the eigenvalues of the original G matrix were negative. Once the number of factors to be retained is established, the reduced-rank covariance matrix, 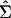 , is obtained by multiplying the matrix of factor loadings by its transpose as in (6).
, is obtained by multiplying the matrix of factor loadings by its transpose as in (6).
Method III: bootstrapping of the eigenvalues of G:
Mezey and Houle (2005) attempted to determine the dimensionality of G by generating a bootstrapped sample of the eigenvalues of the original G matrix. Here, we implement this bootstrap procedure to enable a comparison with the other two methods we present. Bootstrapping was conducted by sampling sires (and all their progeny as blocks) with replacement, generating 1000 estimates of G. The eigenvalues of each G were then calculated and mean eigenvalues and their 95% confidence intervals are presented. Constructing a nonnegative definite matrix from this approach is not possible as it does not allow a determination of the exact dimensions that have received statistical support, a limitation that we discuss in detail below.
Simulation:
To assess the relative effectiveness of the three methods, we conducted a simulation using 100 data sets. For simplicity and to reduce computational times we did not simulate a hierarchical data structure as in the half-sib experiment, but rather each data set consisted of 100 “sires,” each with 6 “offspring,” resulting in 600 offspring for which eight traits were generated. To generate data with known dimensionality at the sire level for each data set, we first generated an 8 × 2 factor matrix, 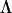 , to represent the factor loadings of two latent variables, with randomly drawn constants in all elements except
, to represent the factor loadings of two latent variables, with randomly drawn constants in all elements except 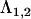 , which was set to zero as the pivot. We then multiplied
, which was set to zero as the pivot. We then multiplied 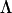 by its transpose (as in factor-analytic modeling, Equation 6) to construct the 8 × 8 “genetic” covariance matrix. One hundred random linear combinations of the first 2 eigenvectors of G, where the random multipliers were drawn from distributions of mean zero and variance equal to the corresponding eigenvalue, were produced to represent 100 sire “breeding values.” One hundred simulated sets of breeding values for eight traits that could be completely described by two different latent variables were generated using this process.
by its transpose (as in factor-analytic modeling, Equation 6) to construct the 8 × 8 “genetic” covariance matrix. One hundred random linear combinations of the first 2 eigenvectors of G, where the random multipliers were drawn from distributions of mean zero and variance equal to the corresponding eigenvalue, were produced to represent 100 sire “breeding values.” One hundred simulated sets of breeding values for eight traits that could be completely described by two different latent variables were generated using this process.
We conducted the simulation on four different approximate levels of heritability: 0.75, 0.50, 0.25, and 0.10. To generate offspring phenotypes with an average heritability of ∼0.75, we generated for each simulated data set 600 (100 sires by six offspring) rows and eight columns of random numbers independently distributed with mean 0 and variance 1.5. The breeding value of each sire was added to six of these random numbers to create six offspring values for that sire. To obtain heritabilities of ∼0.50, 0.25, and 0.10, the diagonals of the “error” covariance matrix were set at 10, 30, and 100, respectively.
We applied Amemiya's method and factor analytic modeling to the same 100 data sets for each heritability level. Bootstrapping was initially applied to the 10 and 75% heritability treatments, and because the results differed little between the two extreme treatments, and to save on computational time, we did not proceed to apply this method to the intermediate heritability treatments. For Amemiya's method, variance components were estimated using least-squares, using Proc GLM in SAS as the data sets were balanced. Factor-analytic modeling was conducted using Proc Mixed in SAS, with an unconstrained covariance structure modeled at the “within-sire” level and the factor-analytic structure at the sire level. Bootstrapping of each data set was conducted by sampling sires with replacement, and 1000 bootstrap replicates of each of the 100 data sets were generated.
RESULTS
Here, we have reanalyzed the data from the half-sib experiment described in Blows et al. (2004). In this experiment 66 sires were each mated to up to three dams, resulting in 367 offspring for which eight contact pheromones were analyzed. To this data set we have applied each of the three methods for determining the dimensionality of the additive genetic variance–covariance matrix. Although this experiment has not been designed to allow an exhaustive search for the presence of very small levels of genetic variance (Mezey and Houle 2005), it serves to illustrate the implementation and relative performance of the three methods in an experiment of a size commonly found in evolutionary studies.
Method I: the dimensionality of the effect space from a linear model:
The model for this experimental design was the standard nested MANOVA model, with dams nested within sires. The mean square matrices 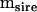 (r = 5.4218),
(r = 5.4218), 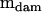 (r = 1.8326), and
(r = 1.8326), and 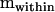 can be found in Table 1. The approach outlined by Amemiya (1985) for such a model differed in one aspect from that presented above for the one-way model. Here, the approach was applied in a two-step process, by first finding that part of the dam-level covariance matrix (
can be found in Table 1. The approach outlined by Amemiya (1985) for such a model differed in one aspect from that presented above for the one-way model. Here, the approach was applied in a two-step process, by first finding that part of the dam-level covariance matrix (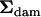 ) that was nonnegative definite and had strong statistical support. By using
) that was nonnegative definite and had strong statistical support. By using 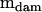 and
and 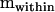 in the above procedure, the ordered eigenvalues of
in the above procedure, the ordered eigenvalues of  were 3.260, 2.464, 2.115, 1.913, 1.726, 1.277, 1.164, and 1.003. Since all λi > 1, Σdam is nonnegative definite, although a number of the eigenvalues are quite small. We began the hypothesis-testing procedure by accepting the null hypothesis that m ≤ 8, and each subsequent null hypothesis was accepted until m ≤ 2, which was rejected (Y = 22.032, P < 0.05), suggesting that
were 3.260, 2.464, 2.115, 1.913, 1.726, 1.277, 1.164, and 1.003. Since all λi > 1, Σdam is nonnegative definite, although a number of the eigenvalues are quite small. We began the hypothesis-testing procedure by accepting the null hypothesis that m ≤ 8, and each subsequent null hypothesis was accepted until m ≤ 2, which was rejected (Y = 22.032, P < 0.05), suggesting that 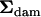 has three dimensions that have statistical support. A new dam-level covariance matrix
has three dimensions that have statistical support. A new dam-level covariance matrix 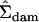 was constructed with a rank of 3, which allowed the construction of
was constructed with a rank of 3, which allowed the construction of 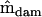 by rearranging (1) to
by rearranging (1) to  , where
, where  .
.
The dimensionality of  (Table 2) was determined by following the same procedure, now using
(Table 2) was determined by following the same procedure, now using  and the Cholesky decomposition of
and the Cholesky decomposition of 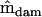 . The ordered eigenvalues of
. The ordered eigenvalues of 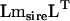 were 3.542, 3.001, 2.580, 1.506, 1.436, 1.046, 0.941, and 0.638. Since two roots were <1, the null hypothesis that m ≤ 6 was accepted. Each subsequent null hypothesis was accepted until m ≤ 1, which was marginally nonsignificant (Y = 24.410, P = 0.059), where the exact probability level was derived from Equation 4 in Amemiya et al. (1990), suggesting that there was evidence for two dimensions of genetic variance in the set of eight pheromones.
were 3.542, 3.001, 2.580, 1.506, 1.436, 1.046, 0.941, and 0.638. Since two roots were <1, the null hypothesis that m ≤ 6 was accepted. Each subsequent null hypothesis was accepted until m ≤ 1, which was marginally nonsignificant (Y = 24.410, P = 0.059), where the exact probability level was derived from Equation 4 in Amemiya et al. (1990), suggesting that there was evidence for two dimensions of genetic variance in the set of eight pheromones.
TABLE 2.
Original (italics, above diagonal) and reduced-rank (non-italics, below diagonal) estimates of the sire covariance matrix (Σsire and 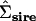 , respectively)
, respectively)
| Z,Z-5,9-C25:2 | Z-9-C25:1 | Z-9-C26:1 | 2-Me-C26 | Z,Z-5,9-C27:2 | 2-Me-C28 | Z,Z-5,9-C29:2 | 2-Me-C30 | |
|---|---|---|---|---|---|---|---|---|
| Z,Z-5,9-C25:2 | 0.06050 | 0.06847 | 0.04430 | 0.04326 | 0.07116 | 0.02292 | 0.05205 | 0.01543 |
| 0.07640 | ||||||||
| Z-9-C25:1 | 0.07032 | 0.12849 | 0.06608 | 0.04943 | 0.07991 | 0.04690 | 0.03170 | 0.04569 |
| 0.06584 | ||||||||
| Z-9-C26:1 | 0.06020 | 0.05558 | 0.05317 | 0.04265 | 0.07090 | 0.02855 | 0.05923 | 0.01684 |
| 0.04746 | ||||||||
| 2-Me-C26 | 0.08862 | 0.09133 | 0.07138 | 0.14902 | 0.07267 | 0.09863 | 0.11609 | 0.07362 |
| 0.18794 | ||||||||
| Z,Z-5,9-C27:2 | 0.10189 | 0.09018 | 0.07971 | 0.08682 | 0.14225 | 0.04074 | 0.08510 | 0.02312 |
| 0.14744 | ||||||||
| 2-Me-C28 | 0.05722 | 0.06034 | 0.04631 | 0.13323 | 0.05168 | 0.06544 | 0.07143 | 0.04935 |
| 0.09535 | ||||||||
| Z,Z-5,9-C29:2 | 0.07255 | 0.06940 | 0.05759 | 0.10705 | 0.08832 | 0.07231 | 0.05139 | 0.04071 |
| 0.07505 | ||||||||
| 2-Me-C30 | 0.04187 | 0.04508 | 0.03403 | 0.10561 | 0.03482 | 0.07615 | 0.05510 | 0.03629 |
| 0.06117 |
The two genetic dimensions supported by the analysis are the first two principal components of the constructed nonnegative definite matrix 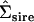 (Table 2) and are displayed in Table 3. The original G matrix presented in Blows et al. (2004) was indefinite with three negative eigenvalues. Here, it can be seen that the two dimensions supported by the current analysis are very similar to the first two principal components of the original G matrix (Table 3). A more formal subspace comparison (Krzanowski 1979; Blows et al. 2004) indicated the two two-dimensional subspaces in Table 3 are indeed very similar. The comparison yields a value of 1.76 for the sum of the eigenvalues of the S matrix (hereon represented as
(Table 2) and are displayed in Table 3. The original G matrix presented in Blows et al. (2004) was indefinite with three negative eigenvalues. Here, it can be seen that the two dimensions supported by the current analysis are very similar to the first two principal components of the original G matrix (Table 3). A more formal subspace comparison (Krzanowski 1979; Blows et al. 2004) indicated the two two-dimensional subspaces in Table 3 are indeed very similar. The comparison yields a value of 1.76 for the sum of the eigenvalues of the S matrix (hereon represented as 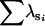 ) defined in Equation 3 in Blows et al. (2004), which in this case ranges from 0 for orthogonal subspaces to 2 for coincident subspaces.
) defined in Equation 3 in Blows et al. (2004), which in this case ranges from 0 for orthogonal subspaces to 2 for coincident subspaces.
TABLE 3.
The first and second principal components (PC) of the original Σsire, the Amemiya (1985) reduced-rank estimate of 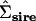 , and the factor-analytic model containing two genetic dimensions
, and the factor-analytic model containing two genetic dimensions
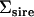
|
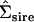
|
FA0(2)
|
||||
|---|---|---|---|---|---|---|
| PC1 | PC2 | PC1 | PC2 | PC1 | PC2 | |
| Z,Z-5,9-C25:2 | 0.27625 | −0.29635 | 0.32778 | 0.26629 | 0.23243 | 0.31928 |
| Z-9-C25:1 | 0.36458 | −0.46562 | 0.31665 | 0.15944 | 0.13215 | 0.18153 |
| Z-9-C26:1 | 0.27934 | −0.24780 | 0.26066 | 0.19619 | 0.25510 | 0.21317 |
| 2-Me-C26 | 0.47941 | 0.53674 | 0.51058 | −0.43718 | 0.53639 | −0.43631 |
| Z,Z-5,9-C27:2 | 0.43758 | −0.37945 | 0.38912 | 0.62990 | 0.44929 | 0.64238 |
| 2-Me-C28 | 0.31447 | 0.33435 | 0.34787 | −0.38635 | 0.36325 | −0.36156 |
| Z,Z-5,9-C29:2 | 0.37784 | 0.19407 | 0.34633 | 0.05226 | 0.42989 | −0.01368 |
| 2-Me-C30 | 0.22311 | 0.22900 | 0.26698 | −0.35395 | 0.23892 | −0.29276 |
Method II: factor-analytic modeling:
Factor-analytic models were run constraining the covariance matrix at the sire level to be from a single dimension to full rank (eight dimensions), and model fitting statistics are displayed in Table 4. Of the eight models, the two-dimensional model, FA0(2), displayed the lowest value of the Akaike information criterion (AIC), suggesting that overall, this was the model of best fit. The series of nested hypothesis tests indicated that moving from two dimensions to one resulted in the first significant increase in the log likelihood (χ2 = 17.4, d.f. = 7, P = 0.015), again suggesting that the two-dimensional model was the favored model. Eigenanalysis of the resulting covariance matrix constrained to be two-dimensional revealed that the two significant genetic dimensions were almost identical to the two dimensions found to have statistical support by Amemiya's approach (Table 3). A formal subspace comparison indicated that the two dimensions from each of these covariance matrices described almost coincident subspaces (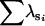 = 1.93 of 2).
= 1.93 of 2).
TABLE 4.
Model fit statistics for the nested series of factor-analytic models testing the dimensionality of the sire-level covariance matrix using REML
| Model fit
|
|||
|---|---|---|---|
| −2 LL | AIC | No. of parameters | |
| FA0(8) | 6296.3 | 6436.3 | 73 |
| FA0(7) | 6296.3 | 6436.3 | 72 |
| FA0(6) | 6296.3 | 6434.3 | 70 |
| FA0(5) | 6296.3 | 6430.3 | 67 |
| FA0(4) | 6296.5 | 6422.5 | 63 |
| FA0(3) | 6298.0 | 6414.0 | 58 |
| FA0(2) | 6308.1 | 6410.1 | 52 |
| FA0(1) | 6325.5 | 6415.5 | 45 |
LL, log likelihood.
Method III: bootstrapping of the eigenvalues of G:
Mean eigenvalues from the bootstrapped sample of each of the eigenvectors are presented in Table 5, along with the fifth percentile value that represents the lower 95% confidence interval in this circumstance as the significance of a variance component from zero is a one-tailed hypothesis. The lower 95% confidence interval does not overlap zero for the first four eigenvectors, indicating that significant genetic variance is present in these four dimensions. The next eigenvector, g5, has a positive mean eigenvalue, but the lower 95% confidence interval overlaps zero and equates to a significance level of P = 0.187, indicating that there is no evidence for genetic variance in this dimension.
TABLE 5.
Mean bootstrap eigenvalue and 5th percentile bootstrap eigenvalue for the half-sib genetic data
| Bootstrapped sample statistics
|
||
|---|---|---|
| Eigenvector | Mean eigenvalue | 5th percentile eigenvalue |
| gmax | 0.523 | 0.209 |
| g2 | 0.147 | 0.095 |
| g3 | 0.069 | 0.038 |
| g4 | 0.024 | 0.007 |
| g5 | 0.006 | −0.002 |
| g6 | −0.005 | −0.020 |
| g7 | −0.032 | −0.065 |
| g8 | −0.088 | −0.200 |
Simulation:
Amemiya's method successfully identified two dimensions at the sire level in 99 of the 100 data sets in the 75% heritability treatment. The sole remaining case was identified as having only a single dimension and was the data set that displayed the most extreme condition index (here defined as the ratio of the first to the second eigenvalue). This method was less effective at lower heritability, with only 22 cases correctly identified as two-dimensional in the 10% heritability treatment. All other cases were identified as one-dimensional, indicating Amemiya's approach consistently underestimated the number of dimensions when heritability was low.
In contrast, factor-analytic modeling was relatively consistent in its ability to identify the correct number of dimensions across heritability treatments (Figure 1), although rejection of the two-dimensional model occurred in at least 30% of cases in each heritability treatment. The behavior of factor-analytic modeling with a change in heritability was more complex than Amemiya's approach, with overestimation of the number of dimensions at high heritability, with increasing underestimation of dimensionality at lower heritabilities (Table 6). One possible explanation for the failure of factor-analytic modeling to identify the correct number of dimensions was if the search algorithm had been captured by a local peak on the likelihood surface. To test this, we reran a subset of cases using the correct factor-analytic structure as the starting values. REML iterated to the same solution as when the default MIVQUE starting values were used, suggesting that the global maximum likelihood had been found in these cases.
Figure 1.

Relationship between heritability and the correct identification of a two-dimensional subspace in the simulation. Open circles, factor-analytic modeling; solid circles, Amemiya's approach.
TABLE 6.
Summary of simulation results
| Dimensions identified
|
||||||||
|---|---|---|---|---|---|---|---|---|
| h2 | Method | Statistic | 0 | 1 | 2 | 3 | 4 | 5 |
| 0.1 | A | n | 8 | 70 | 22 | 0 | 0 | 0 |
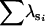 |
0 | 0.98 | 1.95 | — | — | — | ||
| FA | n | 0 | 28 | 61 | 11 | 0 | 0 | |
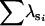 |
— | 0.94 | 1.76 | 1.93 | — | — | ||
| BS | n | 0 | 0 | 0 | 42 | 58 | 0 | |
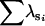 |
— | — | — | 1.68 | 1.86 | — | ||
| 0.25 | A | n | 0 | 29 | 71 | 0 | 0 | 0 |
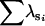 |
— | 0.98 | 1.96 | — | — | — | ||
| FA | n | 0 | 12 | 70 | 18 | 0 | 0 | |
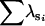 |
— | 0.99 | 1.92 | 1.98 | — | — | ||
| 0.5 | A | n | 0 | 11 | 89 | 0 | 0 | 0 |
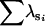 |
— | 0.99 | 1.98 | — | — | — | ||
| FA | n | 0 | 3 | 70 | 27 | 0 | 0 | |
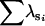 |
— | 0.99 | 1.94 | 1.99 | — | — | ||
| 0.75 | A | n | 0 | 1 | 99 | 0 | 0 | 0 |
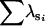 |
— | 0.999 | 1.99 | — | — | — | ||
| FA | n | 0 | 1 | 67 | 32 | 0 | 0 | |
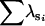 |
— | 0.999 | 1.988 | 1.84 | — | — | ||
| BS | n | 0 | 0 | 0 | 21 | 78 | 1 | |
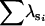 |
— | — | — | 1.996 | 1.994 | 1.997 | ||
A, Amemiya's method; FA, factor analytic modeling; BS, bootstrap method. The Krzanowski subspace comparison scores for estimated vs. sampled G are denoted by 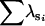 . The number of simulated data sets for which the method of interest identified a given number of dimensions is denoted by n. No method identified more than five dimensions out of the possible eight.
. The number of simulated data sets for which the method of interest identified a given number of dimensions is denoted by n. No method identified more than five dimensions out of the possible eight.
To determine the effectiveness of Amemiya's method and factor-analytic modeling in identifying the correct genetic dimensions, we investigated the ability of the two approaches to identify the correct two-dimensional subspace in the 75% heritability treatment in which Amemiya's approach had a 99% success rate. We compared the two-dimensional subspaces of the estimated positive semidefinite covariance matrices from the two methods with the subspace defined by the first two eigenvectors of the rescaled simulated genetic covariance matrix (to account for orientation changes due to the standardization of the traits) for each data set. Both Amemiya's method and factor-analytic modeling were highly successful in uncovering the correct two-dimensional genetic subspace, with the mean values of 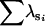 (±SD) being 1.982 ± 0.102 and 1.944 ± 0.106, respectively. Figure 2 shows the relationship between the two methods in their ability to estimate the correct two-dimensional subspace. When factor-analytic modeling incorrectly indicated three dimensions, the two-dimensional subspace recovered by this method was consistently a poorer match to the correct subspace (Figure 2A). Including the third dimension in the subspace comparison (Figure 2B) improved the recovery of the correct subspace to a level comparable to that in Amemiya's method. Therefore, in the data sets in which factor-analytic modeling incorrectly identified three dimensions, the third dimension does in fact contain some proportion of the originally defined two-dimensional genetic subspace. Figure 2C shows that for those cases where factor-analytic modeling correctly indicated that two dimensions existed, there is an almost perfect relationship between the two methods.
(±SD) being 1.982 ± 0.102 and 1.944 ± 0.106, respectively. Figure 2 shows the relationship between the two methods in their ability to estimate the correct two-dimensional subspace. When factor-analytic modeling incorrectly indicated three dimensions, the two-dimensional subspace recovered by this method was consistently a poorer match to the correct subspace (Figure 2A). Including the third dimension in the subspace comparison (Figure 2B) improved the recovery of the correct subspace to a level comparable to that in Amemiya's method. Therefore, in the data sets in which factor-analytic modeling incorrectly identified three dimensions, the third dimension does in fact contain some proportion of the originally defined two-dimensional genetic subspace. Figure 2C shows that for those cases where factor-analytic modeling correctly indicated that two dimensions existed, there is an almost perfect relationship between the two methods.
Figure 2.



Relationship between Amemiya's method and factor-analytic modeling in recovering the original genetic subspace in the simulation. (A) Two-dimensional factor-analytic model. (B) Three-dimensional factor-analytic model. (C) Magnification of region of high subspace recovery scores from A. Simulated data sets that factor-analytic modeling correctly identified as having two dimensions are indicated by solid circles, and those data sets incorrectly identified as having three dimensions are indicated by open circles.
Bootstrapping did not recover the correct number of dimensions in any of the 100 data sets, consistently overestimating the number of dimensions with 21, 78, and 1 cases having three, four, and five dimensions, respectively, at the 75% heritability level (Table 6). At the 10% heritability level bootstrapping again consistently overestimated the rank of G, although the number of dimensions identified decreased overall, with 42 and 58 cases of three and four dimensions, respectively. To assess the performance of bootstrapping in recovering the correct subspace within the dimensions identified as being significant, the mean bootstrap estimate of G was compared to simulated (rescaled, as above) G. Bootstrapping performed better for the higher heritability level, with mean values of 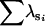 (±SD) being 1.994 ± .017 and 1.781 ± 0.245 at the 75 and 10% heritability levels, respectively.
(±SD) being 1.994 ± .017 and 1.781 ± 0.245 at the 75 and 10% heritability levels, respectively.
DISCUSSION
Determining the dimensionality of G matrices:
The effective dimensionality of a set of traits plays a central role in the genetics of adaptation (Fisher 1930; Orr 2000) and the definition of absolute multivariate genetic constraints (Pease and Bull 1988; Blows and Hoffmann 2005). Here, we have investigated three methods for the determination of the effective dimensionality of a G matrix. Of the three methods, the simulation study indicated that Amemiya's approach performed best at higher heritabilities, but consistently underestimated dimensionality at lower heritabilities and was particularly biased in the 10% heritability treatment. Factor-analytic modeling tended to overestimate the number of genetic dimensions by 1 at higher heritabilities and underestimate at lower heritabilities by the same magnitude in a substantial number of cases. Although the performance of factor analysis in recovering the original genetic dimensions dropped off at the lowest heritability level, the two methods recovered the same two-dimensional subspace when the correct number of dimensions was indicated by factor-analytic modeling for the 25, 50, and 75% heritability treatments, suggesting a close mathematical association between the two approaches. The precise mathematical relationship between the two methods deserves further attention.
The simulation study indicated that bootstrapping will consistently overestimate the dimensionality of the G matrix, which is likely to be a consequence of two related limitations. First, each bootstrapped estimate of the covariance matrix will have a unique set of eigenvectors. Although the first eigenvalue will always be the largest in each replicate, the eigenvectors associated with each of the eigenvalues will be different; how different will depend mainly on the sample size, but even with very large samples the same trait combination (eigenvector) is unlikely to be replicated exactly in any of the bootstrapped samples. Since the eigenvalue is simply an estimate of the genetic variance, this is analogous to estimating the genetic variance of a different trait in each of the bootstrapped replicates, but treating the eigenvalues for hypothesis testing as if they estimated the genetic variance in the same trait each time. Second, if the eigenvalues of the original covariance matrix are distributed in such a way that some are very close in size, the order of the eigenvectors may change in different bootstrapped replicates (Cohn 1999; Peres-Neto et al. 2005), adding to the variation in the trait combinations that will be associated with each ranked eigenvalue. This is likely to be a particular problem with eigenvectors associated with small eigenvalues.
Either of these problems will result in like-sized eigenvalues of bootstrapped replicates being binned together for the calculation of the confidence interval for an estimate of an original eigenvalue, even though each estimate reflects a different trait combination. In this way, confidence intervals will be estimated as being small as the variance in each bin (i.e., first eigenvalue, second eigenvalue, etc.) is determined only by the rank of the eigenvalue for each bootstrapped replicate, rather than by how much genetic variance is present for a particular trait combination in each bootstrapped replicate. Such an approach will tend to uncover as many statistically significant dimensions as there were positive eigenvalues in the original estimate of the G matrix; in our case, five positive eigenvalues were present in the original G matrix, and four dimensions had nonzero eigenvalues determined by bootstrapping. This pattern was also present in the results from the simulation study; in 63 (75% heritability treatment) and 70 (10% heritability treatment) cases, one less dimension than the number of positive eigenvalues was recovered.
Although the factor-analytic approach provides the most flexibility for determining the dimensionality of G from virtually any experimental design, Amemiya's approach may provide an important alternative under some conditions. One such situation is that in practice REML often does not converge with the inclusion of a large number of traits. For laboratory-based quantitative genetic or genomic experiments based on simple breeding designs with relatively balanced data sets, Amemiya's approach supplies a noniterative alternative to determining dimensionality. For example, microarray analysis tends to involve a very large number of gene expression traits. One approach to future microarray experiments will be to conduct these experiments within a breeding design. Such an experimental design will allow the determination of the number of independent genetic factors underlying the typically large number of gene expression changes among treatment groups. With so many traits to be included in a factor-analytic model, convergence problems are likely to be encountered and therefore these experiments may benefit from applying Amemiya's approach.
It is important to note that the approaches outlined above are subject to the usual power constraints due to small sample sizes. Genetic experiments conducted with small sample sizes are likely to be capable of detecting only one or two dimensions that explain a substantial amount of genetic variance. The poor performance of Amemiya's approach at low heritability suggests that this method will be particularly adversely affected by small sample size. Factor-analytic modeling also underestimated the number of dimensions as heritability dropped, with a less dramatic loss of performance than Amemiya's approach as the information content of the data was reduced. Consequently, we recommend these approaches as objective aids in defining a genetic subspace, but they should not be applied as the sole determining factor in subspace selection. In particular, in cases where fewer genetic dimensions included may increase the likelihood of rejecting a null hypothesis of interest (Blows et al. 2004 would be a good example), these approaches should be used with caution.
Are G matrices of full rank?
In our example, eight pheromone traits were shown to be adequately represented by only two underlying genetic dimensions by two of these methods: Amemiya's approach and factor-analytic modeling of the covariance structure at the sire level. The bootstrap method identified four dimensions that had eigenvalues significantly different from zero, but as indicated by the simulation, this is likely to be an overestimate. The sample size in this data set is not sufficient to allow an exhaustive search for dimensions describing small levels of genetic variance, so a definitive conclusion that only two dimensions have genetic variance would be premature. In the only other statistical determination of the rank of G, Mezey and Houle (2005) reported G matrices of full rank for wing shape in D. melanogaster, using the bootstrap approach in an experiment with a very large sample. Since the bootstrap approach is likely to overestimate dimensionality, it is uncertain if G matrices of full rank may be a feature of multivariate descriptions of morphology.
A major advantage of Amemiya's approach and factor-analytic modeling is that both can result in the identification of the reduced subspace for which there is strong statistical support for the presence of genetic variance. It is important to note that in our example, the two dimensions supported by these analyses were not exactly the same as the first two eigenvectors of estimated G, suggesting that directions in the space of G that describe substantial amounts of the additive genetic variance may also be associated with large nonadditive or environmental sources of variance.
In conclusion, determining the dimensionality of G provides an important perspective on the genetic basis of a multivariate suite of traits. Understanding the genetic basis of adaptation will require a multivariate approach, as single traits are rarely under selection in isolation (Lande and Arnold 1983; Schluter and Nychka 1994; Blows and Brooks 2003). In combination with emerging genomic technologies, statistical approaches to determining the number of genetically independent traits may potentially allow an investigation of how many independent genetic changes underlie a response to selection involving complex adaptations.
Acknowledgments
We thank Y. Amemiya, D. Houle, H. Rundle, and four anonymous reviewers for comments on previous drafts of this manuscript. M.W.B. was supported by a grant from the Australian Research Council.
References
- Amemiya, Y., 1985. What should be done when an estimated between-group covariance-matrix is not nonnegative definite. Am. Stat. 39: 112–117. [Google Scholar]
- Amemiya, Y., T. W. Anderson and P. A. W. Lewis, 1990. Percentage points for a test of rank in multivariate components of variance. Biometrika 77: 637–641. [Google Scholar]
- Anderson, T. W., and Y. Amemiya, 1991. Testing dimensionality in the multivariate-analysis of variance. Stat. Probab. Lett. 12: 445–463. [Google Scholar]
- Blows, M. W., and R. Brooks, 2003. Measuring nonlinear selection. Am. Nat. 162: 815–820. [DOI] [PubMed] [Google Scholar]
- Blows, M. W., and A. A. Hoffmann, 2005. A reassessment of genetic limits to evolutionary change. Ecology 86: 1371–1384. [Google Scholar]
- Blows, M. W., S. F. Chenoweth and E. Hine, 2004. Orientation of the genetic variance-covariance matrix and the fitness surface for multiple male sexually selected traits. Am. Nat. 163: 329–340. [DOI] [PubMed] [Google Scholar]
- Calvin, J. A., and R. L. Dykstra, 1991. Maximum-likelihood-estimation of a set of covariance matrices under lower order restrictions with applications to balanced multivariate variance-components models. Ann. Stat. 19: 850–869. [Google Scholar]
- Charlesworth, B., 1990. Optimization models, quantitative genetics, and mutation. Evolution 44: 520–538. [DOI] [PubMed] [Google Scholar]
- Cohn, R. D., 1999. Comparisons of multivariate relational structures in serially correlated data. J. Agric. Biol. Environ. Stat. 4: 238–257. [Google Scholar]
- Dickerson, G. E., 1955. Genetic slippage in response to selection for multiple objectives. Cold Spring Harbor Symp. Quant. Biol. 20: 213–224. [DOI] [PubMed] [Google Scholar]
- Fisher, R. A. S., 1930. The Genetical Theory of Natural Selection. Clarendon Press, Oxford.
- Hill, W. G., and R. Thompson, 1978. Probabilities of non-positive definite between-group or genetic covariance matrices. Biometrics 34: 429–439. [Google Scholar]
- Hine, E., S. Chenoweth and M. W. Blows, 2004. Multivariate quantitative genetics and the lek paradox: genetic variance in male sexually selected traits of Drosophila serrata under field conditions. Evolution 58: 2754–2762. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick, M., and K. Meyer, 2004. Direct estimation of genetic principal components: simplified analysis of complex phenotypes. Genetics 168: 2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, M., D. Lofsvold and M. Bulmer, 1990. Analysis of the inheritance, selection and evolution of growth trajectories. Genetics 124: 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzanowski, W. J., 1979. Between-groups comparison of principal components. J. Am. Stat. Assoc. 74: 703–707. [Google Scholar]
- Krzanowski, W. J., and Y. T. Lai, 1988. A criterion for determining the number of groups in a data set using sum-of-squares clustering. Biometrics 44: 23–34. [Google Scholar]
- Lande, R., 1979. Quantitative genetic-analysis of multivariate evolution, applied to brain-body size allometry. Evolution 33: 402–416. [DOI] [PubMed] [Google Scholar]
- Lande, R., and S. J. Arnold, 1983. The measurement of selection on correlated characters. Evolution 37: 1210–1226. [DOI] [PubMed] [Google Scholar]
- Mathew, T., A. Niyogi and B. K. Sinha, 1994. Improved nonnegative estimation of variance-components in balanced multivariate mixed models. J. Multivar. Anal. 51: 83–101. [Google Scholar]
- McGuigan, K., S. F. Chenoweth and M. W. Blows, 2005. Phenotypic divergence along lines of genetic variance. Am. Nat. 165: 32–43. [DOI] [PubMed] [Google Scholar]
- Meyer, K., and M. Kirkpatrick, 2005. Restricted maximum likelihood estimation of genetic principal components and smoothed covariance matrices. Genet. Sel. Evol. 37: 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezey, J. G., and D. Houle, 2005. The dimensionality of genetic variation for wing shape in Drosophila melanogaster. Evolution 59: 1027–1038. [PubMed] [Google Scholar]
- Orr, H. A., 2000. Adaptation and the cost of complexity. Evolution 54: 13–20. [DOI] [PubMed] [Google Scholar]
- Pease, C. M., and J. J. Bull, 1988. A critique of methods for measuring life-history trade-offs. J. Evol. Biol. 1: 293–303. [Google Scholar]
- Peres-Neto, P. R., D. A. Jackson and K. M. Somers, 2005. How many principal components? Stopping rules for determining the number of non-trivial axes revisited. Comput. Stat. Data Anal. 49: 974–997. [Google Scholar]
- Schluter, D., 1996. Adaptive radiation along genetic lines of least resistance. Evolution 50: 1766–1774. [DOI] [PubMed] [Google Scholar]
- Schluter, D., and D. Nychka, 1994. Exploring fitness surfaces. Am. Nat. 143: 597–616. [Google Scholar]
- Sun, Y. J., B. K. Sinha, D. von Rosen and Q. Y. Meng, 2003. Nonnegative estimation of variance components in multivariate unbalanced mixed linear models with two variance components. J. Stat. Plann. Inf. 115: 215–234. [Google Scholar]
- Thompson, R., B. Cullis, A. Smith and A. Gilmour, 2003. A sparse implementation of the average information algorithm for factor analytic and reduced rank variance models. Aust. N. Z. J. Stat. 45: 445–459. [Google Scholar]