

Abstract
Amyloid peptide (Aβ) aggregates, derived from initial β-site proteolytic processing of the amyloid precursor protein (APP), accumulate in the brains of Alzheimer's disease patients. The plasmin-generating cascade appears to serve a protective role in the central nervous system since plasmin-mediated proteolysis of APP utilizes the α site, eventually generating nontoxic peptides, and plasmin also degrades Aβ. The conversion of plasminogen to plasmin by tissue-type plasminogen activator in the brain is negatively regulated by plasminogen activator inhibitor type-1 (PAI-1) resulting in attenuation of plasmin-dependent substrate degradation with resultant accumulation of Aβ. PAI-1 and its major physiological inducer TGF-β1, moreover, are increased in models of Alzheimer's disease and have been implicated in the etiology and progression of human neurodegenerative disorders. This review highlights the potential role of PAI-1 and TGF-β1 in this process. Current molecular events associated with TGF-β1-induced PAI-1 transcription are presented with particular relevance to potential targeting of PAI-1 gene expression as a molecular approach to the therapy of neurodegenerative diseases associated with increased PAI-1 expression such as Alzheimer's disease.
INTRODUCTION
In patients with Alzheimer's disease (AD), plaques comprised of aggregated β-amyloid peptides (Aβ) accumulate in specific areas of the brain as a consequence of the proteolytic processing of the single-pass transmembrane amyloid precursor protein (APP) [1]. These Aβ deposits trigger prolonged inflammation, neuronal death, and progressive cognitive decline [2]. Aβ peptides are derived from APP by β-site cleavage by an aspartic protease (BACE) producing a membrane-bound COOH-terminal C99 fragment followed by a complex proteolytic event (involving presenilin and nicastrin) at the C99 transmembrane-localized γ position [3–5]. An alternative APP processing pathway also exists in which membrane-proximal (α-site) cleavage by matrix metalloproteinases (TACE, ADAM 10) replaces β position utilization producing a membrane-anchored C83 fragment. Subsequent γ-site processing of the C83 product results in generation of the nontoxic p3 peptide [3, 6].
The broad-spectrum protease plasmin also degrades Aβ [7–9] and activation of plasmin decreases Aβ peptide levels [10]. Plasmin-mediated proteolysis of APP, moreover, appears to involve the α site (either as a direct or indirect target) resulting in decreased Aβ production, thus suggesting a protective role for the plasmin cascade in the central nervous system. Indeed, plasmin levels in the brains of AD patients are considerably reduced [10] further supporting a causal relationship between deficient activity of the plasmin-generating proteolytic system and accumulation of Aβ in the progression of AD.
PLASMIN-ACTIVATING SYSTEM IN ALZHEIMER'S DISEASE
Several members of the serine protease inhibitor (SERPIN) superfamily exhibit neurotrophic, neuroprotective, or neuropathophysiologic activities depending on the specific cell type and pathways involved [11]. These include SERPINF1, SERPINI1 (neuroserpin), SERPINE1 (plasminogen activator inhibitor type-1; PAI-1), SERPINE2 (nexin-1), and SERPINA3 [11]. PAI-1, in particular, has multifunctional roles in the central nervous system as it both maintains neuronal cellular structure and initiates signaling through the ERK pathway [12]. PAI-1 directly influences the plasmin-dependent pericellular proteolytic cascade by regulating the conversion of plasminogen to plasmin by urokinase- and tissue-type plasminogen activators (uPA/tPA) (Figure 1).
Figure 1.

tPA and uPA convert plasminogen to the active, broad-spectrum, protease plasmin both at the cell surface and in the immediate pericellular space. Plasmin, in turn, degrades target substrates (eg, APP, Aβ) directly as well as indirectly through downstream activation of matrix metalloproteinases (MMPs). Inhibition of MMP activity (GM6001, TIMP) has confirmed their participation in plasmin-initiated proteolysis. Most importantly, this cascade is effectively attenuated by overexpression (or exogenous addition) or PAI-1 which blocks tPA and uPA catalysis inhibiting, thereby, plasmin generation.
PAI-1 immunoreactivity in the central nervous system of AD patients was associated with senile plaques and ghost tangle structures [13] consistent with the earlier colocalization of PA and PAI-1 in plaque structures [14] which are sites of sustained inflammation [15]. Recent findings in Tg2576 and TgCRN8 transgenic mice, engineered to express brain-targeted Swedish mutant Aβ and the double Swedish/V717F mutant Aβ, respectively, under control of the hamster prion promoter and exhibit age-dependent Aβ plaque development (at 12 and 3 months, resp) as well as cognitive deficiencies [16], established that tPA activity was significantly decreased compared to controls [17]. This decline correlated with corresponding increases in PAI-1 expression specifically in areas of the brain where tPA activity was reduced (hippocampus, amygdala). Since direct Aβ peptide injection increased PAI-1 expression and whereas Aβ removal from the hippocampal region required both tPA and plasminogen, it appears that a functional tPA-plasmin axis is required for Aβ clearance [17]. While PAI-1 may be neuroprotective in specific acute injury settings (eg, tPA-triggered neuronal apoptosis) [18], chronically elevated PAI-1 levels likely promote Aβ accumulation by inhibiting plasmin-dependent degradation (Figure 1).
CONTROLS ON TGF-β1 TARGET GENES: THE PAI-1 MODEL
Several reports described elevated TGF-β1 levels in brain biopsies from patients with Parkinson's disease, AD, and stroke [20–22]. This growth factor is likely to influence the onset and progression of AD at several levels. Increased brain expression of TGF-β1 correlates with Aβ angiopathy, and transgenic mice that overexpress TGF-β1 in astrocytes exhibit early onset Aβ deposition [23]. TGF-β1, moreover, induces astrocyte APP expression through a TGF-β1-responsive AGAC Smad-binding element in the APP promoter; subsequent Aβ production, moreover, was enhanced by TGF-β1 signaling [24]. Since the PAI-1 gene is also transcriptionally upregulated by TGF-β1 [19, 25], the coordinate overexpression of PAI-1 and increased Aβ generation in response to elevated TGF-β1 in the brains of AD patients may dispose to disease progression [26]. Collectively, these findings raise the possibility that targeting TGF-β1-inducible genes (eg, PAI-1, APP) may have therapeutic benefit in the setting of AD.
The regulation of TGF-β1-activated genes (ie, PAI-1) is largely transcriptional [19, 25, 27, 28] with the PAI-1 gene subject to complex combinatorial expression controls involving the major transcription effectors p53, Sp1, and members of the MYC family [19, 29, 30]. Prominent TGF-β-response elements in the human PAI-1 promoter include the hexanucleotide E box motif (5′-CACGTG-3′; as in the PE1, PE2, HRE-2 sites) and closely related sequences recognized by the basic helix-loop-helix/leucine zipper (bHLH-LZ) transcription factors of the MYC family (eg, MYC, MAX, TFE3, USF-1, and USF-2) [31–36]. This E box element likely functions, therefore, as a “platform” for recruitment of both positive and negative regulators of PAI-1 expression [37–39]. Recent UV crosslinking and tethered DNA affinity chromatographic analyses identified the bHLH-LZ protein upstream stimulatory factor-1 (USF-1) as a major PAI-1 E box-recognition factor [40]. Specific E box mutations that ablate USF-1 binding to a PAI-1 target deoxyoligonucleotide probe (CA → TC) effectively attenuated TGF-β1-stimulated PAI-1 promoter-driven CAT reporter activity [36]. The human PAI-1 promoter, however, harbors several additional TGF-β-responsive elements, including three E box-adjoining Smad sequences located just 5′ of the PE2 site [32, 33, 35, 41]. Since an engineered two-base-pair mutation CACGTG → CAATTG in a serum-responsive PAI-1 E box attenuated growth state as well as TGF-β1-dependent transcription [36], this same dinucleotide substitution was incorporated into a luciferase reporter construct bearing the immediate 806 base pairs of the human PAI-1 5′ upstream region. Initial truncation approaches did, in fact, confirm that a major (albeit not the only) TGF-β1-responsive element resided within the most proximal 606 base pairs of the human PAI-1 promoter [19]. Specific disruption of the PE2 region E box element by site-directed mutagenesis significantly attenuated TGF-β1-mediated PAI-1 transcription (Figure 2) [19]. The consequences of USF binding to the PAI-1 PE2 E box site may be more complex, however, than simple motif occupancy. Indeed, certain E box-recognition factors including USF-1 and USF-2 effectively induce DNA bending. Cooperative interactions between Sp1 and/or p53 with USF proteins, for example, may be dependent on USF-initiated modifications to DNA conformation allowing distally spaced factors important in expression control to interact with resulting effects on PAI-1 transcription [42].
Figure 2.

Topography of the PAI-1p806-Luc reporter construct illustrating the two (PE1 and PE2) E box sequences. Site-directed mutagenesis and luciferase assays clearly indicated that an intact E box at the PE2 site is required for maximal TGF-β1-induced PAI-1 transcription in human epithelial cells [19].
SEQUENCE REQUIREMENTS FOR USF OCCUPANCY OF THE PE2 REGION E BOX MOTIF
Chromatin immunoprecipitation recently confirmed that the PAI-1 PE2 E box site is an in vivo USF target motif [38]. Since an intact consensus PE2 region E box sequence is necessary for a maximal transcriptional response to growth factors [19], it was important to identify any additional requirements for PE2 E box-occupancy that might influence site residence including the Smad-binding AGAC elements implicated in TGF-β1-dependent APP expression [24]. PE1 and PE2 probes recognition appeared dependent solely on an intact 5′-CACGTG-3′ motif since nuclear factor binding to individual PE1 and PE2 target constructs was successfully blocked by short double-stranded deoxyoligonucleotides containing a consensus E box flanked by non-PAI-1 sequences whereas a mutant E box (5′-CAATTG-3′) “bait” failed to compete [19]. It was important, however, to confirm these results using site-specific mutants within the context of native PAI-1 promoter sequences (eg, the PE2 region backbone) in order to assess the potential contributions of the Smad-binding elements, E box flanking nucleotides (such as the AAT trinucleotide “spacer” between the PE2 E box and the first upstream Smad site), and the CACGTG motif to nuclear protein binding (Figure 2). A recent study established that the major protein/DNA interactions in the PE2 segment were, in fact, E box-dependent and did not require accessory sites since mutation of all three Smad-binding sites (AGAC → CTTG) or removal of the ATT spacer did not affect USF occupancy of the PE2 region E box [19]. While the CACGTG “core” is a target for occupancy by at least seven members of the bHLH-LZ transcription factor family (USF-1, USF-2, c-MYC, MAX, TFE3, TFEB, TFII-I), USF proteins have a preference for C or T at the −4 position in the presence of MgCl2 [43]. Indeed, the human PAI-1 gene has a T at the −4 site of the PE2 region E box as well as a purine at +4 and −5 and a pyrimidine at +5 (A−5T−4C−3A−2C−1G+1T+2G+3G+4C+5), all of which facilitate USF binding [43]. Chromatin immunoprecipitation of the PE2 region E box site in the human PAI-1 gene, moreover, indicated a dynamic occupancy by USF subtypes (USF-1 versus USF-2) as a function of growth state [44]. This motif was clearly a platform for USF-1 binding in quiescent cells. An exchange of PE2 E box USF-1 homodimers with USF-2 homo- or USF-1/USF-2 heterodimers, furthermore, closely correlated with PAI-1 gene activation. This switch may well determine the transcriptional status of the PAI-1 gene in quiescent versus growth factor-stimulated culture conditions [38, 45]. Site occupancy and transcriptional activity additionally require conservation of the PE2 core E box structure as the CACGTG → CACGGA and TCCGTG dinucleotide substitutions (in the rat gene) [36] and a CACGTG → CAATTG or TCCGTG replacement (in the human gene), with retention of PAI-1 flanking sequences, resulted in loss of both competitive binding and growth factor-dependent reporter activity [19, 44]. The CACGTG → TCCGTG mutation is particularly relevant since bHLH proteins with E box-recognition activity have a conserved glutamate important for interaction with the first two nucleotides (CA) in the E box motif [46]. These data are also consistent with the known hexanucleotide preference (CACGTG or CACATG) of USF proteins [39, 47, 48]. To further dissect the role of USF in TGFβ1-mediated PAI-1 transcription in vivo, a dominant-negative USF construct (A-USF) was implemented for molecular genetic targeting [19]. A-USF effectively titers away functional USF proteins by forming highly stable interactions with native USF proteins; such USF/A-USF heterodimers, however, are unable to bind DNA due to replacement (in the A-USF construct) of the basic DNA-binding residues with an acidic domain [49]. A-USF transfection effectively attenuated TGF-β1-induced PAI-1 expression establishing the importance of USF family transcription factors in PAI-1 gene control [19, 50, 51].
MAPPING THE TGF-β1-INDUCED PAI-1 SIGNALING AND TRANSCRIPTIONAL CONTROL NETWORKS: OPPORTUNITIES FOR THERAPEUTIC INTERVENTION
The molecular mechanisms associated with the TGF-β1-initiated E box-dependent PAI-1 gene control and the collateral Smad-mediated APP induction/TGF-β-directed Aβ processing in specific central nervous system cell types remain to be clarified. The available data, however, clearly suggest that these two responses to TGF-β1 are linked in the pathophysiology of human neurodegenerative disease. It has become apparent, moreover, that PAI-1 overexpression is a likely major contributory if not a causative event in AD progression. Our current understanding of the pathways utilized by the TGF-β1 to stimulate the PAI-1 transcription (summarized in Figure 3) indicate that this growth factor activates a kinase cascade, at least partially as a function of epidermal growth factor receptor mobilization (either through the release of the appropriate ligands or the direct receptor transactivation), involving MEK, ERK1/2, and perhaps p38 [49–51]. Pharmacological approaches, use of dominant-negative constructs, and kinase assays suggest that src family kinases and ras GTPase are upstream of MEK-ERK-p38 in this model of induced PAI-1 expression [50, 51]. MAP kinases, in turn, interact with nuclear transcription factors including members of the USF family that, once phosphorylated, bind as dimers to E box motifs in the PAI-1 promoter to modulate gene expression [19, 26, 38, 40, 50, 51]. Genetic perturbation of PAI-1 synthesis in specific areas of the brain (with dominant-negative USF or PAI-1 antisense vectors) or delivery of PAI-1 neutralizing antibodies may effectively stimulate uPA- and/or plasmin-dependent target substrate degradation (eg, Aβ) or at least attenuate the rate of Aβ accumulation (Figures 1 and 3). The continued identification of regulatory points in the PAI-1 expression control network (Figure 3) and recent identification of TGF-β1-response sites (as well as the involved nuclear factors) in the APP and PAI-1 promoters [19, 38, 40, 52] may provide new molecular targets for the therapy of neurodegenerative syndromes associated with PAI-1 upregulation. Indeed, specific SERPINS (including PAI-1) have already been suggested as potential novel therapeutic targets for stroke and cerebral ischemia [12]. The TGF-β1 gene, furthermore, is also USF-regulated [53] suggesting that interference with USF-dependent transcriptional events may have widespread therapeutic implications.
Figure 3.
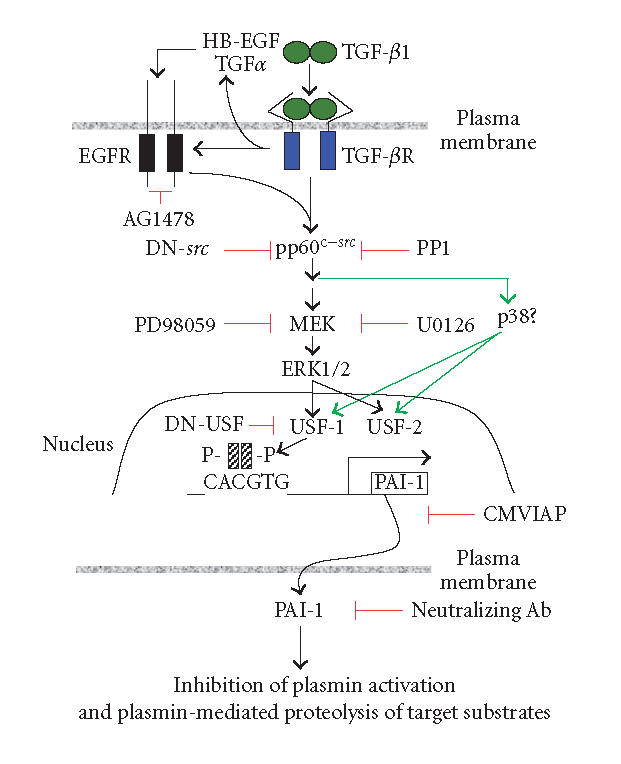
Pathways involved in the regulation of PAI-1 expression and function in response to TGF-β1 stimulation. Positve influences are depicted in black arrows; effective inhibitors defined pharmacologically or by use of dominant-negative constructs (DN) are highlighted in red (detailed in [50]). CMVIAP = PAI-1 antisense expression vector.
ACKNOWLEDGMENT
This work was supported by NIH Grant GM57242.
References
- 1.Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochemical and Biophysical Research Communications. 1984;122(3):1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399(6738 suppl):A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 3.Parvathy S, Hussain I, Karran EH, Turner AJ, Hooper NM. Cleavage of Alzheimer's amyloid precursor protein by α-secretase occurs at the surface of neuronal cells. Biochemistry. 1999;38(30):9728–9734. doi: 10.1021/bi9906827. [DOI] [PubMed] [Google Scholar]
- 4.Vassar R, Bennett BD, Babu-Khan S, et al. β-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 5.Yu G, Nishimura M, Arawaka S, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature. 2000;407(6800):48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 6.Periz G, Fortini ME. Proteolysis in Alzheimer's disease: can plasmin tip the balance? EMBO Reports. 2000;1(6):477–478. doi: 10.1093/embo-reports/kvd124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tucker HM, Kihiko M, Caldwell JN, et al. The plasmin system is induced by and degrades amyloid-β aggregates. Journal of Neuroscience. 2000;20(11):3937–3946. doi: 10.1523/JNEUROSCI.20-11-03937.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Nostrand WE, Porter M. Plasmin cleavage of the amyloid β-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry. 1999;38(35):11570–11576. doi: 10.1021/bi990610f. [DOI] [PubMed] [Google Scholar]
- 9.Wnendt S, Wetzels I, Gunzler WA. Amyloid β peptides stimulate tissue-type plasminogen activator but not recombinant prourokinase. Thrombosis Research. 1997;85(3):217–224. doi: 10.1016/s0049-3848(97)00006-6. [DOI] [PubMed] [Google Scholar]
- 10.Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG. Brain plasmin enhances APP α-cleavage and Aβ degradation and is reduced in Alzheimer's disease brains. EMBO Reports. 2000;1(6):530–535. doi: 10.1093/embo-reports/kvd107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silverman GA, Bird PI, Carrell RW, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Journal of Biological Chemistry. 2001;276(36):33293–33296. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- 12.Vivien D, Buisson A. Serine protease inhibitors: novel therapeutic targets for stroke? Journal of Cerebral Blood Flow and Metabolism. 2000;20(5):755–764. doi: 10.1097/00004647-200005000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Hino H, Akiyama H, Iseki E, et al. Immunohistochemical localization of plasminogen activator inhibitor-1 in rat and human brain tissues. Neuroscience Letters. 2001;297(2):105–108. doi: 10.1016/s0304-3940(00)01679-7. [DOI] [PubMed] [Google Scholar]
- 14.Rebeck GW, Harr SD, Strickland DK, Hyman BT. Multiple, diverse senile plaque-associated proteins are ligands of an apolipoprotein e receptor, the α2-macroglobulin receptor/ low-density-lipoprotein receptor - related protein. Annals of Neurology. 1995;37(2):211–217. doi: 10.1002/ana.410370212. [DOI] [PubMed] [Google Scholar]
- 15.McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Research Reviews. 1995;21(2):195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 16.Hsiao K, Chapman P, Nilsen S, et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 17.Melchor JP, Pawlak R, Strickland S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-β (Aβ) degradation and inhibits Aβ-induced neurodegeneration. Journal of Neuroscience. 2003;23(26):8867–8871. doi: 10.1523/JNEUROSCI.23-26-08867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flavin MP, Zhao G, Ho LT. Microglial tissue plasminogen activator (tPA) triggers neuronal apoptosis in vitro. GLIA. 2000;29(4):347–354. [PubMed] [Google Scholar]
- 19.Allen RR, Qi L, Higgins PJ. Upstream stimulatory factor regulates E box-dependent PAI-1 transcription in human epidermal keratinocytes. Journal of Cellular Physiology. 2005;203(1):156–165. doi: 10.1002/jcp.20211. [DOI] [PubMed] [Google Scholar]
- 20.Agid Y. Parkinson's disease: pathophysiology. Lancet. 1991;337(8753):1321–1324. doi: 10.1016/0140-6736(91)92989-f. [DOI] [PubMed] [Google Scholar]
- 21.Flanders KC, Lippa CF, Smith TW, Pollen DA, Sporn MB. Altered expression of transforming growth factor-β in Alzheimer's disease. Neurology. 1995;45(8):1561–1569. doi: 10.1212/wnl.45.8.1561. [DOI] [PubMed] [Google Scholar]
- 22.Lehrmann E, Kiefer R, Finsen B, Diemer NH, Zimmer J, Hartung HP. Cytokines in cerebral ischemia: expression of transforming growth factor beta-1 (TGF-β1) mRNA in the postischemic adult rat hippocampus. Experimental Neurology. 1995;131(1):114–123. doi: 10.1016/0014-4886(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 23.Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E. Chronic overproduction of transforming growth factor-β1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. American Journal of Pathology. 2000;156(1):139–150. doi: 10.1016/s0002-9440(10)64713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lesne S, Docagne F, Gabriel C, et al. Transforming growth factor-β1 potentiates amyloid-β generation in astrocytes and in transgenic mice. Journal of Biological Chemistry. 2003;278(20):18408–18418. doi: 10.1074/jbc.M300819200. [DOI] [PubMed] [Google Scholar]
- 25.Boehm JR, Kutz SM, Sage EH, Staiano-Coico L, Higgins PJ. Growth state-dependent regulation of plasminogen activator inhibitor type-1 gene expression during epithelial cell stimulation by serum and transforming growth factor-β1. Journal of Cellular Physiology. 1999;181(1):96–106. doi: 10.1002/(SICI)1097-4652(199910)181:1<96::AID-JCP10>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 26.Lesne S, Blanchet S, Docagne F, et al. Transforming growth factor-β1-modulated cerebral gene expression. Journal of Cerebral Blood Flow and Metabolism. 2002;22(9):1114–1123. doi: 10.1097/00004647-200209000-00009. [DOI] [PubMed] [Google Scholar]
- 27.Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nature Genetics. 2001;29(2):117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 28.Kutz SM, Hordines J, McKeown-Longo PJ, Higgins PJ. TGF-β1-induced PAI-1 gene expression requires MEK activity and cell-to-substrate adhesion. Journal of Cell Science. 2001;114(pt 21):3905–3914. doi: 10.1242/jcs.114.21.3905. [DOI] [PubMed] [Google Scholar]
- 29.Kunz C, Pebler S, Otte J, von der Ahe D. Differential regulation of plasminogen activator and inhibitor gene transcription by the tumor suppressor p53. Nucleic Acids Research. 1995;23(18):3710–3717. doi: 10.1093/nar/23.18.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parra M, Jardi M, Koziczak M, Nagamine Y, Munoz-Canoves P. p53 Phosphorylation at serine 15 is required for transcriptional induction of the plasminogen activator inhibitor-1 (PAI-1) gene by the alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine. Journal of Biological Chemistry. 2001;276(39):36303–36310. doi: 10.1074/jbc.M103735200. [DOI] [PubMed] [Google Scholar]
- 31.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. The EMBO Journal. 1998;17(11):3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hua X, Liu X, Ansari DO, Lodish HF. Synergistic cooperation of TFE3 and Smad proteins in TGF-β-induced transcription of the plasminogen activator inhibitor-1 gene. Genes & Development. 1998;12(19):3084–3095. doi: 10.1101/gad.12.19.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hua X, Miller ZA, Wu G, Shi Y, Lodish HF. Specificity in transforming growth factor β-induced transcription of the plasminogen activator inhibitor-1 gene: interactions of promoter DNA, transcription factor μE3, and Smad proteins. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(23):13130–13135. doi: 10.1073/pnas.96.23.13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kietzmann T, Roth U, Jungermann K. Induction of the plasminogen activator inhibitor-1 gene expression by mild hypoxia via a hypoxia response element binding the hypoxia-inducible factor-1 in rat hepatocytes. Blood. 1999;94(12):4177–4185. [PubMed] [Google Scholar]
- 35.Riccio A, Pedone PV, Lund LR, Olesen T, Olsen HS, Andreasen PA. Transforming growth factor β1-responsive element: closely associated binding sites for USF and CCAAT-binding transcription factor-nuclear factor I in the type 1 plasminogen activator inhibitor gene. Molecular and Cellular Biology. 1992;12(4):1846–1855. doi: 10.1128/mcb.12.4.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White LA, Bruzdzinski C, Kutz SM, Gelehrter TD, Higgins PJ. Growth state-dependent binding of USF-1 to a proximal promoter E box element in the rat plasminogen activator inhibitor type 1 gene. Experimental Cell Research. 2000;260(1):127–135. doi: 10.1006/excr.2000.5001. [DOI] [PubMed] [Google Scholar]
- 37.Grinberg AV, Kerppola T. Both Max and TFE3 cooperate with Smad proteins to bind the plasminogen activator inhibitor-1 promoter, but they have opposite effects on transcriptional activity. Journal of Biological Chemistry. 2003;278(13):11227–11236. doi: 10.1074/jbc.M211734200. [DOI] [PubMed] [Google Scholar]
- 38.Qi L, Higgins PJ. Use of chromatin immunoprecipitation to identify E box-binding transcription factors in the promoter of the growth state-regulated human PAI-1 gene. Recent Research Development in Molecular Biology. 2003;1:1–12. [Google Scholar]
- 39.Samoylenko A, Roth U, Jungermann K, Kietzmann T. The upstream stimulatory factor-2a inhibits plasminogen activator inhibitor-1 gene expression by binding to a promoter element adjacent to the hypoxia-inducible factor-1 binding site. Blood. 2001;97(9):2657–2666. doi: 10.1182/blood.v97.9.2657. [DOI] [PubMed] [Google Scholar]
- 40.Providence KM, White LA, Tang J, Gonclaves J, Staiano-Coico L, Higgins PJ. Epithelial monolayer wounding stimulates binding of USF-1 to an E-box motif in the plasminogen activator inhibitor type 1 gene. Journal of Cell Science. 2002;115(pt 19):3767–3777. doi: 10.1242/jcs.00051. [DOI] [PubMed] [Google Scholar]
- 41.Riccio A, Lund LR, Sartorio R, et al. The regulatory region of the human plasminogen activator inhibitor type-1 (PAI-1) gene. Nucleic Acids Research. 1988;16(7):2805–2824. doi: 10.1093/nar/16.7.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumari D, Gabrielian A, Wheeler D, Usdin K. The roles of Sp1, Sp3, USF1/USF2 and NRF-1 in the regulation and three-dimensional structure of the Fragile X mental retardation gene promoter. The Biochemical Journal. 2005;386(pt 2):297–303. doi: 10.1042/BJ20041124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bendall AJ, Molloy PL. Base preferences for DNA binding by the bHLH-Zip protein USF: effects of MgCl2 on specificity and comparison with binding of Myc family members. Nucleic Acids Research. 1994;22(14):2801–2810. doi: 10.1093/nar/22.14.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qi L, Allen RR, Lu Q, et al. PAI-1 Transcriptional regulation during the G0 → G1 transition in human epidermal keratinocytes. Journal of Cellular Biochemistry. doi: 10.1002/jcb.20885. In press. [DOI] [PubMed] [Google Scholar]
- 45.Ghosh AK, Datta PK, Jacob ST. The dual role of helix-loop-helix-zipper protein USF in ribosomal RNA gene transcription in vivo. Oncogene. 1997;14(5):589–594. doi: 10.1038/sj.onc.1200866. [DOI] [PubMed] [Google Scholar]
- 46.Fisher F, Goding CR. Single amino acid substitutions alter helix-loop-helix protein specificity for bases flanking the core CANNTG motif. The EMBO Journal. 1992;11(11):4103–4109. doi: 10.1002/j.1460-2075.1992.tb05503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ismail PM, Lu T, Sawadogo M. Loss of USF transcriptional activity in breast cancer cell lines. Oncogene. 1999;18(40):5582–5591. doi: 10.1038/sj.onc.1202932. [DOI] [PubMed] [Google Scholar]
- 48.Littlewood TD, Evan GI. Transcription factors 2: helix-loop-helix. Protein Profile. 1995;2(6):621–702. [PubMed] [Google Scholar]
- 49.Galibert MD, Carreira S, Goding CR. The Usf-1 transcription factor is a novel target for the stress-responsive p38 kinase and mediates UV-induced Tyrosinase expression. The EMBO Journal. 2001;20(17):5022–5031. doi: 10.1093/emboj/20.17.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kutz SM, Higgins CE, Samarakoon R, et al. TGF-β1-induced PAI-1 expression is E box/USF-dependent and requires EGFR signaling. Experimental Cell Research. doi: 10.1016/j.yexcr.2005.12.027. In press. [DOI] [PubMed] [Google Scholar]
- 51.Samarakoon R, Higgins CE, Higgins SP, Kutz SM, Higgins PJ. Plasminogen activator inhibitor type-1 gene expression and induced migration in TGF-β1-stimulated smooth muscle cells is pp60c−src /MEK-dependent. Journal of Cellular Physiology. 2005;204(1):236–246. doi: 10.1002/jcp.20279. [DOI] [PubMed] [Google Scholar]
- 52.Docagne F, Gabriel C, Lebeurrier N, et al. Sp1 and Smad transcription factors co-operate to mediate TGF-β-dependent activation of amyloid-β precursor protein gene transcription. The Biochemical Journal. 2004;383(pt 2):393–399. doi: 10.1042/BJ20040682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weigert C, Brodbeck K, Sawadogo M, Haring HU, Schleicher ED. Upstream stimulatory factor (USF) proteins induce human TGF-β1 gene activation via the glucose-response element-1013/-1002 in mesangial cells: up-regulation of USF activity by the hexosamine biosynthetic pathway. The Journal of Biological Chemistry. 2004;279(16):15908–15915. doi: 10.1074/jbc.M313524200. [DOI] [PubMed] [Google Scholar]