

Abstract
River systems are vulnerable to natural and anthropogenic habitat fragmentation and will often harbor populations deviating markedly from simplified theoretical models. We investigated fine-scale population structure in the sedentary river fish Cottus gobio using microsatellites and compared migration estimates from three FST estimators, a coalescent maximum-likelihood method and Bayesian recent migration analyses. Source-sink structure was evident via asymmetry in migration and genetic diversity with smaller upstream locations emigration biased and larger downstream subpopulations immigration biased. Patterns of isolation by distance suggested that the system was largely, but not entirely, in migration-drift equilibrium, with headwater populations harboring a signal of past colonizations and in some cases also recent population bottlenecks. Up- vs. downstream asymmetry in population structure was partly attributable to the effects of flow direction, but was enhanced by weirs prohibiting compensatory upstream migration. Estimators of migration showed strong correspondence, at least in relative terms, especially if pairwise FST was used as an indirect index of relative gene flow rather than being translated to Nm. Since true parameter values are unknown in natural systems, comparisons among estimators are important, both to determine confidence in estimates of migration and to validate the performance of different methods.
ESTIMATION of gene flow is a key to understanding and predicting human impacts on natural populations. Traditionally, the principal indirect estimator of gene flow has been FST, which in Wright's (1951) infinite island model equates explicitly with the effective number of migrants (Nm) via FST ≈ 1/(4Nm + 1). The island model makes many simplifying assumptions, and inference of gene flow from FST has attracted severe criticism (Bossart and Prowell 1998; Whitlock and McCauley 1999). Moreover, the impacts of deviations from the island model have primarily considered FST as an inbreeding (F) coefficient, rather than more widely used estimators of FST that are based on pairwise allele-frequency variation (Neigel 2002). Modern coalescent-based maximum-likelihood estimators of gene flow and Bayesian estimators of recent migration are potentially more accurate because they make fewer assumptions and estimate all parameters simultaneously. While contemporary studies seldom attempt to assess gene flow solely via FST-based analysis, the accuracy of modern estimators of migration in natural systems exhibiting complex population structures is poorly understood.
Therefore, the application of migration estimators that rely on different assumptions in empirical studies of natural populations is important to give insights into their relative performance and to assess how confidently parameter estimates can be interpreted.
Rivers appear to be particularly problematical systems for the implementation of traditional population genetic analyses. Migration will typically take place only along a dendritic habitat network that, with simple theoretical models, might be best approximated by the linear stepping-stone model, in which migrants are exchanged only between immediate neighbors in a one-dimensional chain of subpopulations (Kimura and Weiss 1964). Although the relationship between FST and Nm appears to be quite robust to distance-limited dispersal (Slatkin and Barton 1989), near unidirectional flow of water creates a force that promotes movement of organisms along corridors downstream, setting conditions for asymmetric migration and resultant isolation of headwater habitats (Fraser et al. 2004). Unequal migration constitutes a major deviation from the symmetry of both the island and stepping-stone models, with both theory and simulations predicting that bias in FST can ensue (Whitlock and McCauley 1999; Wakeley 2001; Wilkinson-Herbots and Ettridge 2004). Isolated populations are expected to be more vulnerable to extinction due to inbreeding or demographic effects (Saccheri et al. 1998; Morita and Yamamoto 2002; Fagan et al. 2005). Thus, headwater populations might be prone to show cycles of decline and recovery and might never attain migration-drift equilibrium—a critical assumption for most analytical methods if gene flow estimates are to reflect ongoing, rather than historical, processes. Asymmetry in population size, migration, and the potential for local extinction may be enhanced by human activities that fragment rivers, such as the creation of barriers (e.g., by construction of weirs and culverts) or pollution if these primarily impact active upstream movement. Most previous studies of the effects of human-made barriers have focused on migratory salmonids (Meldgaard et al. 2003; Yamamoto et al. 2004; Van Houdt et al. 2005; Wofford et al. 2005) and little is known about impacts on potentially more vulnerable species with limited dispersal and fecundity. Riverine fishes, particularly sedentary species exhibiting habitat specialization, are thus useful models to examine the following applied and fundamental questions: (1) How are natural populations impacted by human activities? and (2) How congruent are results from different estimators of migration in natural population structures deviating markedly from theoretical models?
The bullhead or river sculpin, Cottus gobio, is a widely distributed, but locally endangered, freshwater fish. Bullheads require clean, cool, well-oxygenated waters, with moderate flow rates and at least partial coverage of river beds with reasonably large stones to serve as breeding sites (Knaepkens et al. 2002a). Such ideal habitat is often patchily distributed along river courses with associated disjunctive distribution of bullheads. Adult movement of C. gobio is very limited. Tagging experiments have shown that individuals typically move <10 m during the course of 1 year with only occasional movements of up to 100 m (Downhower et al. 1990; Knaepkens et al. 2004). Fecundity is moderate, with a maximum of a few hundred eggs deposited at a male's nest site by one to six females (our personal observations). These specialized habitat requirements, coupled with limited dispersal and reproductive potential, suggest that bullhead populations will be sensitive to environmental changes, such as intentional or accidental human activities that fragment river systems. In light of such potential vulnerability, knowledge of the spatial structure and connectivity of subpopulations, and of the resulting consequences for population stability, is important for river management.
In this article we compare several estimators of genetic diversity and migration in a study of sources of habitat fragmentation in C. gobio. In spite of multiple deviations from simple theoretical population models, including marked asymmetries in migration and subpopulation sizes, contrasting analytical methods demonstrate an adverse impact of anthropogenic barriers within the river system.
MATERIALS AND METHODS
Sample sites and geographic information:
Our study focused on the River Rye and its tributaries, a river system located in Northeast England. Analysis of long-term monitoring data by the United Kingdom Environment Agency revealed that C. gobio is widely distributed across most of the Rye catchment (Figure 1). In the upper parts of the Rye and its tributaries, bullhead habitat is semicontinuous, comprising lengthy stretches interspersed with relatively small sections of unsuitable habitat (≤50 m) and C. gobio was found consistently and at high density in all potential sample sites. Farther downstream, distributions became patchier and high-density sites were interspersed with unsuitable habitat covering several hundred meters or more. In only one case, however, were bullheads absent for a distance >10 km. Samples of 18–40 bullheads, mostly adults, were taken from 24 sites across the River Rye catchment area by electro-fishing from a river stretch of 50–100 m/site. Sample sites were chosen to cover all main tributaries that contained bullheads and sites of high bullhead densities within tributaries. (Figure 1). A caudal fin clip from each individual was stored in 98% ethanol for genetic analysis. Geographic distances between sample sites were estimated as river distances using Environment Agency databases. As a measure of the relative position of each sample site within the river system, we used “upstream size” (Hänfling and Brandl 1998b), defined as the total length of all suitable river habitats above each site (i.e., catchment length); thus, upstream populations have a lower upstream size than their downstream counterparts. The location of weirs—potential anthropogenic obstructions to bullhead movement—were obtained from Environment Agency databases and maps (D. Hopkins, personal communication) and confirmed by personal observations and landowner surveys. A total of 20 weirs were identified (Figure 1) and, although data on their ages were not available, landowners and Environment Agency records confirmed that weirs have been present in the River Rye catchment for at least 200 years. Since the generation time of bullheads is ∼1–2 years (Nolte et al. 2005), this represents ∼100–200 bullhead generations. It is therefore reasonable to assume that weirs represent a potentially relevant influence on both long- and short-term gene flow.
Figure 1.—

Distribution of sample sites in the River Rye catchment. Shaded areas represent a near-continuous distribution of suitable habitat; hatched areas indicate a more patchy habitat distribution. Latitudes and longitudes: top (of box): 54°24′ N; bottom: 54°10′ N; left: 01°13′ W; right: 00°49′ W. Pie charts represent allelic richness at each sample site as a proportion of overall number of alleles.
Microsatellite genotyping and variability:
DNA was extracted from fin tissue using a standard “salting-out” method (Bruford et al. 1992). All individuals were genotyped at seven highly polymorphic microsatellite loci (Englbrecht et al. 1999) using an ALFExpress automated sequencer (Amersham-Pharmacia). PCR conditions followed Englbrecht et al. (1999), with minor modifications to annealing temperatures. Three or four internal size standards were used for each locus to permit reliable scoring of amplified fragments using FRAGMENT MANAGER 1.2 (Amersham-Pharmacia). Additionally, the same two individuals were run on all gels to facilitate cross-referencing among gels. Genotypes were checked for scoring errors that might be attributable to stutter products, large allele dropout, or the presence of null alleles, using the software MICRO-CHECKER v.2.2 (Van Oosterhout et al. 2004). Allele frequencies at each locus and for each sample were tested for conformity to Hardy–Weinberg equilibrium (as either an excess or deficit of homozygotes to maximize the power of tests) and for linkage disequilibrium among all combinations of loci using GENEPOP version 3.4 with 100 batches of 5000 iterations (Raymond and Rousset 1995). Provided that loci are unlinked, each can represent an independent test of a single hypothesis, such as a test of deviation from H–W (or population differentiation; see below), and, to combine probabilities, we applied the Bernoulli equation: P = [N!/(N − K)!K!] · αK(1 − α)N−K, where N is the number of tests and K is the number below the designated type I error rate, α (Moran 2003), which we set at 0.05. Following the terminology of Kinnison et al. (2002), we refer to this as the “binomial likelihood method” (LM). Since the LM always requires repeatability of individually significant results across tests to yield an overall P < 0.05, it has the desirable property of not being influenced by single extremely low P-values in contrast with the commonly applied Fisher's method. Genetic variability of samples was estimated as unbiased expected heterozygosity (He; Nei 1978) and allelic richness (Â)—the number of alleles, estimated by rarefaction, if all samples are equal to the smallest sample size—using FSTAT 2.9.3.2 (Goudet 2001).
Equilibrium conditions:
Two independent procedures were performed to test whether the set of populations from the River Rye had attained regional migration-drift equilibrium. The first procedure calculated the relative likelihood of a model of immigration–drift–equilibrium vs. a model of drift (without equilibrium) for the system as a whole, using a Markov chain Monte Carlo (MCMC) approach implemented in the program 2mod (Ciofi et al. 1999; software available at http://www.rubic.rdg.ac.uk/∼mab/software.html). In 2mod the variable I indicates which of the alternative models is most likely across all populations for each iteration. The MCMC search was carried out twice for 105 iterations with the first 3 × 104 discarded as burn-in—I values proved to be wholly consistent in each run of 2mod. Our second procedure was based on the graphical isolation by distance (IBD) method of Hutchison and Templeton (1999) that interprets the relationship between pairwise FST and distance in terms of outcomes expected with differing degrees of drift and migration. Specifically, Hutchison and Templeton (1999) predicted that a strong IBD relationship throughout the sampled range, with increasing scatter as stochastic drift gradually becomes more important than migration, is compatible with regional migration-drift equilibrium (type I relationship), whereas strong IBD at shorter distances that decays to a lack of detectable IBD at larger scales suggests a system exhibiting only local-scale equilibrium (type IV relationship; Hutchison and Templeton 1999). Initially we used Mantel tests implemented by the software ZT (Bonnet and Van de Peer 2002) with 100,000 randomizations to test the overall significance of the FST vs. distance relationship and saved residuals from a regression to test the relationship between scatter and distance, again using a Mantel test. However, since type IV relationships may be cryptic, we also split the data into distinct 15-km windows, which proved to be close to the minimum required to obtain enough data points for a reasonable signal of IBD, and within each window the FST vs. distance slope was analyzed separately. For the longer-distance pairwise comparisons in our study, FST might be impacted by mutation. Therefore, we also analyzed IBD relationships via log Nm vs. log distance, since, at high levels of differentiation, Nm [calculated from FST ≈ 1/(4Nm + 1)] is less sensitive to mutation than FST (Slatkin 1993).
Mutation-drift equilibirum was examined by testing for evidence of recent population bottlenecks [within the last ∼0.2–4Ne generations; Luikart and Cornuet (1998) using the coalescent approach of Cornuet and Luikart (1996) implemented in BOTTLENECK (Piry et al. 1999]. A recent simulation study (Williamson-Natesan 2005) has shown that this test performed better than an alternative approach of Garza and Williamson (2001) under conditions that are probable for bullhead populations studied using microsatellites: small prebottleneck diversity and a two-phase model of microsatellite evolution. The Wilcoxon signed-ranks procedure was used to test whether observed heterozygosity exceeded that expected at mutation-drift equilibrium under two more realistic models of microsatellite evolution: the stepwise mutation model (SMM) and a two-phase mutation model with 70% single-step mutations (TPM).
Population structure:
To determine whether our samples might contain individuals that were first generation (F0) immigrants from unsampled populations, we used the Bayesian assignment procedure of Rannala and Mountain (1997), as implemented in GENECLASS 2 (Piry et al. 2004). Since our specific task was to identify F0 immigrants, we used Paetkau et al.'s (2004) method to compute probabilities from 10,000 simulated genotypes. This creates a test distribution of simulated individuals by drawing haplotypes, rather than alleles, from the observed data and thus preserves the partial linkage disequilibrium present in genotypes that have immigrant ancestry, but are not F0 immigrants (Paetkau et al. 2004). Weir and Cockerham's (1984) method for estimating pairwise FST was calculated using GENEPOP 3.4. To compute an estimate of FST as a population-specific inbreeding coefficient (F), we used two different methods. FVit was calculated using the method of Vitalis and Couvet (2001a) by the software ESTIM 1.2 (Vitalis and Couvet 2001b) and F2mod by the software 2mod (Ciofi et al. 1999). Both methods estimate F from the probability that two genes share a common ancestor within a subpopulation, but while FVit is estimated from analytical formulas, F2mod is estimated from a coalescent model using a MCMC approach. We took the mean of F2mod from two independent runs since estimates were highly correlated between runs (Pearson correlation, R = 0.97, P < 0.001). Probabilities of allelic (genic) homogeneity among populations were computed using the exact χ2 test in GENEPOP 3.4 (using 100 batches of 5000 iterations), with the LM technique (see above) used to obtain a combined probability across loci.
Long-term effective population size and migration:
Ratios of migration rate to mutation rate (M) and θ (= 4Neμ; Ne is effective population size and μ is mutation rate) were calculated simultaneously for each pair of subpopulations using the coalescent-based maximum-likelihood (LMAX) method implemented in MIGRATE 2.03 (Beerli and Felsenstein 1999, 2001). MIGRATE 2.03 assumes an n-island model at mutation–migration-drift equilibrium with values of M and θ constant over time. Since such constancy is generally unrealistic, M and θ are best viewed as long-term parameter estimates. MIGRATE uses a MCMC search algorithm to find the set of model parameters that maximizes the likelihood of observing the data. Due to the computational demands of MIGRATE, it proved necessary to limit the number of sample sites to 20 by excluding three locations from small tributaries of the River Seven (SuBe, NoBe, and HoBe) and one sample from the upper Hodge Beck (HoCT). These sites were chosen since we expected their levels of genetic exchange with the rest of the system to be relatively low, although we have also examined the possible effect of their omission (see below). The Brownian motion model was used as an approximation of the stepwise mutation model, and following initial trials, search criteria for the MCMC sampler were set to 20 short chains of 20,000 steps and 3 long chains of 200,000 steps. Searches were combined across all long chains (Geyer 1994) and a static heating scheme with four temperatures (1°, 1.5°, 3°, and 6°) was used (Geyer and Thompson 1994). MCMC sampling was carried out independently for each of the seven loci on separate computers and results were combined using the PERLscript program distributed with the MIGRATE package. The resulting combined file was used as the input for a separate run to obtain LMAX values and confidence intervals across all loci using the “Fast” option. To obtain reliable parameter estimates, three subsequent runs were performed using the LMAX estimates from the previous run as start values. Convergence was accepted if >95% of the confidence intervals for all parameters overlapped between runs. This criterion was achieved after the third run, at which point confidence intervals for all θ estimates, and for 96% of M estimates, overlapped with those from the previous run.
Estimates of gene flow between sample sites were calculated as Nm using four approaches. First, 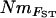 estimates were calculated from pairwise FST-values via FST ≈ 1/(4Nm + 1) (Wright 1951). Second,
estimates were calculated from pairwise FST-values via FST ≈ 1/(4Nm + 1) (Wright 1951). Second, 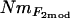 estimates were calculated, again using Wright's (1951) equation, from the population-specific F-coefficients computed by 2mod (Ciofi et al. 1999). Third,
estimates were calculated, again using Wright's (1951) equation, from the population-specific F-coefficients computed by 2mod (Ciofi et al. 1999). Third, 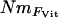 estimates were calculated in the same way using F-values computed by Estim 1.2 (Vitalis and Couvet 2001b). Fourth, values of NmMIG were calculated for each population by multiplying MIGRATE's LMAX estimates of M by θ/4.
estimates were calculated in the same way using F-values computed by Estim 1.2 (Vitalis and Couvet 2001b). Fourth, values of NmMIG were calculated for each population by multiplying MIGRATE's LMAX estimates of M by θ/4.
Recent migration rates:
Patterns of very recent migration (over the last few generations) were investigated using the Bayesian admixture analysis (with the predefined populations option) in BAPS 3.1 (Corander et al. 2004). BAPS estimates the optimal posterior mode of the proportion of an individual's multilocus genotype that is represented by each population. Comparison of the likelihood of the modal solution with the likelihood of the individual being restricted to pure ancestry gives the posterior probability for the hypothesis of no recent immigrant ancestry. Using a critical probability level of 0.05, migration rates (mBAPS) were calculated as the sum of genomic admixture proportions (either in total or from a specific population for pairwise comparisons) divided by the population sample size.
Testing the effect of unsampled populations:
Although our sampling scheme was reasonably comprehensive, it is likely that unsuitable habitats such as deep, slow-flowing pools and lack of substrate impose geographic fragmentation on a scale smaller than that of our sampling grid. Consequently, it is possible that not all genetically distinct demes were sampled. All of our estimators of total immigration as well as the pairwise estimates of NmMIG and mBAPS are potentially sensitive to such unsampled subpopulations. We explored such potential sampling effects by comparing the results from the full data set with those obtained using a subset of 12 samples (supplemental Appendix 2 at http://www.genetics.org/supplemental/).
Hypothesis testing:
First, we examined the interrelationships among several population genetic summary statistics, and also the geographical variable upstream size, using Spearman rank correlations—since a number of relationships were expected to be nonlinear—using SPSS v 11.5. Second, using ZT (Bonnet and Van de Peer 2002), simple and partial Mantel tests were used to analyze the correlations among pairwise matrices of migration estimators and their relationships with pairwise geographic matrices, river distance, and the presence of weirs with 100,000 randomizations. Third, we examined the impact of weirs and distance on upstream and downstream migration using the directional estimates of long-term and recent immigration from MIGRATE and BAPS. Only samples from within the same tributary were used in this analysis because otherwise an unambiguous definition of upstream vs. downstream direction could not be made. Moreover, since Mantel tests must be applied to complete matrices, only a subset of sites that were connected in linear fashion could be used. Thus our analysis used one such linear subset, which comprised the River Seven sample sites (SeRF, SeTh, SeRo, SeHa, SeAs, SeSi, SeBB, SeBF, also including RyNB); other possible subsets contained too few sample sites that were not separated by weirs. Simple and partial Mantel tests were performed separately using upstream migration or downstream migration as dependent variables, with geographic distance and presence of weirs as independent variables. For pairwise comparisons involving these River Seven populations, the effect of weirs on the number of downstream-biased vs. upstream-biased migration estimates was tested directly using a contingency χ2 test. Finally, the effect of distance and number of separating weirs on the change in alleic richness among neighboring River Seven sample sites was investigated via partial correlations (in SPSS v 11.5) with each predictor variable in turn held constant.
RESULTS
Exploratory data analysis:
The seven microsatellite loci were highly polymorphic with between 9 and 44 alleles across all sample sites. MICRO-CHECKER revealed no evidence of scoring errors for any locus in any population. From a total of 504 tests, 22 exhibited significant evidence for linkage disequilibrium at a critical level of P < 0.05. This is slightly fewer than expected by chance as type I errors (25), and no tests were significant following sequential Bonferroni correction. Since populations can be regarded as independent tests for linkage disequilibrium between loci, it was possible to combine probabilities. No pairing of loci exhibited more than three test results below P < 0.05 (minimum LM P > 0.08). From 168 one-tailed tests, each for excess or deficiency of homozygotes, 6 and 4, respectively, exhibited P < 0.05. No more than one result of P < 0.05 for homozygote excess or deficit occurred in any single population across loci (LM P > 0.25), and no more than two of P < 0.05 for any locus across populations (LM P > 0.20). Thus, we detected no evidence for scoring errors, linkage disequilibrium, or deviation from H–W equilibrium in our data.
Population structure:
Using a critical P of 0.05 for first-generation (F0) immigrant analysis, 15 individuals from 777 could not be assigned to any of the sampled locations; however, since the binomial probability of identifying only 15 immigrants by chance with a true type I error rate of 0.05 is P < 0.001, the power to detect F0 immigrants appears to be low. To address this problem, we reset the critical P of the test to a new nominal level that yielded the number of migrants predicted by the upper binomial confidence limit (of 51, at a nominal critical P of 0.145). Moreover, to maintain a relatively liberal test, we made a Bonferroni adjustment of this critical P of 0.145 according to the sample size per population, rather than the overall sample size. At the Bonferroni-adjusted critical value of P = 0.0045 we identified only one migrant from outside the system—in the most downstream site, RyNB. Thus, while caveats regarding the power of the test must still be noted, it seems unlikely that migrants from outside the sampled sites will have had a substantial impact on our analyses.
There was marked genetic differentiation across all samples with a global FST of 0.27 (±0.023 standard error) and pairwise FST estimates between 0.00 and 0.67 (supplemental Appendix 1 at http://www.genetics.org/supplemental/). Population differentiation was near universal among sampling locations with only 4 of 276 pairwise comparisons not significant (supplemental Appendix 1 at http://www.genetics.org/supplemental/). Differentiation was observed at very fine scales. For example, the comparison between SeAs and HoBe, which were located within the River Seven and a small tributary, respectively, yielded an FST estimate of 0.07 (P < 0.001) despite being only a few hundred meters apart.
Mutation-drift equilibrium:
There were no significant tests for population bottlenecks under the most conservative mutation model, the SMM, although the test for SeRF approached significance (P = 0.055). However, four bottleneck tests were significant under the more realistic TPM (SeRF, P < 0.05; SeTh, P < 0.05; HoBe, P < 0.05; SuBe, P < 0.01), with the test for NoBe close to a significant level (P = 0.055). Therefore, while we did not detect evidence of deviation from mutation-drift equilibrium in most populations, five sample sites located upstream or in small tributaries show significant (or marginally significant) evidence of recent population decline.
Isolation by distance and migration-drift equilibrium:
In every individual iteration of two runs (70,000/run), the indicator variable I (estimated by 2mod) suggested that a migration-drift equilibrium model was more likely than a nonequilibrium drift-only model. Moreover, within the river system as a whole there was a strong relationship between FST and distance (Mantel test, R = 0.53, P < 0.001) and increasing residual variation about the FST-distance regression with distance (Mantel test R = 0.23, P < 0.05). While such an IBD pattern appears consistent with regional migration-drift equilibrium as predicted by Hutchison and Templeton's (1999) type I model, closer inspection of the FST-distance plot suggested that the relationship was not wholly linear with a possible change in slope between 40 and 50 km. Indeed, when split into four nonoverlapping distance windows (three of 15 km and a final one of 35 km; Figure 2), we identified strong and very similar patterns of IBD to 45 km, but no FST-distance relationship in the final window (45–80 km). Since data points arising from a pairwise matrix are not independent, the significance of slopes was not tested directly, but instead we divided the data into four groups of six sample sites on the basis of their average separation from all other sites (Table 1). Consistent with the pattern of slopes in Figure 2, the IBD relationship was significantly positive in the first three distance groupings, but not significant in the distance window with the highest average pairwise distance (Table 1). Thus, the river system appeared to show local migration-drift equilibrium to a scale of ∼45 km but had not attained equilibrium at longer distances. Since the maximum distance between sampling points within any individual river is 45.5 km (RaBe and RyNB; see Figure 1), evidence for nonequilibrium conditions arose primarily from comparisons among rivers, particularly among upstream locations.
Figure 2.—

Isolation by distance and approach to migration-drift equilibrium for FST. Separate regression lines are plotted for each 15-km window until 45 km and then from 45 to 80 km. The number of pairwise comparisons in each window are: 69, 72, 73, and 62, respectively, from left to right. The final window is shown with open symbols to highlight the change in relationship.
TABLE 1.
Distance characteristics (mean, min, max) of the windows partitioned to analyze variation in the correlation (Pearson's R) between FST and distance
| Window | Mean | Min | Max | R | P | Sites |
|---|---|---|---|---|---|---|
| 1 | 9.6 | 2.7 | 17.5 | 0.75 | 0.031 | RyNB SeBB SeBF RyWN DoSH SeSi |
| 2 | 19.6 | 0.2 | 37.2 | 0.77 | 0.001 | DoKM SeAs HoBe HoHC SuBe SeHa |
| 3 | 37.5 | 0.6 | 47.1 | 0.42 | 0.035 | DoLo RyDu BogG HOCT SeRo NoBe |
| 4 | 46.5 | 2.6 | 79.3 | 0.10 | 0.346 | SeTh RyCr DoHF SeRF RyHa RaBe |
Probabilities are from Mantel tests.
Evidence of past colonizations:
The shape of the residuals about the regression line in the final window of Figure 2 is of interest because of an apparent decline with distance. We examined this nonequilibrium region in more detail via the framework of Slatkin (1993), who showed that the slope of log 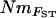 vs. log distance in a one-dimensional stepping-stone model can change progressively from negative to zero or positive, with decreasing residual scatter, depending on time since colonization. Two extreme distance windows were partitioned: (1) short-distance comparisons (maximum pairwise separation of 15 km and a mean of 9 km) comprising the five most downstream sites in the system and (2) long-distance comparisons (minimum pairwise separation of 30 km and a mean of 51 km) comprising the most upstream site in each major river. There was a striking contrast in slopes between the distance windows, with a very strong negative correlation between log
vs. log distance in a one-dimensional stepping-stone model can change progressively from negative to zero or positive, with decreasing residual scatter, depending on time since colonization. Two extreme distance windows were partitioned: (1) short-distance comparisons (maximum pairwise separation of 15 km and a mean of 9 km) comprising the five most downstream sites in the system and (2) long-distance comparisons (minimum pairwise separation of 30 km and a mean of 51 km) comprising the most upstream site in each major river. There was a striking contrast in slopes between the distance windows, with a very strong negative correlation between log 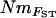 and log distance for short-distance comparisons (and no significant trend in residuals) yet a similarly strong positive correlation for long-distance comparisons, coupled with significantly decreasing residual variation (Figure 3). Therefore, short-distance comparisons among downstream sites show an IBD relationship expected at equilibrium in a stepping-stone population model. Long-distance comparisons among extreme upstream sites, on the other hand, apparently carry a strong historical colonization signal, consistent with Slatkin's (1993) predictions for a recent expansion scenario in a linear stepping-stone model.
and log distance for short-distance comparisons (and no significant trend in residuals) yet a similarly strong positive correlation for long-distance comparisons, coupled with significantly decreasing residual variation (Figure 3). Therefore, short-distance comparisons among downstream sites show an IBD relationship expected at equilibrium in a stepping-stone population model. Long-distance comparisons among extreme upstream sites, on the other hand, apparently carry a strong historical colonization signal, consistent with Slatkin's (1993) predictions for a recent expansion scenario in a linear stepping-stone model.
Figure 3.—

Plot of log 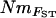 against log river distance. Pairwise comparisons between two groups of sample sites comprise the five most downstream locations (yielding short-distance comparisons) and the five most upstream locations in separate rivers (yielding long-distance comparisons). R is the correlation coefficient; *Mantel tests, P < 0.05.
against log river distance. Pairwise comparisons between two groups of sample sites comprise the five most downstream locations (yielding short-distance comparisons) and the five most upstream locations in separate rivers (yielding long-distance comparisons). R is the correlation coefficient; *Mantel tests, P < 0.05.
Effects of unsampled ghost populations on parameter estimates:
Full details of our investigation, which compared results from four different analytical methods with those obtained using a subset of 12 sample sites, are given in supplemental Appendix 2 at http://www.genetics.org/supplemental/. Measures of total immigration (NmMIG and mBAPS), inbreeding coefficients (FVit and F2mod), and θ all showed a very strong correlation between full and reduced data sets, as did pairwise mBAPS and, to a somewhat lesser extent, NmMIG. However, while the F-coefficients and θ were not consistently higher or lower in the reduced data set compared to the full data set, total NmMIG and mBAPS showed a significant difference between data sets with the former ∼40% higher, and the latter 40% lower in the reduced data set. While pairwise mBAPS values were not consistently higher or lower in the reduced data set compared to the full data set, pairwise estimates of NmMIG differed by 176% in the reduced data set, with 89% of values higher than those in the full data set. Thus for our study system, unsampled ghost populations appear to have little potential to affect estimates of F-coefficients, θ, and pairwise mBAPS, and a moderate, although directional, impact on total mBAPS and NmMIG. However, since pairwise NmMIG values were on average substantially higher in the reduced data set, both ghost populations and the four sites intentionally omitted from analysis (see materials and methods) could have exerted a nontrivial quantitative impact on results.
Genetic diversity, population isolation, and migration asymmetry:
Genetic diversity, whether measured as  or He, varied about fivefold across the sample sites, as did estimates of θ (Table 2). Measures of genetic diversity ( and He) and θ were strongly interrelated, and each was significantly correlated with upstream size (Table 3), indicating that upstream locations displayed lower genetic diversity and Ne than their downstream counterparts. The inbreeding coefficients F2mod and FVit showed strong inverse correlations with θ, Â, He, and upstream size (Table 3), demonstrating that smaller, less genetically diverse upstream populations were also more genetically isolated. A mechanism potentially contributing to these patterns was evident from both immigrant gene flow, NmMIG, and recent immigration rates, mBAPS, both of which were reduced to more upstream populations (Tables 2 and 3). Clear evidence for systematic source-sink population structure was provided by patterns of immigration–emigration asymmetry (Im–Em), a measure of the degree to which a population is a donor or a recipient of migrants. Im–Em increased strongly with upstream size (Table 3); i.e., upstream populations tended to be donors and downstream populations to be recipients (Figure 4).
TABLE 2.
Characteristics of each sample site
| Site | n | Upstream size | He | Â | θ | F2mod | FVit | NmMIG | mBAPS |
|---|---|---|---|---|---|---|---|---|---|
| RaBe | 33 | 1.50 | 0.54 | 4.55 | 0.32 | 0.24 | 0.23 | 4.08 | 0.03 |
| RyHa | 35 | 4.00 | 0.50 | 6.15 | 0.46 | 0.11 | 0.29 | 4.30 | 0.06 |
| RyCr | 31 | 24.19 | 0.59 | 7.25 | 0.48 | 0.09 | 0.16 | 5.83 | 0.17 |
| RyDu | 35 | 31.09 | 0.60 | 7.48 | 0.51 | 0.07 | 0.14 | 8.05 | 0.25 |
| RyWN | 37 | 48.52 | 0.63 | 9.19 | 0.65 | 0.05 | 0.11 | 6.03 | 0.15 |
| BogG | 35 | 1.00 | 0.28 | 2.59 | 0.22 | 0.67 | 0.61 | 1.36 | 0 |
| DoHF | 40 | 2.50 | 0.38 | 2.86 | 0.27 | 0.38 | 0.46 | 2.07 | 0.04 |
| DoLo | 35 | 11.70 | 0.44 | 3.87 | 0.29 | 0.26 | 0.38 | 2.92 | 0.15 |
| DoKM | 40 | 19.91 | 0.54 | 5.18 | 0.41 | 0.18 | 0.23 | 6.03 | 0.21 |
| HoCT | 34 | 2.00 | 0.14 | 1.65 | NA | 0.52 | 0.80 | NA | 0 |
| HoHC | 34 | 7.40 | 0.19 | 2.20 | 0.20 | 0.41 | 0.73 | 1.13 | 0.06 |
| DoSH | 35 | 40.29 | 0.64 | 7.76 | 0.53 | 0.07 | 0.09 | 7.73 | 0.24 |
| SeRF | 18 | 0.50 | 0.51 | 2.86 | 0.17 | 0.42 | 0.28 | 2.68 | 0.04 |
| SeTh | 26 | 3.10 | 0.56 | 3.62 | 0.22 | 0.28 | 0.21 | 3.66 | 0.27 |
| SeRo | 38 | 6.20 | 0.52 | 3.44 | 0.32 | 0.29 | 0.26 | 6.24 | 0.20 |
| NoBe | 23 | 0.50 | 0.54 | 3.36 | NA | 0.30 | 0.23 | NA | 0.22 |
| SeHa | 36 | 11.30 | 0.52 | 3.70 | 0.33 | 0.26 | 0.27 | 5.16 | 0.37 |
| SeAs | 36 | 14.10 | 0.55 | 3.80 | 0.34 | 0.38 | 0.22 | 4.96 | 0.11 |
| HoBe | 35 | 0.50 | 0.57 | 3.95 | NA | 0.22 | 0.19 | NA | 0.09 |
| SuBe | 23 | 4.50 | 0.65 | 4.18 | NA | 0.28 | 0.07 | NA | 0.09 |
| SeSi | 33 | 29.20 | 0.63 | 4.84 | 0.45 | 0.15 | 0.11 | 6.30 | 0.32 |
| SeBF | 34 | 34.19 | 0.68 | 8.20 | 0.69 | 0.05 | 0.03 | 11.17 | 0.30 |
| SeBB | 22 | 36.89 | 0.73 | 8.65 | 0.53 | 0.04 | −0.03 | 9.27 | 0.40 |
| RyNB | 29 | 158.86 | 0.74 | 9.66 | 0.85 | 0.02 | −0.05 | 14.85 | 0.47 |
Values are: sample size (n), upstream size and estimates of genetic diversity (He, Â), relative effective population size (θ), population inbreeding coefficients (F2mod, FVit), total immigrant gene flow (NmMIG), and total recent immigration rate (mBAPS).
TABLE 3.
Spearman rank correlation coefficients (below diagonal; probabilities above) between estimators of genetic diversity, inbreeding coefficients, and migration for each sample site, and the environmental predictor variable upstream size
| Upstream size | He | Â | θ | F2mod | FVit | NmMIG | mBAPS | Im–EmMIG | Im–EmBAPS | |
|---|---|---|---|---|---|---|---|---|---|---|
| Upstream size | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | |
| He | 0.658 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | |
| Â | 0.787 | 0.832 | <0.001 | <0.001 | <0.001 | <0.001 | <0.01 | <0.001 | <0.001 | |
| θ | 0.890 | 0.842 | 0.957 | <0.001 | <0.001 | <0.001 | <0.01 | 0.000 | <0.001 | |
| F2mod | −0.773 | −0.795 | −0.971 | −0.943 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | |
| FVit | −0.655 | −0.999 | −0.827 | −0.847 | 0.789 | <0.001 | <0.001 | <0.001 | <0.001 | |
| NmMIG | 0.831 | 0.892 | 0.837 | 0.887 | −0.852 | −0.897 | <0.001 | <0.001 | <0.001 | |
| mBAPS | 0.676 | 0.706 | 0.606 | 0.626 | −0.680 | −0.709 | 0.806 | <0.001 | <0.001 | |
| Im–EmMIG | 0.792 | 0.881 | 0.792 | 0.785 | −0.821 | −0.886 | 0.914 | 0.807 | <0.001 | |
| Im–EmBAPS | 0.743 | 0.753 | 0.691 | 0.764 | −0.750 | −0.755 | 0.859 | 0.863 | 0.850 |
Figure 4.—

Immigration–emigration asymmetry in (a) long-term gene flow (NmMIG) and (b) recent migration rate (mBAPS). Bars show the mean of differences between immigrant and emigrant estimates per population, ±1 standard error. Samples sites are ordered (left to right) by upstream size, such that the more downstream locations are to the right (see Figure 1 for locations).
Comparison between estimators of migration:
Gene flow estimated from F2mod showed a strong correlation with gene flow estimated as NmMIG (Figure 5). While the range of values was very similar, the relationship was nonlinear, with 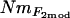 much lower than NmMIG values for populations with low Nm, but correspondingly more closely related at higher levels of Nm (Figure 5).
much lower than NmMIG values for populations with low Nm, but correspondingly more closely related at higher levels of Nm (Figure 5). 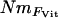 values showed a similar pattern, although Nm could not be estimated from the negative estimates of FVit for two of the populations. Although highly significant, the correlation between pairwise matrices of
values showed a similar pattern, although Nm could not be estimated from the negative estimates of FVit for two of the populations. Although highly significant, the correlation between pairwise matrices of 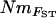 and NmMIG was moderate (Mantel test, R = 0.33, P < 0.001) and weak between
and NmMIG was moderate (Mantel test, R = 0.33, P < 0.001) and weak between 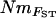 and mBAPS (R = 0.19, P < 0.05). However, the relationship between pairwise matrices of FST and NmMIG or mBAPS was much stronger (Mantel tests, R = −0.76, P < 0.001 and R = −0.53, P < 0.001, respectively), suggesting that the lower congruence of
and mBAPS (R = 0.19, P < 0.05). However, the relationship between pairwise matrices of FST and NmMIG or mBAPS was much stronger (Mantel tests, R = −0.76, P < 0.001 and R = −0.53, P < 0.001, respectively), suggesting that the lower congruence of 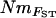 and NmMIG resulted from the translation of pairwise FST values to Nm. All pairwise estimates of
and NmMIG resulted from the translation of pairwise FST values to Nm. All pairwise estimates of 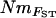 were higher than corresponding NmMIG estimates (supplemental Appendix 1 at http://www.genetics.org/supplemental/), as was global
were higher than corresponding NmMIG estimates (supplemental Appendix 1 at http://www.genetics.org/supplemental/), as was global 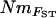 (0.68) by comparison to the overall average NmMIG (0.28). Pairwise directional estimates of scaled migration rates MMIG were quite well correlated with mBAPS for both immigration (R = 0.49, P < 0.001, Mantel test) and emigration (R = 0.54, P < 0.001) and particularly so for the average of immigration and emigration for each pair of populations (Mantel test, R = 0.71, P < 0.001). However, Figure 6 suggests that mBAPS declined much more sharply with distance than MMIG.
(0.68) by comparison to the overall average NmMIG (0.28). Pairwise directional estimates of scaled migration rates MMIG were quite well correlated with mBAPS for both immigration (R = 0.49, P < 0.001, Mantel test) and emigration (R = 0.54, P < 0.001) and particularly so for the average of immigration and emigration for each pair of populations (Mantel test, R = 0.71, P < 0.001). However, Figure 6 suggests that mBAPS declined much more sharply with distance than MMIG.
Figure 5.—

Comparison of Nm calculated from inbreeding coefficient (F) estimates for each population (F2mod and FVit) via F ≈ 1/(1 + 4Nm) with total estimated immigration from MIGRATE (NmMIG). A quadratic line is fitted to the 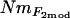 data (R2 = 0.91), which provided a better fit than a linear function (R2 = 0.78). †x-axis positions of missing data points from
data (R2 = 0.91), which provided a better fit than a linear function (R2 = 0.78). †x-axis positions of missing data points from 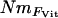 , where FVit estimates were negative and thus Nm was inestimable.
, where FVit estimates were negative and thus Nm was inestimable.
Figure 6.—

Variation in pairwise migration rates with increasing distance between locations. (a) MMIG is the estimated long-term migration rate/mutation rate; a logarithmic curve (R2 shown) provided a better fit than a linear regression (R2 = 0.27). (b) mBAPS is the estimated recent migration rate; again, a linear regression provided a poorer fit (R2 = 0.29) than the fitted logarithmic curve.
Flow direction, weirs, and distance as determinants of migration:
To examine whether distance and weirs affected migration in the river system as a whole, we applied simple and partial Mantel tests to the pairwise matrices of NmMIG, mBAPS, and FST. Distance and weirs showed significant correlations with all metrics in both simple and partial Mantel tests (Table 4a). The preceding analysis had the advantage of including all or most sample sites, but had limited power to estimate the relative impacts of weirs and pairwise distance on gene flow because of relatively few sites that were not separated by weirs. Moreover, the action of flow direction on migration might be an important determinant of the relationships between upstream size and population genetic characteristics, but could be assessed unambiguously only among sites within the same river. Therefore, we compared estimates of upstream vs. downstream NmMIG for sites within the linearly arranged chain of populations from the River Seven and RyNB. In addition, with a maximum pairwise distance of 31.5 km (mean = 13.8 km) sample sites along the River Seven were likely to be near local migration-drift equilibrium. Downstream-biased migration was significantly more frequent than expected by chance for both NmMIG (χ2 = 13.4, P < 0.001) and mBAPS (χ2 = 6.8, P < 0.01). However, as Figure 7a illustrates, detection of a downstream bias in NmMIG was dependent on the presence of a weir between sites (Fisher's exact test, P < 0.01). Figure 7b suggests a similar effect of weirs on asymmetry for mBAPS, and although this was marginally nonsignificant (Fisher's exact test, 0.05 < P < 0.10), fewer data were available because of some cases of zero mBAPS estimates in both flow directions. Weirs reduced upstream migration to a greater extent than distance, for both NmMIG and mBAPS, although distance remained independently significant, or almost so, for both estimators (Table 4b). By contrast, distance had little independent effect on downstream migration for NmMIG, and the negative impact of weirs, although significant, was considerably less than for upstream migration (Table 4b). For mBAPS, on the other hand, the effect of distance on downstream migration was much greater than that of weirs (Table 4b).
TABLE 4.
Pearson correlations between pairwise distance and the presence of a weir separating sample sites and (a) pairwise estimates of migration or population differentiation for all sample sites and (b) directional migration estimates in the River Seven
| Simple Mantel test
|
Partial Mantel test
|
|||
|---|---|---|---|---|
| Distance | Weir | Distance | Weir | |
| a. | ||||
| NmMIG | −0.56*** | −0.51*** | −0.46*** | −0.39*** |
| mBAPS | −0.54*** | −0.40*** | −0.46*** | −0.25** |
| FST | 0.53*** | 0.35*** | 0.46*** | 0.18* |
| b. | ||||
| NmMIG | ||||
| Upstream | −0.60*** | −0.79*** | −0.37* | −0.70*** |
| Downstream | −0.31* | −0.48** | −0.09 | −0.39* |
| mBAPS | ||||
| Upstream | −0.47*** | −0.58** | −0.26a | −0.44** |
| Downstream | −0.40* | −0.16 | −0.37* | 0.05 |
***P < 0.001; **P < 0.01; *P < 0.05.
0.05 < P < 0.10.
Figure 7.—

Upstream vs. downstream (a) long-term gene flow and (b) recent migration in a linear chain of nine samples sites from the River Seven (shown in Figure 8). Pairwise comparisons separated by at least one intervening weir are coded by open symbols; those without an intervening weir by solid symbols. The diagonal line in each shows a 1:1 upstream:downstream relationship.
A possible consequence of these relationships on genetic diversity is illustrated in Figure 8. Change in  between neighboring sample sites in the River Seven was significantly correlated with the number of weirs (partial correlation with neighbor distance held constant, R = 0.85, P < 0.05), but showed no independent correlation with neighbor distance (partial correlation with number of weirs between neighbors held constant, R = 0.09, NS).
Figure 8.—

Change in allelic richness (Â) in relation to distance between neighboring populations (measured from the top of the River Seven) and intervening weirs (vertical lines) in a linear chain of sample sites from the River Seven. Points show the mean  across microsatellite loci with standard errors obtained by jackknifing over loci.
DISCUSSION
Distribution of genetic diversity:
The consistently high, and significant, levels of genetic differentiation even at a small geographical scale (>0.5 km) indicated that bullhead populations of the River Rye catchment are highly fragmented despite a near-continuous distribution of suitable habitat across the study area. This is consistent with field data that indicate a low individual dispersal capacity of bullheads (Downhower et al. 1990; Knaepkens et al. 2004) and marked genetic (allozyme) differentiation among neighboring river systems (Hänfling and Brandl 1998a). Furthermore, there was a striking level of correlation among nearly all measures of genetic diversity and migration/gene flow and also with upstream size—a measure of position within the river system. Therefore, in relative terms at least, many genetic characteristics of bullhead populations within the river system could be predicted from consideration of a small number of measures or, in the absence of specific genetic data, by consideration of the upstream location of a population. Upstream samples exhibited significantly lower levels of genetic diversity and lower effective population sizes and receive fewer immigrants than their downstream counterparts. A strong relationship between upstream size and allelic richness was previously described when comparing bullhead populations from distinct river systems across Europe, including one site (RyWN) from the present data set (R = 0.76, P < 0.001; (Hänfling et al. 2002). With a near identical correlation, this study strongly suggests that the result in Hänfling et al. (2002) was not simply attributable to large-scale variation among long-diverged populations, which otherwise might be most parsimoniously explained by historical processes.
Scale of equilibrium:
Even at the small geographic scale of sampling in this study, the strong and predictable pattern in the distribution of genetic diversity could be generated either through historical processes (e.g., past upstream colonization) or by ongoing ecological processes (e.g., downstream-biased gene flow). Genetic signals of past colonization events are expected to disappear over time as populations approach migration-drift equilibrium. Two independent tests—the strength of isolation by distance, the comparison of drift vs. migration + drift models (in the software 2mod), and additionally, the strong correlation between long-term and contemporary migration—each suggested that bullhead populations from the River Rye catchment are to a large extent in migration-drift equilibrium. Nevertheless, close examination of the FST-distance relationship revealed a pattern consistent with the Hutchison and Templeton (1999) type IV graphical model, suggesting that while much of the system is in migration-drift equilibrium, long-distance comparisons involving upstream sites will reflect historical, rather than contemporary, gene flow. This finding highlights the importance of careful examination of evidence for migration-drift equilibrium throughout different parts of a natural study system. Acceptance of more general evidence such as that of 2mod, which tests only extreme scenarios (equilibrium or not), can result in heterogeneity of equilibrium conditions being overlooked. Indeed, where a stepping-stone population model seems most applicable, it is perhaps wise to consider lack of migration-drift equilibrium as the null hypothesis, at least when considering sites at the edges of a distribution, because the time to approach equilibrium will often be extremely long (Slatkin 1993; Efremov 2004)
Evidence for extinction-recolonization dynamics:
Bottleneck analysis indicated that at least four locations have experienced a recent population decline. All were in sites with low upstream size, either in small tributaries or in the upper part of the catchment, suggesting that reductions in population sizes might occur primarily at the upstream margins of the river system. While the low power of all bottleneck tests (Williamson-Natesan 2005) means that we cannot rule out the possibility that downstream sites have also experienced recent declines, our a priori expectation was that isolated upstream sites would be more prone to such events. Such bottlenecks might reflect either in situ decline of preexisting populations or founder events arising from colonizations. Evidence for colonizations came from comparison of correlations between log 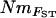 and log distance for extreme upstream and extreme downstream locations. As predicted by Slatkin's (1993) coalescent simulations, the negative relationship expected at equilibrium was strong among downstream locations, but reversed (i.e., strongly positive) among upstream locations, which also, as predicted, showed declining residual variation with distance. Slatkin's (1993) simulations suggest that such a positive slope will persist for only a very short time in a one-dimensional stepping-stone model (<10 generations) before decaying to a more temporally stable IBD state of approximately zero slope. Although the timescale of Slatkin's predictions might be affected by deviations from his stepping-stone range expansion model, such a strong colonization signal is likely of recent origin and thus more likely reflects recolonizations of previously occupied areas, rather than a novel range expansion.
and log distance for extreme upstream and extreme downstream locations. As predicted by Slatkin's (1993) coalescent simulations, the negative relationship expected at equilibrium was strong among downstream locations, but reversed (i.e., strongly positive) among upstream locations, which also, as predicted, showed declining residual variation with distance. Slatkin's (1993) simulations suggest that such a positive slope will persist for only a very short time in a one-dimensional stepping-stone model (<10 generations) before decaying to a more temporally stable IBD state of approximately zero slope. Although the timescale of Slatkin's predictions might be affected by deviations from his stepping-stone range expansion model, such a strong colonization signal is likely of recent origin and thus more likely reflects recolonizations of previously occupied areas, rather than a novel range expansion.
It might seem puzzling that only one (SeRF) of the five populations representing the upstream extremities of our sampling showed evidence for a bottleneck, and none for linkage disequilibrium, which might also be expected. However, extreme upstream locations were genetically impoverished, even to the extent that some loci were monomorphic. Consequently, the power to detect both bottlenecks and linkage disequilibrium (see, for example, Zaykin et al. 1995 for the latter) is likely to have been particularly low for upstream locations. Alternatively, if Slatkin's predicted timescale is applicable, post-bottleneck heterozygosity may have had insufficient time to recover for a bottleneck signal to be detected (≈0.2 Ne generations; Luikart and Cornuet 1998).
Natural vs. anthropogenic determinants of source-sink structure:
Relative Ne (i.e., θ) and immigration estimates showed a strong correlation with upstream size. Gene flow was strongly biased downstream with lower Ne upstream populations serving as net producers of migrants and higher Ne downstream populations as net recipients. Such asymmetric gene flow could be the result of the passive downstream drift created by the essentially unidirectional water movement in riverine environments. Therefore, one might expect such a pattern to be a natural consequence of living in a river habitat. All else being equal, this should prevent long-term persistence of riverine populations, a phenomenon known as the “drift paradox” (Müller 1954) that has puzzled ecologists for >50 years. Most solutions proposed to solve the drift paradox indicate that either active or passive compensatory upstream migration is required to prevent a species boundary from shifting downstream (Hershey et al. 1993; Speirs and Gurney 2001; Humphries and Ruxton 2002; Pachepsky et al. 2005). Our analysis of data from the River Seven showed that upstream migration was much more strongly impacted by the presence of weirs than downstream migration. Moreover, stepwise changes in allelic richness among these neighboring populations was unrelated to distance among sites but strongly correlated with the number of weirs. Although the populations of the River Seven are within a distance window that suggests that they could be near migration-drift equilibrium, the good agreement between results for long-term gene flow, NmMIG (which assume equilibrium), and for recent migration rates, mBAPS (which do not), adds considerable strength to our conclusions. The only disagreement between estimators concerned the relative effects of distance and weirs on downstream migration, which may result from the different timescale that each estimator can detect (see below).
Our results suggest that bullhead populations may be able to at least partly compensate for downstream drift by active upstream migration, provided this is not prohibited by barriers. In turn, this suggests that anthropogenic obstructions are a major source of habitat fragmentation and enhance the isolation and low genetic diversity of upstream bullhead populations. Such anthropogenic impacts might threaten population persistence in at least three ways. First, fragmentation might increase the risk of stochastic population extinction, which is particularly high in hierarchical dendritic habitats such as rivers (Fagan 2002; Morita and Yokota 2002). Second, a “washout” effect caused by prevention of the upstream movement necessary to overcome passive drift could lead to a gradual shift of distribution boundaries downstream (Speirs and Gurney 2001). Third, inbreeding depression caused by genetic isolation might lower survival probabilities of upstream individuals and further diminish population sizes (Saccheri et al. 1998; Brook et al. 2002). Previous work suggests that inbreeding depression occurs in isolated Belgian bullhead populations (Knaepkens et al. 2002b), and we are currently investigating whether the same may apply in some of the upstream populations of the River Rye system.
Recent results from the grayling Thymallus thymallus (Meldgaard et al. 2003) and the cutthroat trout Oncorhynchus clarki clarki (Wofford et al. 2005) have shown, respectively, a correlation between numbers of weirs and FST and between numbers of isolating barriers (some anthropogenic) and genetic diversity (He, Â). Although neither article investigated migration as directly as this study, or the interaction between weirs and flow direction, the general comparability of results from these highly mobile, fecund salmonids and a sedentary, low-fecundity sculpin (C. gobio) suggests that anthropogenic barriers could have substantial negative impacts on river fish in general.
Congruence among estimators of migration:
The population structure of bullheads in the River Rye system is characterized by distance-limited dispersal with directional barriers to migration, systematic asymmetry in Nm and Ne, deviation from equilibrium, and perhaps also extinction–recolonization dynamics. Consequently, our study system bears little resemblance to Wright's (1951) infinite island model upon which the FST:Nm relationship is based. Although inference of gene flow from FST seems quite robust to a number of deviations from the island model, including moderate numbers of subpopulations and distance-limited dispersal (reviewed by (Neigel 2002), systematic asymmetry in Ne, m, or Nm can be problematic for estimation of 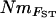 (Whitlock and McCauley 1999; Wakeley 2001; Wilkinson-Herbots and Ettridge 2004). Moreover, FST can be biased downward by mutation (Rousset 1996), which can be considered negligible only if m ≫ μ (where m and μ are mutation and migration rates, respectively). Even at the fine geographic scale of sampling in our study, migration between populations was often very limited and MIGRATE's estimates of M (= m/μ) ranged from 0.2 to 13.2 (supplemental Appendix 1 at http://www.genetics.org/supplemental/). Such estimates are plausible when highly polymorphic microsatellites are used to screen individuals from a highly differentiated system and suggest that mutation might have impacted some of our estimates of FST. By contrast, MIGRATE incorporates mutation, albeit using a simple model of microsatellite evolution and estimates relative m in both directions among populations and also relative Ne for each population. Consequently, since MIGRATE is less constrained by assumptions than
(Whitlock and McCauley 1999; Wakeley 2001; Wilkinson-Herbots and Ettridge 2004). Moreover, FST can be biased downward by mutation (Rousset 1996), which can be considered negligible only if m ≫ μ (where m and μ are mutation and migration rates, respectively). Even at the fine geographic scale of sampling in our study, migration between populations was often very limited and MIGRATE's estimates of M (= m/μ) ranged from 0.2 to 13.2 (supplemental Appendix 1 at http://www.genetics.org/supplemental/). Such estimates are plausible when highly polymorphic microsatellites are used to screen individuals from a highly differentiated system and suggest that mutation might have impacted some of our estimates of FST. By contrast, MIGRATE incorporates mutation, albeit using a simple model of microsatellite evolution and estimates relative m in both directions among populations and also relative Ne for each population. Consequently, since MIGRATE is less constrained by assumptions than 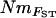 , MIGRATE parameter estimates are often considered likely to be more accurate, at least provided reasonably convergent estimates can be obtained (as was the case for our study). Unfortunately, because of its computational demands, tests of MIGRATE have been limited to date, and although Beerli and Felsenstein (1999) reported good performance in their initial article describing the model, recent tests using simulated mtDNA sequence data by Abdo et al. (2004) showed reasonable accuracy in the estimation of relative Ne, but often substantial inaccuracy in estimates of M. The generality of Abdo et al.'s (2004) results is not clear, especially for microsatellite data, but they highlight the importance of comparing parameter estimates from MIGRATE with those obtained from other methods, since in natural systems true parameter values are unknown.
, MIGRATE parameter estimates are often considered likely to be more accurate, at least provided reasonably convergent estimates can be obtained (as was the case for our study). Unfortunately, because of its computational demands, tests of MIGRATE have been limited to date, and although Beerli and Felsenstein (1999) reported good performance in their initial article describing the model, recent tests using simulated mtDNA sequence data by Abdo et al. (2004) showed reasonable accuracy in the estimation of relative Ne, but often substantial inaccuracy in estimates of M. The generality of Abdo et al.'s (2004) results is not clear, especially for microsatellite data, but they highlight the importance of comparing parameter estimates from MIGRATE with those obtained from other methods, since in natural systems true parameter values are unknown.
In contrast to the strong correlations between different population-specific estimates of total immigrant Nm, pairwise estimates of NmMIG (average of immigration and emigration) were only moderately well correlated with pairwise 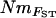 (calculated from Weir and Cockerham's estimator of FST), and all pairwise
(calculated from Weir and Cockerham's estimator of FST), and all pairwise 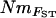 estimates were lower than corresponding NmMIG values. This contrasted with the population-specific Nm estimates from F2mod and FVit, which were always lower than the total NmMIG, and might reflect a much greater sensitivity to mutation for FST calculated from pairwise allele-frequency variation than FST calculated as an inbreeding coefficient (Neigel 2002). We are not aware of any previous comparisons of gene flow estimates from F2mod or FVit with those from MIGRATE, but the relationships proved interesting since estimates shared a similar range but became progressively less congruent as estimated Nm decreased (see Figure 5). One possible explanation for this pattern is that, by contrast to MIGRATE, neither method for estimating F allows for asymmetry in Ne, which can inflate F (Wakeley 2001) and consequently would deflate Nm. Such underestimation will become more pronounced as populations show a higher drift:immigration balance and could be important in our system where Ne and immigration appear to co-vary. Alternatively, while it is possible that NmMIG values were overestimated—either because of ghost populations (see below) or because of nonequilibrium signals of past migration—it is not clear why they should have become progressively less so relative to those from the F-coefficients, which also rely on equilibrium.
estimates were lower than corresponding NmMIG values. This contrasted with the population-specific Nm estimates from F2mod and FVit, which were always lower than the total NmMIG, and might reflect a much greater sensitivity to mutation for FST calculated from pairwise allele-frequency variation than FST calculated as an inbreeding coefficient (Neigel 2002). We are not aware of any previous comparisons of gene flow estimates from F2mod or FVit with those from MIGRATE, but the relationships proved interesting since estimates shared a similar range but became progressively less congruent as estimated Nm decreased (see Figure 5). One possible explanation for this pattern is that, by contrast to MIGRATE, neither method for estimating F allows for asymmetry in Ne, which can inflate F (Wakeley 2001) and consequently would deflate Nm. Such underestimation will become more pronounced as populations show a higher drift:immigration balance and could be important in our system where Ne and immigration appear to co-vary. Alternatively, while it is possible that NmMIG values were overestimated—either because of ghost populations (see below) or because of nonequilibrium signals of past migration—it is not clear why they should have become progressively less so relative to those from the F-coefficients, which also rely on equilibrium.
In our study system, and probably many others where the scale of equilibrium was tested less explicitly, the assumption of migration-drift equilibrium was not wholly fulfilled. Given this limitation of equilibrium-based methods such as MIGRATE, comparisons with nonequilibrium estimators of contemporary migration, such as BAPS, may be informative. Migration estimates from MIGRATE and BAPS were well correlated whether estimated as totals per population or as either directional or nondirectional (averaged) pairwise estimates. The estimators diverged somewhat when patterns of variation across pairwise distance among populations were compared, with mBAPS declining more sharply with distance than MMIG. This difference might be largely attributable to a persistent signal of past colonization in longer distance NmMIG estimates, but could also be partly due to the greater capability of MIGRATE than BAPS to detect indirect migration events in a population structure resembling a stepping-stone model.
How important were ghost populations?
Few studies of natural systems will be able to sample all subpopulations that might exchange migrants. While the estimates produced by traditional measures of pairwise FST and population genetic diversity are independent of the sampling scheme, this cannot be assumed to be true for modern methods that estimate all parameters of interest simultaneously. Yet there have been few tests to date of the impact of unsampled ghost populations (as defined by Slatkin 2005). Recently, both Beerli (2004) and Slatkin (2005) have tested the sensitivity of MIGRATE parameter estimates to unsampled populations but came to different conclusions, with Beerli suggesting a limited effect, but Slatkin suggesting that impacts were difficult to predict, but could be large with substantial immigration from ghost populations.
Our results from evaluating potential impacts of ghost populations illustrate a rather extreme example since we removed up to half of the sample sites. Indeed, the analysis used to detect first-generation immigrants suggested that few immigrants could not be assigned to one of the populations that we had sampled. Consequently, ghost populations probably had a limited effect on most of our results since they most likely represented “intermediate” subpopulations in a linear stepping-stone model. Nevertheless the apparent sensitivity of the different estimators was interesting. Estimators of F-coefficients were not impacted strongly by use of a reduced data set. This was anticipated for the method of Vitalis and Couvet (2001a,b), but was far less certain for that implemented by 2mod (Ciofi et al. 1999). The effects on MIGRATE's estimates of θ and Nm differed, with θ well correlated and not directionally biased between the data sets, but total NmMIG and particularly pairwise NmMIG estimates showing a significant upward bias. Since, even for our full MIGRATE data set, we omitted 4 of the 24 populations to reduce run times, it is likely that our estimates of migration parameters were upward biased to some extent. Use of reduced data sets for MIGRATE is quite commonplace in the literature because of computational demands, but our analysis suggests that resulting parameter estimates may well be biased. Sampling effects on BAPS were quite different from those for MIGRATE, with a significant downward bias to total immigration rate estimates but virtually no effect on pairwise estimates. This rather surprising result implies that immigrant genotypes, or portions thereof, were not reallocated to the “next best” population if their most probable source is omitted, although we suspect that this result could be contingent on the level of differentiation of the study system. Our very brief analysis of ghost population impacts must be regarded as very preliminary in any general sense. However, the problem is very poorly researched and we hope that our results will stimulate further work on an important issue for modern genetic analysis.
Conclusion:
There are two major conclusions from this study. The first is that by blocking compensatory countercurrent migration anthropogenic barriers have a major impact on C. gobio gene flow and the consequent distribution of genetic diversity within the River Rye system. It seems likely that these results will hold not only for bullheads in other rivers systems but also for other headwater riverine species, since all will encounter the ubiquitous force of near unidirectional river flow to some extent. While further investigation is desirable, the potential negative consequences of anthropogenic barriers for population persistence seem clear. We therefore suggest that river managers carefully balance the utility of waterway obstructions against their consequences for conservation of genetic diversity. Second, we found a high degree of concordance among the several estimators of migration and genetic diversity. Even the least congruent estimator—pairwise Nm values calculated from Weir and Cockerham's (1984) FST estimator—became far more so if pairwise FST were used as an index of relative gene flow, rather than being translated into Nm. Such agreement between methods suggests that traditional, maximum-likelihood, and nonequilibrium estimators of diversity and migration parameters all performed well for bullheads exhibiting complex natural population structure, deviating in manifold ways from simplified theoretical models. In addition to strengthening our conclusion, we concur with Neigel's (2002) view that it remains important to apply traditional and modern estimators, both to provide a comparative link with past work and to facilitate tests of computationally intensive modern methods, deviations from the assumptions of which are far less well understood than those for FST.
Acknowledgments
We are grateful to Ian Cowx, Shaun McGinnty, Jon Harvey, David Hopkins, Bob Trigg (sampling); Maggy Harley and Gary Carvalho (laboratory); Peter Beerli, Mark Beaumont, Africa Gomez, and Hilde Wilkinson-Herbots (analysis and discussion). We also thank Robin Waples and an anonymous reviewer for some particularly insightful comments on an earlier version of the manuscript that led to our examination of ghost population impacts and the scale of equilibrium. This work was funded by a European Union Marie Curie Fellowship to B.H. and United Kingdom Natural Environment Research Council grant NER/A/S/2001/01171.
References
- Abdo, Z., K. A. Crandall and P. Joyce, 2004. Evaluating the performance of likelihood methods for detecting population structure and migration. Mol. Ecol. 13: 837–851. [DOI] [PubMed] [Google Scholar]
- Beerli, P., 2004. Effect of unsampled populations on the estimation of population sizes and migration rates between sampled populations. Mol. Ecol. 13: 827–836. [DOI] [PubMed] [Google Scholar]
- Beerli, P., and J. Felsenstein, 1999. Maximum-likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 152: 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli, P., and J. Felsenstein, 2001. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 98: 4563–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet, E., and Y. Van de Peer, 2002. zt: a software tool for simple and partial Mantel tests. J. Stat. Software 7: 1–12. [Google Scholar]
- Bossart, J. L., and D. P. Prowell, 1998. Genetic estimates of population structure and gene flow: limitations, lessons and new directions. Trends Ecol. Evol. 13: 202–206. [DOI] [PubMed] [Google Scholar]
- Brook, B. W., D. W. Tonkyn, J. J. Q'Grady and R. Frankham, 2002. Contribution of inbreeding to extinction risk in threatened species. Conserv. Ecol. 6(1): 16. (http://www.ecologyandsociety.org/vol6/iss1/art16)
- Bruford, M. W., O. Hanotte, J. F. Y. Brookfield and T. Burke, 1992. Single locus and multilocus DNA fingerprinting, pp. 227–229 in Molecular Genetic Analysis of Populations: A Practical Approach, edited by A. R. Hoelzel. IRL Press, Oxford.
- Ciofi, C., M. A. Beaumont, I. R. Swingland and M. W. Bruford, 1999. Genetic divergence and units for conservation in the Komodo dragon Varanus komodoensis. Proc. R. Soc. Lond. Ser. B Biol. Sci. 266: 2269–2274. [Google Scholar]
- Corander, J., P. Waldmann, P. Marttinen and M. J. Sillanpaa, 2004. BAPS 2: enhanced possibilities for the analysis of genetic population structure. Bioinformatics 20: 2363–2369. [DOI] [PubMed] [Google Scholar]
- Cornuet, J. M., and G. Luikart, 1996. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downhower, J. F., P. Lejeune, P. Gaudin and L. Brown, 1990. Movements of the Chabot (Cottus gobio) in a small stream. Pol. Arch. Hydrobiol. 37: 119–126. [Google Scholar]
- Efremov, V. V., 2004. The rate of approach to the equilibrium value of F-ST in island and one-dimensional stepping-stone models of migration. Russ. J. Genet. 40: 1041–1045. [PubMed] [Google Scholar]
- Englbrecht, C. C., C. R. Largiader, B. Hänfling and D. Tautz, 1999. Isolation and characterization of polymorphic microsatellite loci in the European bullhead Cottus gobio L-(Osteichthyes) and their applicability to related taxa. Mol. Ecol. 8: 1966–1969. [DOI] [PubMed] [Google Scholar]
- Fagan, W. F., 2002. Connectivity, fragmentation, and extinction risk in dendritic metapopulations. Ecology 83: 3243–3249. [Google Scholar]
- Fagan, W. F., C. Aumann, C. M. Kennedy and P. J. Unmack, 2005. Rarity, fragmentation, and the scale dependence of extinction risk in desert fishes. Ecology 86: 34–41. [Google Scholar]
- Fraser, D. J., C. Lippe and L. Bernatchez, 2004. Consequences of unequal population size, asymmetric gene flow and sex-biased dispersal on population structure in brook charr (Salvelinus fontinalis). Mol. Ecol. 13: 67–80. [DOI] [PubMed] [Google Scholar]
- Garza, J. C., and E. G. Williamson, 2001. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 10: 305–318. [DOI] [PubMed] [Google Scholar]
- Geyer, C., 1994. Estimating Normalizing Constants and Reweighting Mixtures in Markov Chain Monte Carlo. Technical report no. 568 R(4). University of Minnesota, Minneapolis.
- Geyer, C., and E. A. Thompson, 1994. Annealing Markov chain Monte Carlo with Applications to Ancestral Inference. Technical report no. 589 R(1). University of Minnesota, Minneapolis.
- Goudet, J., 2001. FSTAT: A Program to Estimate and Test Gene Diversities and Fixation Indices, Version 2.9.3. http://www.unil.ch/izea/softwares/fstat.html.
- Hänfling, B., and R. Brandl, 1998. a Genetic differentiation of the bullhead Cottus gobio L. across watersheds in central Europe: evidence for two taxa. Heredity 80: 110–117. [Google Scholar]
- Hänfling, B., and R. Brandl, 1998. b Genetic variability, population size and isolation of distinct populations in the freshwater fish Cottus gobio L. Mol. Ecol. 7: 1625–1632. [Google Scholar]
- Hänfling, B., B. Hellemans, F. A. M. Volkaert and G. R. Carvalho, 2002. Late glacial history of the cold-adapted freshwater fish Cottus gobio, revealed by microsatellites. Mol. Ecol. 11: 1717–1729. [DOI] [PubMed] [Google Scholar]
- Hershey, A. E., J. Pastor, B. J. Peterson and G. W. Kling, 1993. Stable isotopes resolve the drift paradox for baetis mayflies in an arctic river. Ecology 74: 2315–2325. [Google Scholar]
- Humphries, S., and G. D. Ruxton, 2002. Is there really a drift paradox? J. Anim. Ecol. 71: 151–154. [Google Scholar]
- Hutchison, D. W., and A. R. Templeton, 1999. Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 53: 1898–1914. [DOI] [PubMed] [Google Scholar]
- Kimura, M., and G. H. Weiss, 1964. The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 49: 561–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnison, M. T., P. Bentzen, M. J. Unwin and T. P. Quinn, 2002. Reconstructing recent divergence: evaluating nonequilibrium population structure in New Zealand chinook salmon. Mol. Ecol. 11: 739–754. [DOI] [PubMed] [Google Scholar]
- Knaepkens, G., L. Bruyndoncx, L. Bervoets and M. Eens, 2002. a The presence of artificial stones predicts the occurrence of the European bullhead (Cottus gobio) in a regulated lowland river in Flanders (Belgium). Ecol. Freshwater Fish 11: 203–206. [Google Scholar]
- Knaepkens, G., D. Knapen, L. Bervoets, B. Hänfling, E. Verheyen et al., 2002. b Genetic diversity and condition factor: a significant relationship in Flemish but not in German populations of the European bullhead (Cottus gobio L.). Heredity 89: 280–287. [DOI] [PubMed] [Google Scholar]
- Knaepkens, G., L. Bruyndoncx and M. Eens, 2004. Assessment of residency and movement of the endangered bullhead (Cottus gobio) in two Flemish rivers. Ecol. Freshwater Fish 13: 317–322. [Google Scholar]
- Luikart, G., and J. M. Cornuet, 1998. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv. Biol. 12: 228–237. [Google Scholar]
- Meldgaard, T., E. E. Nielsen and V. Loeschcke, 2003. Fragmentation by weirs in a riverine system: a study of genetic variation in time and space among populations of European grayling (Thymallus thymallus) in a Danish river system. Conserv. Genet. 4: 735–747. [Google Scholar]
- Moran, M. D., 2003. Arguments for rejecting the sequential Bonferroni in ecological studies. Oikos 100: 403–405. [Google Scholar]
- Morita, K., and S. Yamamoto, 2002. Effects of habitat fragmentation by damming on the persistence of stream-dwelling charr populations. Conserv. Biol. 16: 1318–1323. [Google Scholar]
- Morita, K., and A. Yokota, 2002. Population viability of stream-resident salmonids after habitat fragmentation: a case study with white-spotted charr (Salvelinus leucomaenis) by an individual based model. Ecol. Modell. 155: 85–94. [Google Scholar]
- Müller, K., 1954. Investigations on the organic drift in North Swedish streams. Inst. Freshwater Res. Rep. 34: 133–148. [Google Scholar]
- Nei, M., 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neigel, J. E., 2002. Is F-ST obsolete? Conserv. Genet. 3: 167–173. [Google Scholar]
- Nolte, A. W., J. Freyhof, K. C. Stemshorn and D. Tautz, 2005. An invasive lineage of sculpins, Cottus sp (Pisces, Teleostei) in the Rhine with new habitat adaptations has originated from hybridization between old phylogeographic groups. Proc. R. Soc. Lond. B Biol. Sci. 272: 2379–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachepsky, E., F. Lutscher, R. M. Nisbet and M. A. Lewis, 2005. Persistence, spread and the drift paradox. Theor. Popul. Biol. 67: 61–73. [DOI] [PubMed] [Google Scholar]
- Paetkau, D., R. Slade, M. Burden and A. Estoup, 2004. Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol. Ecol. 13: 55–65. [DOI] [PubMed] [Google Scholar]
- Piry, S., G. Luikart and J. M. Cornuet, 1999. BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 90: 248–249. [Google Scholar]
- Piry, S., A. Alapetite, J. M. Cornuet, D. Paetkau, L. Baudouin et al., 2004. GENECLASS2: a software for genetic assignment and first-generation migrant detection. J. Hered. 95: 536–539. [DOI] [PubMed] [Google Scholar]
- Rannala, B., and J. L. Mountain, 1997. Detecting immigration by using multilocus genotypes. Proc. Natl. Acad. Sci. USA 94: 9197–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond, M., and F. Rousset, 1995. GENEPOP (version 1.2): a population genetic software for exact tests and ecumenicism. J. Hered. 86: 248–249. [Google Scholar]
- Rousset, F., 1996. Equilibrium values of measures of population subdivision for stepwise mutation processes. Genetics 142: 1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccheri, I., M. Kuussaari, M. Kankare, P. Vikman, W. Fortelius et al., 1998. Inbreeding and extinction in a butterfly metapopulation. Nature 392: 491–494. [Google Scholar]
- Slatkin, M., 1993. Isolation by distance in equilibrium and non-equilibrium populations. Evolution 39: 53–65. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 2005. Seeing ghosts: the effect of unsampled populations on migration rates estimated for sampled populations. Mol. Ecol. 14: 67–73. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., and N. H. Barton, 1989. A comparison of 3 indirect methods for estimating average levels of gene flow. Evolution 43: 1349–1368. [DOI] [PubMed] [Google Scholar]
- Speirs, D. C., and W. S. C. Gurney, 2001. Population persistence in rivers and estuaries. Ecology 82: 1219–1237. [Google Scholar]
- Van Houdt, J. K. J., J. Pinceel, M. C. Flamand, M. Briquet, E. Dupont et al., 2005. Migration barriers protect indigenous brown trout (Salmo trutta) populations from introgression with stocked hatchery fish. Conserv. Genet. 6: 175–191. [Google Scholar]
- Van Oosterhout, C., W. F. Hutchinson, D. P. M. Wills and P. Shipley, 2004. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4: 535–538. [Google Scholar]
- Vitalis, R., and D. Couvet, 2001. a Estimation of effective population size and migration rate from one- and two-locus identity measures. Genetics 157: 911–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis, R., and D. Couvet, 2001. b ESTIM 1.0: a computer program to infer population parameters from one- and two-locus gene identity probabilities. Mol. Ecol. Notes 1: 354–356. [Google Scholar]
- Wakeley, J., 2001. The coalescent in an island model of population subdivision with variation among demes. Theor. Popul. Biol. 59: 133–144. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., and C. C. Cockerham, 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- Whitlock, M. C., and D. E. McCauley, 1999. Indirect measures of gene flow and migration: F-ST not equal 1/(4Nm+1). Heredity 82: 117–125. [DOI] [PubMed] [Google Scholar]
- Wilkinson-Herbots, H. M., and R. Ettridge, 2004. The effect of unequal migration rates on F-ST. Theor. Popul. Biol. 66: 185–197. [DOI] [PubMed] [Google Scholar]
- Williamson-Natesan, E. G., 2005. Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv. Genet. 6: 551–562. [Google Scholar]
- Wofford, J. E. B., R. E. Gresswell and M. A. Banks, 2005. Influence of barriers to movement on within-watershed genetic variation of coastal cutthroat trout. Ecol. Appl. 15: 628–637. [Google Scholar]
- Wright, S., 1951. The genetical structure of populations. Ann. Eugen. 15: 323–354. [DOI] [PubMed] [Google Scholar]
- Yamamoto, S., K. Morita, I. Koizumi and K. Maekawa, 2004. Genetic differentiation of white-spotted charr (Salvelinus leucomaenis) populations after habitat fragmentation: spatial-temporal changes in gene frequencies. Conserv. Genet. 5: 529–538. [Google Scholar]
- Zaykin, D., L. Zhivotovsky and B. S. Weir, 1995. Exact tests for association between alleles at arbitrary numbers of loci. Genetica 96: 169–178. [DOI] [PubMed] [Google Scholar]