

Abstract
Properties of genes underlying variation in complex traits are largely unknown, especially for variation that segregates within populations. Here, we evaluate allelic effects, cis and trans regulation, and dominance patterns of transcripts that are genetically variable in a natural population of Drosophila melanogaster. Our results indicate that genetic variation due to the third chromosome causes mainly additive and nearly additive effects on gene expression, that cis and trans effects on gene expression are numerically about equal, and that cis effects account for more genetic variation than do trans effects. We also evaluated patterns of variation in different functional categories and determined that genes involved in metabolic processes are overrepresented among variable transcripts, but those involved in development, transcription regulation, and signal transduction are underrepresented. However, transcripts for proteins known to be involved in protein–protein interactions are proportionally represented among variable transcripts.
WITHIN-population genetic variability is a major source of phenotypic differences among individuals and is the raw material for evolution, yet very little is known about the characteristics of genes underlying this variation. Further, it is not clear what evolutionary processes maintain within-population variation in the face of natural selection and genetic drift (Barton and Turelli 1989; Lynch et al. 1998; Charlesworth and Hughes 2000; Turelli and Barton 2004), and models to explain the maintenance of variation depend on parameters that have been difficult to estimate (Charlesworth and Hughes 2000).
Addressing these questions depends on measuring the effects and interactions of genes that segregate within natural populations (Mackay 2001). However, reliable and unbiased data have been difficult to obtain because it is not usually possible to measure phenotypic effects of segregating alleles, except in rare cases of Mendelian segregation of discrete phenotypes. Quantitative trait loci (QTL) mapping studies can estimate allelic effects; however, most QTL have not been resolved to the level of individual loci, and most studies have investigated between- rather than within-population variation (Glazier et al. 2002).
Oligonucleotide microarrays provide a novel tool for investigating within-population variation because they allow the simultaneous sampling of large numbers of phenotypes (mRNA abundance of thousands of different transcripts). A high proportion of these phenotypes exhibit significant within-population variation, even when only moderate numbers of individuals are sampled (Oleksiak et al. 2002; Townsend et al. 2003; Wayne et al. 2004). We therefore combined this technology with quantitative genetic experiments to measure patterns of genetic and phenotypic variation, cis and trans regulation, and dominance for >18,000 genes in adult male Drosophila melanogaster. We found that cis-regulatory effects predominate for transcripts with high levels of genetic variation and that variable transcripts tend to be involved in metabolic processes, but not in developmental or regulatory processes. We also found that most transcripts exhibited within-locus additivity or near additivity with respect to mRNA abundance, and few displayed dominance or overdominance.
MATERIALS AND METHODS
Experimental flies:
We obtained third-chromosome substitution lines of D. melanogaster from J. Leips (University of Maryland, Baltimore County) for this experiment. Within each substitution line, flies were identically homozygous for all genes on the third chromosome (C3), and each line contained a different wild-type C3 derived from a single natural population in Raleigh, North Carolina (De Luca et al. 2003). All other chromosomes were identical across lines and were derived from the highly inbred SAM stock (Lyman et al. 1996). Thus, these isogenic lines differed only with respect to allelic variation of genes on C3. We checked homozygosity of the isogenic lines by typing each line for a set of seven variable microsatellite loci (four on C3 and three on C2). All were homozygous within each line.
We measured transcript abundance in equal-aged adult males from six different isogenic lines. We also intercrossed three of these lines (lines 33, 83, and 483) in all possible combinations to produce F1 flies that were hybrids for alleles on C3. Because all lines contained identical sex chromosomes, reciprocal crosses (e.g., 33 females × 83 males and 83 females × 33 males) produced flies that were identical for the nuclear genome. To eliminate differences between parental and F1 lines due to maternal effects, we pooled offspring from reciprocal crosses and mixed them in equal numbers before extracting RNA. These pooled F1 flies were designated as lines 33 × 83, 33 × 483, and 83 × 483.
For each isogenic and F1 line, we reared offspring from replicate vials produced from two independent sets of parents (block A and block B) to produce true biological replicates. We housed block A flies in one incubator and block B flies in another; both blocks were reared and collected at the same time. Parents of experimental flies were reared from constant-density vials (7 males and females per vial), and males used for expression analysis were reared at a constant larval density of 25 per vial. Over a 2-day period (days 0 and 1), we collected males within 8 hr of eclosion and kept them in single-sex vials at low density (10 males per vial) until day 4 when all males were 3–4 days posteclosion. Because we wanted to assay males with appropriate experience for their age, we housed males with females from a laboratory strain (e/e on Ives outbred background) beginning at day 4 and continuing until they were preserved for mRNA extraction on day 8. These mating vials were established at a density of 3 males and 3 females per vial, and four independent vials were maintained per genotype per block.
On day 8, we chose males from each genotype and block for extraction of mRNA, for a total of 18 extractions. For each extraction, we pooled six males chosen from the four mating vials in the appropriate block. Two males were chosen from each of two mating vials and one male from each of the two other vials. Males were flash frozen in random order between 13:00 and 15:00 hr CST. We extracted RNA from all block A flies on the same day and from all block B flies on the next day. Standard Trizol protocols were used for extraction and labeling with the MessageAmp aRNA kit. We hybridized labeled mRNA to Affymetrix Drosophila 2.0 GeneChips according to manufacturer's protocols and scanned them with an Affymetrix GeneArray Scanner at the University of Illinois Affymetrix Core Facility.
Transcript abundance and genetic variation:
Affymetrix Drosophila 2.0 GeneChip contains probes for 18,769 transcripts with 14 probes per transcript. Probe-level intensity values were obtained for each array from MAS 5.0, and all probe intensity values were standardized to the mean value for the array. We determined that 14,298 transcripts were detectable in experimental flies by comparing signals from perfect-match (PM) probes to those from mismatch probes across all arrays using a Wilcoxon sign-rank test (n ranged from 162 to 213). This is probably a liberal test for transcript presence, but we deemed this appropriate because it includes more genes in the entire analysis than a more conservative test, so the false discovery rate (FDR, the expected proportion of significant results that are false positives; Benjamini and Hochberg 1995) calculations for the remainder of the tests we applied are conservative. We used a cutoff of P < 0.04 for determining that a transcript was present, for an FDR of 0.05.
For subsequent analyses, we used only the mean-standardized PM intensity values from the 14,298 detectable transcripts (Chu et al. 2002). The consistency of probe-level measures from independent replicates of the same genotype was high (Pearson's r = 0.94), indicating high repeatability of expression measurements. We tested for significant variation in mRNA abundance among the C3 substitution lines, using gene-specific mixed linear models of the form Log2(PM) = μ + L + P + L × P + B(L) + e, where L was the line effect, P the probe effect, and B(L) the effect of block nested within line. L and P were fixed effects and B(L) was random to provide the error term for L. These models were implemented in SAS PROC MIXED V. 9.1 (SAS Institute 2002). Probe-level values were deleted as outliers if they had external Studentized residuals >3. After outlier removal, 13,423 of the tested transcripts (94%) had residuals that were normally distributed (Shapiro–Wilk P > 0.05). For genes with significant among-line variation, we calculated genetic variance in transcript abundance (VG) as the among-line variance component from a model that included the fixed effect of probe and random effects of line and block within line. To compare genetic variation among transcripts with different abundance levels, we calculated coefficients of genetic variation (CVG) as 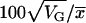 , where
, where  was the mean abundance of the transcript.
was the mean abundance of the transcript.
Cis and trans regulatory effects:
Because the only allelic differences between C3 substitution lines were attributable to genes on C3, all genetic variation in transcript abundance was due to sequence variation on C3. Variable expression of genes on other chromosomes must have been due to trans-acting effects of alleles on C3. This does not imply that the variable genes on other chromosomes have no cis-regulatory mechanisms, only that cis effects would not have contributed to genetic variation in our experiment. Variation in abundance of C3 transcripts could be caused either by cis- or by trans-acting sequence differences, but the proportion subject to trans regulation can be estimated from the number of variable transcripts on the other chromosomes. We therefore obtained chromosomal locations of genes producing each transcript with significant genetic variation, to determine the contributions of cis- and trans-regulatory effects. Chromosomal locations were obtained from the Affymetrix database on February 3, 2005 (Liu et al. 2003).
We used logistic regression to determine if the probability of a transcript occurring on C3 was significantly related to the magnitude of genetic variation as measured by CVG. However, the logistic regression assumes that all transcripts are independent, so the test might be too liberal. We therefore applied a conservative contingency-table test (Wayne et al. 2004), by dividing the 2329 variable transcripts into thirds (tertiles) on the basis of their CVG values and using χ2-tests to determine if chromosomes were equally represented among tertiles (Table 1).
TABLE 1.
Chromosomal location of transcripts in each tertile of genetic coefficient of variation
| Tertilea | C3 total | Cis | Trans | X | C2 | Other | Total | Mean CVGb |
|---|---|---|---|---|---|---|---|---|
| 1 | 639 | 540 | 99 | 39 | 81 | 17 | 776 | 5.1 |
| 2 | 574 | 414 | 160 | 40 | 153 | 9 | 776 | 2.2 |
| 3 | 470 | 226 | 244 | 92 | 203 | 12 | 777 | 1.0 |
| Totalc | 1683 | 1180 | 503 | 171 | 437 | 38 | 2329 | 3.0 |
Tertile 1 comprises transcripts with the most genetic variation in expression; tertile 3 comprises those with the least.
The mean standardized genetic variance for each tertile is the square of the mean CVG.
Expected number of false positives is less than two in all categories.
To determine the total number of cis- vs. trans-regulated transcripts, we also calculated the expected proportion of C3 genes that were trans regulated, assuming that the locations of trans-regulatory variants are randomly distributed relative to their target genes, which is consistent with data from yeast (Brem et al. 2002; Yvert et al. 2003). We based the estimate of trans-regulated C3 genes on the proportion of trans-regulated genes on the other major chromosomes in D. melanogaster (C2 and X).
Functional classification and interacting transcripts:
To test for under- or overrepresentation of functional categories among genetically variable transcripts, we used the Affymetrix gene ontology (GO) mining tool (Liu et al. 2003). The database contains biological process annotation for 6895 genes, of which 1024 were variable among genotypes in our experiment; it contains molecular function annotation for 7240 genes, of which 1086 were variable. We tested the following GO functional categories for over- or underrepresentation among genetically variable genes: “development,” “regulation,” “metabolism,” and “cell communication” for biological process and “catalytic (enzymatic) activity,” “signal transduction,” “regulation of transcription,” and “structural molecule” for molecular function (Rifkin et al. 2003). We tested for disproportional representation of functional categories by comparing the frequency of each category among genetically variable transcripts to its expected frequency among all detectable transcripts. We used χ2-tests for this analysis, because of the potential for nonindependence among transcripts in the same functional category. A category was judged to under- or overrepresented if P < 0.01. Less than 1 false positive is expected under this criterion.
We also compared variable and nonvariable transcripts with respect to evidence for interactions among the protein products of the genes. Giot et al. (2003) reported >20,000 interactions between 7048 proteins encoded in the D. melanogaster genome. They were able to ascribe high, intermediate, or low confidence to these interactions, with high confidence assigned to 4780 interactions involving 4679 proteins. We compared genetically variable and nonvariable transcripts to the entire list of interacting proteins and, separately, to the high-confidence list and tested for nonrandom associations using a two-by-two contingency table analysis.
Dominance:
To calculate dominance effects, we compared mRNA abundance in isogenic lines with that of the F1 hybrid offspring they produced. We first determined which transcripts were variable among the isogenic and F1 lines that we used in crosses: 33, 83, 483, 33 × 83, 33 × 483, and 83 × 483, using the linear model described above. We found that 905 transcripts were significantly variable among these lines at P < 0.013 (FDR of 0.2). For these 905 transcripts, we then determined if there was significant variation among lines that composed a cross (e.g., pairwise differences between lines 33, 83, and 33 × 83), using ESTIMATE statements in PROC MIXED and a P-value cutoff of 0.05. If there were pairwise differences, we calculated the dominance of transcript abundance as d/a, where a is half the difference in abundance between the isogenic parental lines and d is the difference between the F1 hybrid and the mean of the parental lines (Falconer and Mackay 1996; Gibson et al. 2004). If transcript abundance in the hybrid is exactly intermediate to that in the parental lines, d = 0; thus d/a = 0 indicates within-locus additivity and |d/a| = 1 indicates complete dominance. If |d/a| > 1, overdominance (heterosis) of transcript abundance is indicated, meaning the hybrid falls outside the range of phenotypes spanned by the parents.
We determine if mRNA abundance in the two parental lines was significantly different using ESTIMATE statements in PROC MIXED, equivalent to testing 2a > 0. We also determined if the hybrid was significantly different from the mean of its parental lines, equivalent to testing |d| > 0 and indicating significant deviation from within-locus additivity. Type III F-tests for both effects had 2 numerator d.f. and 3 denominator d.f. This is an appropriate test for dominance if the dependent variable is linearly related to transcript abundance. Benchmark trials have shown that log2(PM) is linearly related to log2(abundance) (Cope et al. 2004) with slope estimates between 0.67 and 0.87. For the range of values we observed (4.8 < log2(PM) < 11.6), log2(PM) and abundance are nearly linearly related. We also calculated dominance by assuming a linear relationship between PM and abundance; the results were nearly identical. We therefore report only results obtained on the log scale because of better distributional properties of all variables on this scale.
To determine if transcripts displayed significant overdominance, we calculated 80 and 95% confidence intervals for a and d using ESTIMATE statements within PROC MIXED. If upper and lower limits for d and a did not overlap, we concluded that there was statistical support for overdominance.
RESULTS
Cis and trans regulatory effects:
Of the 14,298 detectable transcripts, 2329 (16.3%) showed significant variation among isogenic lines at P < 0.01 (FDR = 0.06, supplemental Table 1 at http://www.genetics.org/supplemental/). The CVG values for these genes ranged from 19.0 to 0.3. Genes located on C3 were substantially overrepresented among genetically variable transcripts: C3 accounted for 44% (6338) of all detectable transcripts and for 72% (1683) of the genetically variable transcripts (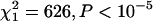 ). This pattern is consistent with substantial cis regulation of genetically variable transcripts.
). This pattern is consistent with substantial cis regulation of genetically variable transcripts.
C3 was also overrepresented among the transcripts exhibiting the most variation when the analysis is limited to the 2329 transcripts with significant genetic variation (Figure 1). The probability of a transcript occurring on C3 was positively related to CVG (likelihood ratio 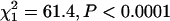 ), while the probability of a transcript occurring on the other chromosomes was negatively related to CVG (X chromosome
), while the probability of a transcript occurring on the other chromosomes was negatively related to CVG (X chromosome 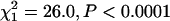 ; chromosome 2
; chromosome 2 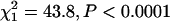 ) (Table 1). Using the tertile method, the representation of C3 was significantly heterogeneous (
) (Table 1). Using the tertile method, the representation of C3 was significantly heterogeneous (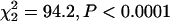 ), with 82% of transcripts in the highest tertile occurring on that chromosome, compared to 74% in the middle tertile and 60% in the bottom tertile (Table 1).
), with 82% of transcripts in the highest tertile occurring on that chromosome, compared to 74% in the middle tertile and 60% in the bottom tertile (Table 1).
Figure 1.—

Coefficient of genetic variation (CVG) in transcript abundance, vs. the probability that the transcript is encoded by a gene on C3. The probabilities were calculated from a logistic regression analysis.
Of 5410 C2 transcripts that were detectable in our study, 81 (1.5%) were genetically variable and in the top tertile of all CVG values, 153 (2.8%) were variable and in the middle tertile, and 203 (3.8%) were variable and in the bottom tertile (supplemental Table 1 at http://www.genetics.org/supplemental/). The proportions were very similar for the 2250 detectable X chromosome transcripts: 39 (1.7%) were in the top, 40 (1.8%) were in the middle, and 92 (4.1%) were in the bottom tertile of all genetically variable transcripts. Taking the proportion of trans-regulated genes on X and C2 as the proportion of detectable transcripts that are expected to be trans regulated by genes on C3 and applying these proportions to the detectable C3 transcripts, we expect 0.0156 × 6338 = 99 C3 transcripts in the top tertile, 0.0125 × 6338 = 160 in the middle tertile, and 0.0385 × 6338 = 244 in the bottom tertile of genetically variable transcripts to be trans regulated by other genes on C3 (Table 1).
Over all tertiles, cis regulation thus accounts for 1180/2329 = 51% of the genetically variable transcripts. However, it accounts for 70% of all transcripts in the top tertile of genetic variation, 53% of transcripts in the middle tertile, and 29% of those in the bottom tertile (Table 1). Because the mean standardized genetic variance (CVG)2 for transcripts in the top tertile is 5.4 times higher than that for transcripts in the middle tertile and 26 times higher than that for transcripts in the bottom tertile (Table 1), we conclude that cis effects are responsible for substantially more genetic variance in gene expression than are trans effects and consequently that cis effects are larger on average than are trans effects (Meiklejohn et al. 2003; Wayne et al. 2004).
To determine if variation in mRNA sequences among inbred lines was a potential source of bias in this analysis (Hsieh et al. 2003), we filtered the 2329 genetically variable transcripts on the basis of a measure of the average between-line correlation of hybridization signals within an individual probe: Cronbach's α (SAS Institute 2002). For each gene on each array, we sequentially deleted probes with the largest values of external Studentized residuals (rS) from the original linear model until the value of Cronbach's α exceeded 0.90. We filtered all genes that did not achieve this level of α after deleting probes with rS ≤ 1.0. This left 1712 genes with known chromosomal locations, of which 112 were on C1, 327 were on C2, and 1273 were on C3. Thus, nearly the same proportion of genes occurs on C3 as on the unfiltered list of variable genes. Sequence variation at probe sites apparently did not bias our estimate of the proportion of cis- and trans-regulated transcripts.
Functional classification and interacting transcripts:
Classification of genetically variable transcripts into biological process categories indicated that developmental, cell communication, and regulatory processes were significantly underrepresented, compared to their frequency among all transcripts in our sample (Table 2). Metabolic processes were overrepresented, but not significantly so. Two molecular function categories were also underrepresented: signal transduction and transcription regulation. Structural molecules were also underrepresented, but the deviation was nonsignificant by the conservative contingency-table analysis we used. Catalytic activity was the only molecular function category that was overrepresented.
TABLE 2.
Gene ontology (GO) categories under- or overrepresented among genetically variable transcripts
| Gene ontology category | Genetically variable | Present in adult males | 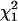 |
Pa |
|---|---|---|---|---|
| Biological process | ||||
| All genes annotated for biological process | 1024 | 6248 | ||
| Underrepresented categories | ||||
| Development | 180 | 1568 | 17.4 | <0.0001 |
| Cell communication | 172 | 1446 | 13.5 | 0.0002 |
| Regulation of biological process | 120 | 1069 | 13.9 | 0.0002 |
| Overrepresented categories | ||||
| Metabolism | 706 | 4007 | 1.9 | 0.17 |
| Molecular function | ||||
| All genes annotated for molecular function | 1086 | 6517 | ||
| Underrepresented categories | ||||
| Signal transduction | 113 | 890 | 6.7 | 0.01 |
| Transcription regulator | 79 | 760 | 15.1 | 0.0001 |
| Structural molecule | 87 | 656 | 3.7 | 0.05 |
| Overrepresented categories | ||||
| Catalytic activity | 632 | 3285 | 7.0 | 0.009 |
Significant categories (two-sided P < 0.0001) are indicated in italics.
Calculated by contingency-table analysis, comparing the representation of a category among genetically variable transcripts to its representation among all annotated transcripts present in our sample.
When we compared variable and nonvariable transcripts with respect to the number of interacting proteins, we found no significant associations. Of the 2329 variable transcripts, 1115 are associated with proteins known to be involved in interactions, and 1214 are not. Of the 11,969 transcripts that were detectable in our sample but not genetically variable, 5533 were associated with interacting proteins, and 6436 were not. There is thus no evidence of association between variability and interaction (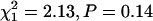 ). When we restricted the analysis to high-confidence protein–protein interactions (Giot et al. 2003), the results were similar: 731 variable transcripts were associated with high-confidence interacting proteins, and 1598 were not, while the numbers for nonvariable transcripts are 3690 of 8279, respectively. Again, there is no evidence of association (
). When we restricted the analysis to high-confidence protein–protein interactions (Giot et al. 2003), the results were similar: 731 variable transcripts were associated with high-confidence interacting proteins, and 1598 were not, while the numbers for nonvariable transcripts are 3690 of 8279, respectively. Again, there is no evidence of association (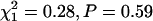 ). Furthermore, the mean number of interactions per transcript is nearly identical for variable and nonvariable transcripts. Variable transcripts average 0.64 (SE = 0.03) interactions per transcript and nonvariable ones average 0.60 (SE = 0.01) interactions.
). Furthermore, the mean number of interactions per transcript is nearly identical for variable and nonvariable transcripts. Variable transcripts average 0.64 (SE = 0.03) interactions per transcript and nonvariable ones average 0.60 (SE = 0.01) interactions.
Dominance of transcript abundance:
We detected at least one pairwise difference between the parental and F1 lines in 1589 cases (646 in cross 33 × 83, 620 in cross 33 × 483, and 323 in cross 83 × 483). Parental lines were significantly different from each other (i.e., a > 0) in 1549 of these cases. The hybrid was significantly different from the mean of the parental lines (i.e., |d| > 0) in only 205 cases. This disparity was not due to differential power of the tests, because they had the same degrees of freedom, median standard errors were not strikingly different (SE[d] = 0.04; SE[a] = 0.03), but medians of the estimates themselves were quite different (median d = 0.06, median a = 0.19). Distributions for a and d are shown in Figure 2, A and B, and all values of standard errors are provided in supplemental Table 2 at http://www.genetics.org/supplemental/.
Figure 2.—



Values of a (A), d (B), and dominance, d/a (C), for transcripts that had at least one pairwise difference in expression between members of a cross. The results for three crosses are shown. Solid areas indicate values for which the parameter was significantly different from 0 (P < 0.05), and open areas indicate nonsignificant estimates. Note the difference in vertical scale for cross 83 × 483.
Distributions of dominance values are shown in Figure 2C. Median values were −0.03 (Wilcoxon test, P > 0.50), 0.08 (P < 0.001), and 0.14 (P < 0.001) for the three crosses, respectively. In each cross, >66% of values fell between −0.5 and +0.5, indicating additivity or intermediate dominance. Overall, 1128 transcripts had d/a values consistent with additivity or intermediate dominance (0 < |d/a| < 0.5), 330 had values consistent with intermediate-to-complete dominance (0.5 < |d/a| < 1), and 131 had values consistent with overdominance (|d/a| > 1).
Of the apparently overdominant transcripts, seven were significantly overdominant using a stringent criterion of nonoverlap of the 95% confidence intervals of d and a; 30 were significant using 80% confidence intervals. A previous study of dominance of mRNA abundance phenotypes used a cutoff of |d/a| > 1.32 on the log2(PM) scale, instead of confidence limits, to indicate overdominance (Gibson et al. 2004). This approach protects against underestimation of the number of overdominant transcripts because of low statistical power. Using this criterion, 88 (3.7%) transcripts displayed overdominance, in broad agreement with the results of Gibson et al. (2004). There were few transcripts with |d/a| > 3, and all were associated with small values of a (Figure 3). Excluding values of a < 0.1, there were no significant associations between a and |d/a| (cross 33 × 83, Pearson's r = −0.04, P = 0.39, N = 546; cross 33 × 483, r = 0.007, P = 0.87, N = 552; crosses 33 × 83 and 83 × 483, r = −0.03, P = 0.62, N = 265).
Figure 3.—

Values of d/a vs. a. Five points are not shown to retain reasonable detail in the graph. These points are, for cross 33 × 83, a, d/a pairs, 0.002, 154; 0.002, 86.8; 0.014, 42.4; 0.004, 40.3; and for cross 33 × 483 a, d/a pairs, 0.012, 22.7. Red symbols indicate transcripts for which d was significantly different from 0; black symbols indicate transcripts for which d was not significant. Horizontal bars show standard errors for a; vertical bars show standard error for d/a (calculated by propagating of error). Note the different scales for a and d/a.
DISCUSSION
Over 16% of the transcripts present in adult males demonstrated significant genetic variance in expression within a single population when only C3 (∼40% of the genome) varied among genotypes. This is only slightly smaller than the proportion of genes showing within-population phenotypic variation in Fundulus (18%, Oleksiak et al. 2002) and/or the proportion of genes varying between inbred lines from different source populations in D. melanogaster (25%, Gibson et al. 2004). This comparison suggests that quantitative genetic variation for mRNA abundance is at least as prevalent within populations as it is between populations of D. melanogaster. Our results indicate that within-population variation in gene expression is due largely to the segregation of many nearly additive alleles, that, numerically, cis effects account for about half the genetically variable transcripts, and that cis effects contribute substantially more to genetic variance in expression that do trans effects. Previous studies of modes of regulation have yielded mixed results, with some reporting a high proportion of trans regulation (Montooth et al. 2003; Yvert et al. 2003; Wayne et al. 2004; Harbison et al. 2005) and some reporting mostly cis (Wittkopp et al. 2004). However, most of these studies evaluated regulation by using between-population or between-species crosses. Trans effects may be more typical of between- than of within-population variation, since selection acting within populations may strictly limit variation in trans-acting genes with multiple downstream effects (Denver et al. 2005).
One previous study of within-population variation in D. simulans also reported evidence for both cis and trans regulation, and found that trans effects were more numerous than cis effects (Wayne et al. 2004), but that cis effects were larger than trans effects (a result also reported by Meiklejohn et al. 2003). Thus there are both intriguing differences and similarities between the two studies. We detected a higher proportion of available transcripts on the Affymetrix chip (76% vs. 56%), a difference that could be explained by our use of a more liberal statistical threshold for transcript presence or by a loss of detectability in the D. simulans study due to hybridizing a different species to D. melanogaster chips or to a difference between first and second-generation Affymetrix arrays (we used the newly available second-generation arrays). We also found a larger proportion of genetically variable transcripts overall (16% vs. 8%). Our lines varied only for C3, which accounts for ∼40% of the D. melanogaster genome, meaning that we would have detected an even higher proportion of variable transcripts had our lines differed for all chromosomes. However, a likely explanation of this difference is that different genetic components of variation were measured in the two studies (additive variance in the previous study vs. variation among homozygous lines in ours). Homozygous genetic variance is expected to exceed additive variation within populations (Charlesworth and Hughes 1996), and experiments confirm this expectation (Hughes 1995a,b; Hughes et al. 2002). Thus our observation of more genetically variable transcripts might simply reflect higher power to detect the larger variances associated with inbred lines.
We also detected an approximately equal numerical representation of cis- than of trans-acting variation in contrast to the study of within-population variation in D. simulans, but like that study, ours indicates that cis effects are on average larger than trans. We could detect only transcript variation that was caused by sequence variation on C3. If patterns of cis and trans regulation differ between chromosomes, and particularly if they differ between sex chromosomes and autosomes, our results would not be representative of sex-linked variation. We know of no direct evidence for such differential patterns of regulation, however. Further, genetic variation on other chromosomes would be likely to induce some additional trans effects on C3, although interchromosomal epistatic interactions could also increase the level of cis-acting variation. Studies comparing chromosome extraction lines directly to whole-genome inbred lines from the same population could be used to address this issue. Finally, the two sets of results are reconcilable if genes that are highly variable within D. melanogaster (mostly cis regulated) are also characterized by greater sequence divergence between species and are therefore less likely to be detected in cross-species hybridizations. Evidence for a positive correlation of within-species variation and between-species divergence has come from studies of genes involved in reproduction (Begun et al. 2000) and cell-surface proteins (Lazzaro 2005); evaluation of the genomewide pattern awaits genome sequencing of other Drosophila species.
Functional analysis of genetically variable transcripts provides insight into the selective constraints that operate on within-population variation. Transcripts involved in metabolic/catalytic processes tend to be genetically variable, while those involved in development, transcriptional regulation, and signal transduction do not. Metabolic transcripts may be more variable than developmental and regulatory ones because they are subject to weaker purifying selection or because variation is actively maintained in metabolic enzymes by heterozygote advantage, genotype–environment interaction, or other forms of balancing selection. Strong purifying selection on developmental and regulatory transcripts may be common if small deviations in transcript abundance disrupt highly integrated developmental and regulatory pathways. Conversely, balancing selection mediated by a variable environment could be particularly relevant to metabolic enzymes, which mediate interactions directly between the organism and its environment.
Genetic variation is not disproportionately represented among transcripts that produce proteins that interact with other proteins. Because transcriptional, translational, and metabolic proteins are all enriched among those engaged in protein–protein interactions (Giot et al. 2003), and metabolic genes are likely to be genetically variable, but regulatory genes are not, this result is not too surprising. However, our results do suggest that protein interaction itself does not constrain or enhance within-population genetic variation.
Dominance patterns indicate that overdominance is rare, and within-locus additivity is typical of segregating variation in transcript abundance. Over 97% of variable transcripts had significant expression differences between two homozygous parents, but only 13% had F1 values that deviated significantly from the midpoint of parental values. There is no obvious relationship between |d/a| and a, as might be expected if genes with large differences between parental lines tend to be dominant or overdominant. This preponderance of additivity contrasts with results from a recent study of crosses between inbred lines derived from different source populations (Gibson et al. 2004). In our study, 71% of dominance values fell between −0.5 and +0.5, while only 15% of values fell in that range in the previous study (calculated from supplemental information for males in Gibson et al. 2004). It might be argued that this difference derives from pooling of reciprocal crosses in our experiment, since Gibson et al. detected differences between F1 reciprocal males in 9% of transcripts tested. However, that study crossed inbred lines that differed at all chromosomes. Male flies from reciprocal crosses thus had different X (and Y) chromosomes, and this could account for differential expression in reciprocal males and for some cases of apparent dominance in males in that experiment. In our study, all lines had the same X, C2, and Y chromosomes, so males from reciprocal crosses had identical genotypes and should have identical expression patterns, aside from maternal effects.
Another possible contributor to different dominance patterns in the two studies is that different filtering criteria were used. Gibson et al. (2004) included a transcript in all dominance calculations if at least one significant pairwise difference was detected among their 12 genotype/sex categories, even though any given dominance value was calculated from only 3 of the 12 categories. We used a more stringent criterion and calculated dominance values only for transcripts showing significant differences among the three genotypes actually used in the dominance calculation.
Because these experimental differences are unlikely to completely account for the large differences in dominance patterns in the two studies, we believe that the discrepancy reflects real differences in the genetic architecture of the lines used. The most obvious difference is that we investigated a random sample of within-population variation, while Gibson et al. used crosses between inbred lines derived from two different source populations (Ore-R and 2b). Although there appears to be limited molecular population structure in D. melanogaster outside of Africa (Dieringer et al. 2005), substantial population differentiation has been found among non-African populations for many quantitative traits (De Jong and Bochdanovits 2003; Schmidt et al. 2005), even on a microgeographic scale (Wayne et al. 2005). In addition, neutral markers have been shown to drastically underestimate population structure for adaptive traits in other species (Karhu et al. 1996). We therefore suggest that differences in the genetic architecture of nonneutral quantitative traits might depend critically on the level at which the variation is investigated (i.e., between vs. within populations).
Alternately, the process of inbreeding itself might lead to different genetic architectures. The lines used by Gibson et al. were produced by many generations of brother–sister mating, which provides an extended opportunity for selection to operate. Furthermore, one of the lines, 2b, underwent selection for low male mating ability during the course of inbreeding. Our lines were produced by sampling third chromosomes directly from a natural population using balancer chromosomes. Third chromosomes that were homozygous lethal or that had very low homozygous fitness would have been removed by this process, but selection was otherwise minimized, and most genetic variation would have been preserved in these lines.
Characterizing effects and interactions of alleles that segregate in natural populations is critical for understanding the genetic basis of phenotypic variation. Our results indicate that the genetic architecture of within-population variation might be different from that observed between populations and that there is a real need for more information on within-population variation. Ultimately, accurate descriptions of variation at both levels will be required for a complete understanding of the causes of genetic and phenotypic diversity.
Acknowledgments
We thank and C. Hartway, J. Leips, C. Milling, C. Whitfield, M. Whitlock, and two anonymous reviewers for insightful comments on manuscript drafts and C. Wilson for conducting the Affymetrix hybrizations and scanning. This work was supported by National Science Foundation awards DEB-0296177 (K.A.H.) and DEB-0092554 (K.N.P.) and by the University of Illinois Urbana Campus Research Board (C.E.C., K.A.H, and K.N.P.).
References
- Barton, N. H., and M. Turelli, 1989. Evolutionary quantitative genetics: how little do we know. Annu. Rev. Genet. 23: 337–370. [DOI] [PubMed] [Google Scholar]
- Begun, D. J., P. Whitley, B. L. Todd, H. M. Waldrip-Dail and A. G. Clark, 2000. Molecular population genetics of male accessory gland proteins in Drosophila. Genetics 156: 1879–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57: 289–300. [Google Scholar]
- Brem, R. B., G. Yvert, R. Clinton and L. Kruglyak, 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., and K. A. Hughes, 1996. Age-specific inbreeding depression and components of genetic variance in relation to the evolution of senescence. Proc. Natl. Acad. Sci. USA 93: 6140–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., and K. A. Hughes, 2000. The maintenance of genetic variation in life history traits, pp. 369–391 in Evolutionary Genetics from Molecules to Morphology, edited by R. S. Singh and C. B. Krimbas. Cambridge University Press, Cambridge, UK.
- Chu, T. M., B. Weir and R. Wolfinger, 2002. A systematic statistical linear modeling approach to oligonucleotide array experiments. Math. Biosci. 176: 35–51. [DOI] [PubMed] [Google Scholar]
- Cope, L. M., R. A. Irizarry, H. A. Jaffee, Z. J. Wu and T. P. Speed, 2004. A benchmark for affymetrix GeneChip expression measures. Bioinformatics 20: 323–331. [DOI] [PubMed] [Google Scholar]
- De Jong, G., and Z. Bochdanovits, 2003. Latitudinal clines in Drosophila melanogaster: body size, allozyme frequencies, inversion frequencies, and the insulin-signalling pathway. J. Genet. 82: 207–223. [DOI] [PubMed] [Google Scholar]
- De Luca, M., N. V. Roshina, G. L. Geiger-Thornsberry, R. F. Lyman, E. G. Pasyukova et al., 2003. Dopa decarboxylase (Ddc) affects variation in Drosophila longevity. Nat. Genet. 34: 429–433. [DOI] [PubMed] [Google Scholar]
- Denver, D. R., K. Morris, J. T. Streelman, S. K. Kim, M. Lynch et al., 2005. The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nat. Genet. 37: 544–548. [DOI] [PubMed] [Google Scholar]
- Dieringer, D., V. Nolte and C. Schlotterer, 2005. Population structure in African Drosophila melanogaster revealed by microsatellite analysis. Mol. Ecol. 14: 563–573. [DOI] [PubMed] [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics. Longman, Essex, UK.
- Gibson, G., R. Riley-Berger, L. Harshman, A. Kopp, S. Vacha et al., 2004. Extensive sex-specific nonadditivity of gene expression in Drosophila melanogaster. Genetics 167: 1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giot, L., J. S. Bader, C. Brouwer, A. Chaudhuri, B. Kuang et al., 2003. A protein interaction map of Drosophila melanogaster. Science 302: 1727–1736. [DOI] [PubMed] [Google Scholar]
- Glazier, A. M., J. H. Nadeau and T. J. Aitman, 2002. Finding the genes that underlie complex traits. Science 298: 2345–2349. [DOI] [PubMed] [Google Scholar]
- Harbison, S., S. Chang, K. Kamdar and T. Mackay, 2005. Quantitative genomics of starvation stress resistance in Drosophila. Genome Biol. 6: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, W. P., T. M. Chu, R. D. Wolfinger and G. Gibson, 2003. Mixed-model reanalysis of primate data suggests tissue and species biases in oligonucleotide-based gene expression profiles. Genetics 165: 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, K. A., 1995. a The evolutionary genetics of male life-history characters in Drosophila melanogaster. Evolution 49: 521–537. [DOI] [PubMed] [Google Scholar]
- Hughes, K. A., 1995. b The inbreeding decline and average dominance of genes affecting male life-history characters in Drosophila melanogaster. Genet. Res. 65: 41–52. [DOI] [PubMed] [Google Scholar]
- Hughes, K. A., J. A. Alipaz, J. M. Drnevich and R. M. Reynolds, 2002. A test of evolutionary theories of senescence. Proc. Natl. Acad. Sci. USA 99: 14286–14291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhu, A., P. Hurme, M. Karjalainen, P. Karvonen, K. Karkkainen et al., 1996. Do molecular markers reflect patterns of differentiation in adaptive traits of conifers? Theor. Appl. Genet. 93: 215–221. [DOI] [PubMed] [Google Scholar]
- Lazzaro, B. P., 2005. Elevated polymorphism and divergence in the class C scavenger receptors of Drosophila melanogaster and D. simulans. Genetics 169: 2023–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, G., A. E. Loraine, R. Shigeta, M. Cline, J. Cheng et al., 2003. NetAffx: Affymetrix probesets and annotations. Nucleic Acids Res. 31: 82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman, R. F., F. Lawrence, S. V. Nuzhdin and T. F. C. Mackay, 1996. Effects of single P-element insertions on bristle number and viablility in Drosophila melanogaster. Genetics 143: 277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., L. Latta, J. Hicks and M. Giorgianni, 1998. Mutation, selection, and the maintenance of life-history variation in a natural population. Evolution 52: 727–733. [DOI] [PubMed] [Google Scholar]
- Mackay, T. F. C., 2001. The genetic architecture of quantitative traits. Annu. Rev. Genet. 35: 303–339. [DOI] [PubMed] [Google Scholar]
- Meiklejohn, C. D., J. Parsch, J. M. Ranz and D. L. Hartl, 2003. Rapid evolution of male-biased gene expression in Drosophila. Proc. Natl. Acad. Sci. USA 100: 9894–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montooth, K. L., J. H. Marden and A. G. Clark, 2003. Mapping determinants of variation in energy metabolism, respiration and flight in Drosophila. Genetics 165: 623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiak, M. F., G. A. Churchill and D. L. Crawford, 2002. Variation in gene expression within and among natural populations. Nat. Genet. 32: 261–266. [DOI] [PubMed] [Google Scholar]
- Rifkin, S. A., J. Kim and K. P. White, 2003. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat. Genet. 33: 138–144. [DOI] [PubMed] [Google Scholar]
- SAS Institute, 2002. SAS/STAT User's Guide, Version 9. SAS Institute, Cary, NC.
- Schmidt, P. S., L. Matzkin, M. Ippolito and W. F. Eanes, 2005. Geographic variation in diapause incidence, life-history traits, and climatic adaptation in Drosophila melanogaster. Evolution 59: 1721–1732. [PubMed] [Google Scholar]
- Townsend, J. P., D. Cavalieri and D. L. Hartl, 2003. Population genetic variation in genome-wide gene expression. Mol. Biol. Evol. 20: 955–963. [DOI] [PubMed] [Google Scholar]
- Turelli, M., and N. H. Barton, 2004. Polygenic variation maintained by balancing selection: pleiotropy, sex-dependent allelic effects and G × E interactions. Genetics 166: 1053–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne, M. L., Y.-J. Pan, S. V. Nuzhdin and L. M. McIntyre, 2004. Additivity and trans-acting effects on gene expression in male Drosophila simulans. Genetics 168: 1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne, M. L., A. Korol and T. F. C. Mackay, 2005. Microclinal variation for ovariole number and body size in Drosophila melanogaster in ‘Evolution Canyon’. Genetica 123: 263–270. [DOI] [PubMed] [Google Scholar]
- Wittkopp, P. J., B. K. Haerum and A. G. Clark, 2004. Evolutionary changes in cis and trans gene regulation. Nature 430: 85–88. [DOI] [PubMed] [Google Scholar]
- Yvert, G., R. B. Brem, J. Whittle, J. M. Akey, E. Foss et al., 2003. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat. Genet. 35: 57–64. [DOI] [PubMed] [Google Scholar]