

Abstract
Limited life span and senescence are near-universal characteristics of eukaryotic organisms, controlled by many interacting quantitative trait loci (QTL) with individually small effects, whose expression is sensitive to the environment. Analyses of mutations in model organisms have shown that genes affecting stress resistance and metabolism affect life span across diverse taxa. However, there is considerable segregating variation for life span in nature, and relatively little is known about the genetic basis of this variation. Replicated lines of Drosophila that have evolved increased longevity as a correlated response to selection for postponed senescence are valuable resources for identifying QTL affecting naturally occurring variation in life span. Here, we used deficiency complementation mapping to identify at least 11 QTL on chromosome 3 that affect variation in life span between five old (O) lines selected for postponed senescence and their five base (B) population control lines. Most QTL were sex specific, and all but one affected multiple O lines. The latter observation is consistent with alleles at intermediate frequency in the base population contributing to the response to selection for postponed senescence. The QTL were mapped with high resolution and contained from 12 to 170 positional candidate genes.
LIMITED life span and senescence, the progressive decline in survivorship and reproduction with advancing years, are near-universal characteristics of eukaryotic organisms. Longevity and senescence are complex traits controlled by many interacting quantitative trait loci (QTL) with individually small effects, whose expression is sensitive to the environment. Knowledge of how genetic and environmental factors interact to limit life span and generate variation in life span between individuals, populations, and species is important from two perspectives: human health and evolutionary theory. As the human population rapidly grows older (Martin and Preston 1994), population aging is becoming one of the most important social and health problems facing the next half century (Rowe 1997). The evolutionary puzzle is why organisms age at all and why there is considerable segregating variation for life span in natural populations. Estimates of heritability of life span are of the order of 10–30% (Finch and Tanzi 1997), and it is easy to artificially select for long-lived strains of Drosophila (Luckinbill et al. 1984; Rose 1984; Partridge and Fowler 1992; Zwaan et al. 1995; Sgro and Partridge 1999).
Classical evolutionary explanations for aging are based on the reduction of the strength of natural selection as organisms grow older (Medawar 1952; Williams 1957; Charlesworth 1980). Medawar's (1952) mutation-accumulation hypothesis posits that mutations that are neutral early in life but deleterious later in life will escape the scrutiny of natural selection and cause both aging and variation in aging. The antagonistic pleiotropy hypothesis is based on costs associated with reproduction, usually represented as trade-offs. Under this model, the effects of a gene that are beneficial early in life are detrimental later in life (Williams 1957; Rose and Charlesworth 1980; Kirkwood and Rose 1991; Tower 1996; Kirkwood and Austad 2000). The models are not mutually exclusive. Both models predict that alleles affecting increased longevity will segregate at intermediate frequency in natural populations, although the distribution of allele frequencies under mutation accumulation will also include rare alleles, consistent with neutrality (Watterson 1975). In addition, one expects some fraction of the standing variation for longevity to be attributable to mutation–selection balance, such that rare deleterious alleles will persist in most populations (Houle et al. 1996).
Understanding the genetic architecture of longevity requires that we identify the genes affecting life span, determine what subset of these genes harbors functional allelic variation in nature, and determine whether the molecular polymorphism(s) causing the variation are rare or common (Mackay 2001). Recently, there has been considerable progress toward understanding the environmental factors and genetic pathways that regulate life span across diverse taxa (Guarente and Kenyon 2000; Finch and Ruvkun 2001; Partridge and Gems 2002; Tatar et al. 2003). For example, dietary restriction extends and reproduction shortens life span, and mutations in evolutionarily conserved genes encoding components of the insulin or insulin-like signaling pathway confer increased longevity (reviewed by Finch and Ruvkun 2001). Pleiotropic effects of mutations causing extended longevity often include increased resistance to oxidative and other stresses as well as reduced fertility (Finch and Ruvkun 2001). Evidence that life span is limited by accumulation of toxic superoxide radicals and peroxides comes from observations that mutations in genes required for the detoxification of reactive oxygen species (ROS) are associated with reduced life span (Griswold et al. 1993; Ishii et al. 1998; Wong et al. 1998). Superoxide dismutase/catalase mimetics increase Caenorhabditis elegans life span (Melov et al. 2000), and overexpression of these genes has been associated with increased life span in Drosophila melanogaster (Parkes et al. 1998; Sun et al. 2002). Consistent with the central role of mitochondria in metabolism and the production of ROS, mutational and RNAi inactivation of genes required for mitochondrial function prolongs C. elegans life span (Dillin et al. 2002; Lee et al. 2003). Other pathways directly implicated in the regulation of longevity include intermediary metabolism, sensory perception, serotonin signaling, chromatin silencing, DNA repair, and the heat-shock response (Finch and Ruvkun 2001; Mackay et al. 2005b). Given the plethora of genes for which expression is altered in aging mice (Lee et al. 1999) and Drosophila (Zou et al. 2000; Pletcher et al. 2002), it is becoming clear that a substantial fraction of the genome could affect life span.
In contrast to the wealth of accumulating knowledge regarding the loci and pathways affecting life span, virtually nothing is known about the genes causing naturally occurring variation in longevity. Are these a subset of the genes identified by analysis of mutations and candidate genes implicated from changes of gene expression with aging, or will analysis of naturally occurring variation for life span uncover additional loci and novel pathways? Is the genetic architecture of natural variation in longevity simple, with relatively few genes with large additive effects contributing to most of the variation, or is it more complex, with variation attributable to many interacting loci?
The first step toward understanding the genetic architecture of natural variation in longevity is to map the QTL causing the variation. Several studies mapping QTL affecting life span have been conducted in Drosophila. At least 25 QTL affecting longevity have been identified in a population of 98 recombinant inbred lines derived from two strains (Oregon and 2b) that were not selected for life span, with effects that are highly sex, environment, and genetic background specific (Nuzhdin et al. 1997; Leips and Mackay 2000, 2002; Vieira et al. 2000; Mackay et al. 2005b). However, the strains used to construct these lines represent only a small sample of the genetic diversity affecting natural variation in life span. Replicated lines selected for postponed senescence that have evolved increased longevity as a correlated response (e.g., Luckinbill et al. 1984; Rose 1984) are more representative of naturally segregating variation and are thus valuable material for identifying QTL affecting naturally occurring variation in life span. Curtsinger and Khazaeli (2002) mapped 3 QTL affecting variation in life span between inbred derivatives of lines selected for postponed senescence (Luckinbill and Clare 1985) and their unselected controls; and Forbes et al. (2004) mapped at least 10 QTL affecting divergence in inbred derivatives of the Rose (1984) selection lines.
QTL defined by recombination mapping are not genetic loci, but typically large chromosomal regions containing one or more loci affecting the trait. The challenge is to map these QTL to the level of individual genes. In Drosophila, quantitative complementation tests to deficiencies can be used to map QTL with high resolution (Pasyukova et al. 2000), and positional candidate genes can be identified with quantitative complementation tests to mutations (Long et al. 1996). Application of these approaches showed that single QTL affecting the variation in longevity between Oregon and 2b fractionated into multiple closely linked QTL and that analysis of natural variants with subtle effects indeed reveals novel loci affecting life span not yet implicated by mutational analyses (Pasyukova et al. 2000; DeLuca et al. 2003; Mackay et al. 2005b).
Here, we have used quantitative complementation tests to deficiencies to map QTL on the third chromosome affecting the difference in life span between the five replicate base (B) lines and five replicate old (O) lines selected for postponed senescence by Rose (1984) and colleagues. We identified a minimum of 11 QTL on the third chromosome that affect the difference in life span between the O and B lines, the majority of which had sex-specific effects.
MATERIALS AND METHODS
Drosophila stocks:
The old (O) lines selected for postponed senescence via later reproduction and their unselected control or base (B) lines have been previously described by Rose (1984) and were a generous gift of Philip Service. B and O lines were maintained in 14-day and 70-day generations, respectively, exactly as described in Service and Vossbrink (1996). Currently, O females and males live more than twice as long as their B counterparts (Service et al. 1998). We crossed females from five replicate B (B1 to B5) lines and five O (O1 to O5) lines to males from a panel of 69 overlapping third chromosome deficiency (Df) stocks, maintained against balancer (Bal) chromosomes. The deficiency/balancer stocks were obtained from the Bloomington Drosophila Stock Center (Bloomington, IN) and covered ∼80% of the third chromosome (supplemental Table 1 at http://www.genetics.org/supplemental/) (Figure 1). The deficiency breakpoints were provided by the donors and were not confirmed independently. All stocks were maintained in vials with 10 ml cornmeal–molasses–agar medium, at 25°.
Figure 1.—

The bars depict the 69 chromosome 3 deficiencies used for deficiency complementation mapping of QTL affecting increased longevity. Green bars denote deficiencies that fail to complement QTL affecting life span, and blue bars indicate deficiencies that complement QTL affecting life span.
Quantitative complementation tests with deficiencies:
The mapping method used is a quantitative extension of classical deficiency mapping in Drosophila (Pasyukova et al. 2000). Stocks carrying a deficiency and a balancer chromosome were crossed to each of the five B and five O lines resulting in four classes of F1 flies: Df/O, Df/B, Bal/O, and Bal/B. Virgin flies were collected in 48-hr increments. Three males and three females of the same genotype were then placed in each of four replicate vials and maintained at 25° on 5 ml cornmeal–molasses–agar medium. Flies were transferred to fresh vials every 1–4 days as needed and scored at 2-day intervals for survival time. Deficiencies that failed to complement longevity QTL (see below) were retested using five replicate vials.
Statistical analyses:
A three-way factorial, mixed-model analysis of variance (ANOVA) was used to assess effects. The full model for the initial screen was
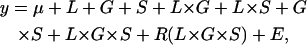 |
where L, G, and S are the fixed cross-classified main effects of line (O1–5, B1–5), genotype (Df vs. Bal), and sex; R is the random effect of replicate vial; and E is the error variance. Reduced ANOVAs were also run for each sex separately. If differences between deficiency O and B genetic backgrounds and balancer O and B backgrounds are constant, yielding a nonsignificant F-statistic (significance at the 5% level) for the L × G or L × G × S interaction term, this is interpreted statistically as no interaction and genetically as quantitative complementation. If differences are not constant and the L × G and/or L × G × S terms are statistically significant, this is interpreted genetically as quantitative failure to complement (Pasyukova et al. 2000).
Because five replicate lines from each B and O population were used, another level of analysis was added to aid in differentiating between common and rare alleles, similar to the above analysis with the addition of a selection term and nesting the line term within selection group. The full model was
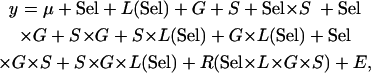 |
where Sel, G, and S are fixed cross-classified main effects of selection group, genotype, and sex; L(Sel) is the random effect of line nested within selection group; R is the random effect of replicate vial; and E is the error variance. Reduced ANOVAs were also run for each sex separately. Significant Sel × G and Sel × G × S terms were indicative of quantitative failure to complement.
We retested deficiencies with significant L × G, L × G × S, Sel × G, and/or Sel × G × S interaction terms, provided they met additional criteria. Failure to complement can be due to allelic or epistatic interactions between the deficiency or balancer chromosomes and the O and B QTL affecting longevity; and the pattern of failure to complement can be attributable to a change in variance or a change of rank order of complementation effects. The pattern of complementation effects expected from allelic failure to complement is that the variance among Df genotypes should be greater than the variance among Bal genotypes; and/or the contrast (Df/O − Df/B) > (Bal/O − Bal/B) is satisfied. These filters eliminate ambiguous results and failure to complement attributable to epistatic interactions between genes on the balancer chromosome and O and B longevity QTL [in which case the variance among Bal genotypes will be greater than the variance among Df genotypes (Geiger-Thornsberry and Mackay 2004; Mackay et al. 2005a), and the contrast will be in the opposite direction].
For deficiencies that were tested twice, the data were pooled over both tests and reanalyzed using the same models described above with the addition of a fixed-effect block term. We then performed reduced ANOVAs to determine which of the O lines contributed to the interaction terms, compared to the average of all the B lines.
All analyses were performed using SAS statistical software (SAS Institute 1998, version 8.2).
RESULTS
Initial screen:
We crossed individuals from five replicate B and five replicate O populations to a panel of 69 deficiency (Df/Bal) stocks uncovering ∼80% of the Drosophila third chromosome to map QTL affecting increased life span that fail to complement deficiencies. The full ANOVA models of the life span data for each of the deficiencies include the cross-classified main effects of line (L), sex (S), and genotype (G) and all interactions between these factors (supplemental Table 2 at http://www.genetics.org/supplemental/). We observed a significant (P < 0.05) line effect for 68 deficiencies—all but one. This is hardly surprising given the large differences in life span between the B and O lines. The genotype term was significant for 47 deficiencies (on average, Df/O and Df/B outlive Bal/O and Bal/B), and the main effect of sex was significant for 36 deficiencies (females live longer than males, in general). Significant L × S interaction terms indicate sex-specific differences in life span that depend on the line in which the deficiency is tested; 24 deficiencies showed significant L × S interactions. Similarly, significant G × S interaction terms denote sex-specific differences in longevity that depend on the genotype; 40 deficiencies showed significant G × S interactions (supplemental Table 2).
A total of 35 deficiencies (51%) exhibited quantitative failure to complement longevity QTL, on the basis of significant L × G and/or L × G × S interaction terms (supplemental Table 2 at http://www.genetics.org/supplemental/). Quantitative failure to complement can be the result of allelism or epistasis. Allelic failure to complement occurs when the deficiency uncovers O and B alleles with quantitatively different effects on life span. Epistatic failure to complement can occur in two ways: (1) when there are interactions between the deficiency and nonhomologous QTL affecting longevity in the B and O lines and (2) when longevity QTL in the B and O lines interact with other life span QTL on the deficiency or balancer chromosomes. Only allelic interactions are interpretable in the context of deficiency mapping (Pasyukova et al. 2000), although the former cause of epistatic failure to complement nonetheless identifies one of the interacting partners. To minimize the confounding effect of epistatic interactions, and also to exclude ambiguous patterns of failure to complement attributable to changes in rank order of complementation effects rather than an increase in variance of Df genotypes compared to Bal genotypes, we conservatively chose deficiencies for retesting on the basis of two additional criteria. We required that the variance in life span among Df genotypes was greater than the variance in life span among Bal genotypes and/or that the (Df/O − Df/B) contrast was greater than the (Bal/O − Bal/B) contrast. Application of these filters excluded 20 deficiencies.
Retest:
We tested the remaining 15 deficiencies again for effects on life span and analyzed the pooled data from both the initial screen and the retests. From the pooled analysis, 12 deficiencies showed significant L × G and/or L × G × S interaction terms (Table 1, Figure 1). For these deficiencies, we performed a separate analysis that included an additional term, selection group, which allowed us to infer response to selection from common alleles that were most likely segregating at intermediate frequencies in the original population and rare alleles that were present at a lower frequency. The logic is that common alleles affecting postponed senescence and increased longevity in the base population would be represented in multiple O lines and give rise to a Sel × G interaction, but that initially rare alleles or new mutations affecting postponed senescence would be sampled in only one of the O lines and would not contribute to a Sel × G interaction unless the effect was very large. Nine of the 12 deficiencies showed a significant Sel × G or Sel × G × S interaction, suggesting that many of the alleles affecting postponed senescence were at intermediate frequencies in the initial base population (Table 1). The differences between the life spans of deficiency and balancer genotypes are illustrated in Figures 2 and 3. Df(3L)lxd6, Df(3R)D605, Df(3R)p-XT103, and Df(3L)vin2 (males) are particularly clear examples of differences in complementation effects between the O and B lines.
TABLE 1.
Summary of P-values for significant deficiencies
| Both sexes
|
Females: | Males: | Both sexes
|
Females: | Males: | |||
|---|---|---|---|---|---|---|---|---|
| Deficiency | L × G | L × G × S | L × G | L × G | Sel × G | Sel × G × S | Sel × G | Sel × G |
| Df(3L)lxd6 | 0.0524 | 0.1075 | 0.5830 | 0.0020 | 0.0254 | 0.3845 | 0.2469 | 0.0660 |
| Df(3R)ea, kniri-1 pp | 0.0002 | 0.3392 | 0.0004 | 0.2028 | 0.3514 | 0.4541 | 0.3099 | 0.5726 |
| Df(3R)D605 | <0.0001 | 0.3478 | 0.1238 | 0.0002 | 0.0087 | 0.0313 | 0.1763 | 0.0014 |
| Df(3L)pbl-X1 | 0.3106 | 0.0021 | 0.2585 | 0.0021 | 0.9640 | 0.1054 | 0.1015 | 0.2416 |
| Df(3R)by62, red1 | <0.0001 | 0.4437 | 0.0212 | 0.0006 | 0.0615 | 0.2158 | 0.1883 | 0.0455 |
| Df(3R)p-XT103, ru1 st1 e1 ca1 | 0.0351 | 0.0062 | <0.0001 | 0.4886 | 0.0160 | 0.0314 | 0.0049 | 0.8945 |
| Df(3L)vin2, ru1 h1 gl2 e4 ca1 | 0.0006 | 0.2286 | 0.0484 | 0.0072 | 0.0131 | 0.1381 | 0.1993 | 0.0066 |
| Df(3L)ZN47, ry506 | <0.0001 | 0.0151 | <0.0001 | 0.0063 | 0.0014 | 0.3099 | 0.0071 | 0.0565 |
| Df(3R)crb87-4, st1 e1 | 0.0070 | 0.1026 | 0.0120 | 0.0670 | 0.0987 | 0.1156 | 0.7904 | 0.0083 |
| Df(3R)awd-KRB, e* ca1 | 0.3589 | 0.0278 | 0.7929 | 0.0011 | 0.0214 | 0.2044 | 0.8053 | 0.0517 |
| Df(3L)HR119 | 0.1412 | 0.0049 | 0.0348 | 0.0315 | 0.0536 | 0.1860 | 0.9732 | 0.0337 |
| Df(3L)Delta1AK, ru1 h1 ry506 sr1 es ca1 | 0.2867 | 0.0034 | 0.0269 | 0.0642 | 0.3980 | 0.5332 | 0.3157 | 0.9491 |
Figure 2.—






Differences in life span (in days) between deficiency and balancer genotypes for deficiencies that fail to complement QTL affecting life span on Chromosome 3L. The plum and blue bars represent females and males, respectively. Error bars represent ±SE. * indicates the O replicate lines that are significantly different from the average of all B replicate lines. (A) Df(3L)HR119. (B) Df(3L)ZN47, ry506. (C) Df(3L)pbl-X1. (D) Df(3L)lxd6. (E) Df(3L)vin2, ru1 h1 gl2 e4 ca1. (F) Df(3L)Delta1AK ru1 h1 ry506 sr1 es ca1.
Figure 3.—






Differences in life span (in days) between deficiency and balancer genotypes for deficiencies that fail to complement QTL affecting life span on Chromosome 3R. The plum and blue bars represent females and males, respectively. Error bars represent ±SE. * indicates the O replicate lines that are significantly different from the average of all B replicate lines. (A) Df(3R)p-XT103, ru1 st1 e1 ca1. (B) Df(3R)by62, red1. (C) Df(3R)ea, kniri-1 pp. (D) Df(3R)crb87-4, st1 e1.(E) Df(3R)D605. (F) Df(3R)awd-KRB, e* ca1.
We inferred sex-specific complementation effects when the L × G × S interaction term was significant and/or only one sex was significant in the analyses of sexes separately. According to these criteria, only 2 of the 12 deficiencies did not have sex-specific effects (Table 1). Further, one of the deficiencies, Df(3L)HR119 did not have a significant L × G term when pooled across sexes, but the L × G × S term and L × G terms from the separate sex analyses were all significant. This suggests sex-antagonistic effects, as was indeed observed (Figure 2).
Longevity QTL:
On the basis of these analyses and the breakpoints of the 12 deficiencies exhibiting failure to complement longevity QTL, we were able to infer that there are at least 11 QTL affecting increased life span on the third chromosome. Ten of these QTL were common to more than one O line. The 11 QTL together span ∼4874.8 kb and include 598 positional candidate genes sequenced to the region (Table 2). None of the QTL regions encompass positional candidate genes that have been previously associated with effects on life span (supplemental Table 3 at http://www.genetics.org/supplemental/). However, many of these genes affect, or are predicted to affect, processes and functions that are known to be associated with variation in longevity: general metabolism, lipid metabolism, defense response, DNA repair, catecholamine biosynthesis, oxidative stress, and protein folding.
TABLE 2.
Significant QTL
| QTL | kb | Genes sequenced to region | Females | Males |
|---|---|---|---|---|
| 63D2; 63E1 | 134.8 | 12 | O1 | O2, O3 |
| 64C13–15; 65C | 1449.4 | 170 | O1, O2, O3, O4 | O1, O2, O5 |
| 65F3; 66B8–9 | 855.5 | 113 | O5 | |
| 67F2–3; 68A2–3 | 116.6 | 13 | O1 | O1, O2, O3, O4, O5 |
| 79E5–F1; 79F2–6 | 226.6 | 23 | O1, O3 | |
| 85A2; 85C1–2 | 557.0 | 86 | O1, O2, O3, O4, O5 | |
| 85E7–F1; 85F6 | 255.3 | 34 | O1, O2 | O1, O2, O4 |
| 88E7–13; 89A1 | 419.5 | 62 | O1, O2, O3, O5 | |
| 95E8–F1; 95F7 | 93.3 | 16 | O2, O4 | |
| 98A1–2; 98A5 | 124.5 | 10 | O1, O2, O4, O5 | |
| 100C; 100D | 642.4 | 59 | O2, O3, O4 | |
| Totals | 4874.8 | 598 |
DISCUSSION
QTL are typically mapped by linkage to molecular markers, but the QTL intervals are usually of the order of ≥10 cM, often embracing hundreds of positional candidate genes (Mackay 2001). The challenge is to map QTL to the level of individual quantitative trait genes, which becomes more feasible if we can refine QTL to smaller regions. In Drosophila, deficiency complementation mapping offers several advantages over recombination mapping to identify QTL (Pasyukova et al. 2000). Deficiency mapping can be directly implemented for outbred selection lines, circumventing the need to construct inbred parental lines for recombination mapping. This is advantageous in terms of economy of effort and avoids the possible effects of inbreeding depression on traits related to fitness. Further, deficiency mapping is readily scalable to multiple lines, which facilitates inferences about frequency of QTL alleles that would otherwise require the construction and analysis of multiple mapping populations. Deficiency mapping is capable of much higher resolution than recombination mapping for an equivalent amount of effort and does not require assessing marker genotypes, and increasing the number of replicate measures per genotype increases the power to detect QTL with small effects. On the other hand, quantitative or qualitative failure to complement deficiencies can be attributable to allelic or epistatic interactions, as noted above. Epistasis between QTL on either the deficiency or balancer chromosome with QTL in the tested strains will generate false positive interactions. Furthermore, deficiency mapping cannot yield estimates of heterozygous and homozygous QTL effects and cannot detect dominant QTL (which would have the same effect in both Deficiency and Balancer genetic backgrounds).
Here, we have used deficiency complementation mapping to fine map QTL on the third chromosome affecting variation in life span between five O lines selected for postponed senescence and five B control lines (Rose 1984). We identified 35 deficiencies (∼51%) exhibiting quantitative failure to complement QTL affecting the difference in longevity between the O and B lines in an initial screen using >16,000 individuals. We conservatively excluded 20 deficiencies from further study as the pattern of complementation effects could be attributable to epistasis or was not unambiguous and retested the remaining 15 deficiencies to eliminate false positives from the initial screen. On the basis of this, we infer a minimum of 11 QTL affecting increased longevity in the lines selected for postponed senescence on the third chromosome alone. Most of these QTL have sex-specific effects. Adding the selection group term to our analysis provides a statistical method for determining if the deficiencies uncover common or rare alleles affecting longevity. Three deficiencies do not show a significant Sel × G or Sel × G × S term (Table 2). Df(3L)pbl-X1 shows a pattern of uncovering a rare allele as significant differences are in only one O line (O5; Figure 2C). Df(3L)Delta1AK shows significant differences in two O lines but the responses in O1 and O3 are not large enough to drive the entire selection group response. Although Df(3R)ea, kniri-1pp shows significant differences in four O lines, it does not show a significant Sel × G or Sel × G × S term. This is most likely a result of the extreme response of a single O line (O3; Figure 2). Nine of the remaining significant deficiencies do show a significant Sel × G or Sel × G × S interaction term and exhibit patterns of uncovering common alleles with three or more O lines responding for each one. Thus, these results are consistent with both the mutation-accumulation (Medawar 1952) and the antagonistic pleiotropy (Williams 1957; Rose and Charlesworth 1980; Kirkwood and Rose 1991; Tower 1996; Kirkwood and Austad 2000) evolutionary models for the maintenance of genetic variation for longevity in natural populations.
Recently, Forbes et al. (2004) constructed three mapping populations using inbred derivatives of three of the O (O3, O4, and O5) and three of the B (B2, B4, and B5) lines and used composite interval mapping (Zeng 1994) to map at least 10 QTL on all three major chromosomes affecting variation in longevity among these strains. Five of these QTL mapped to the third chromosome, with significant effects of essentially the whole chromosome from 61D to 94E. Thus, all but the 3 of our QTL at the tip of 3R overlap those detected by Forbes et al. (2004). The patterns of effects detected in the two experiments were similar, however, as Forbes et al. (2004) also noted sex-specific QTL effects on life span, and there was evidence that many of the same QTL were detected in multiple mapping populations. Sex-specific QTL effects on longevity are observed often (Nuzhdin et al. 1997; Pasyukova et al. 2000; Leips and Mackay 2000, 2002; DeLuca et al. 2003; Mackay et al. 2005b), but not always (see Curtsinger and Khazaeli 2002).
Our estimates of the numbers of QTL affecting increased longevity are minimum estimates. We used stringent criteria for retesting deficiencies that exhibited quantitative failure to complement the life span phenotype of O and B line QTL and cannot exclude the possibility that many of the remaining deficiencies might actually harbor relevant QTL. Further, high-resolution mapping with smaller deficiencies may reveal that each detected QTL fractionates into multiple closely linked QTL (Pasyukova et al. 2000; DeLuca et al. 2003; Mackay et al. 2005b). Nevertheless, these results are consistent with a large number of loci at which alleles increasing longevity segregate at intermediate frequencies in natural populations, accounting for rapid responses to artificial selection for postponed senescence and increased life span (Luckinbill et al. 1984; Rose 1984; Partridge and Fowler 1992; Zwaan et al. 1995; Sgro and Partridge 1999).
Correlated responses of other life history and physiological traits to selection for postponed senescence have been extensively examined in the O and B lines. Young females from O lines have decreased fecundity (Rose 1984) and ovary weight (Rose 1984) relative to those from the B lines, in keeping with the antagonistic pleiotropy theory. Also in keeping with the existence of trade-offs between early and late components of fitness, young males from the O lines were found to have decreased competitive mating success relative to young males from the B lines, but the situation was reversed for old males from these lines (Service 1993). Further, females mated to the B males produce more progeny in the 1- to 5-day period immediately following mating, but females mated to O males produce more progeny in the 6- to 21-day period after mating (Service and Vossbrink 1996). O lines are more resistant to desiccation, starvation, and ethanol vapor stress than the controls (Service et al. 1985). O lines also have a decreased early respiratory rate, increased lipid content, decreased early locomotor activity, increased later locomotor activity, and increased glycogen content and metabolic rate relative to the B lines (Service 1987; Graves et al. 1992). The O lines have evolved increased total development times, mostly attributable to an increase in the larval phase (Chippendale et al. 1994). These observations, in addition to pleiotropic effects of long-lived mutants, suggest what genes and pathways we may expect, a priori, to affect the difference in longevity between the B and O lines.
These results provide the basis for future identification of the genes responsible for increased longevity of O relative to B lines. In particular, the DrosDel and Exelixis deficiencies, which have been generated in coisogenic backgrounds without additional mutations, and have molecularly defined breakpoints (Parks et al. 2004), will be especially valuable for high-resolution mapping of the identified QTL. Further, our ability to utilize quantitative complementation tests of QTL affecting life span to mutations at positional candidate genes to identify new genes and pathways important in the regulation of life span has been enhanced by the Exelixis collection of coisogenic P and piggyBac insertions that has been recently released (Thibault et al. 2004), and the effort to mutate all Drosophila genes is progressing well (Bellen et al. 2004). The O and B lines were derived from the Ives population, which was maintained for >130 generations in the laboratory with 14-day discrete generations (Rose 1984). The Ives population was thus selected for high early reproduction, and the constellation of gene frequencies at loci affecting longevity in this population may not be representative of gene frequencies segregating in a natural population not adapted to life in the laboratory. Therefore, we ultimately need to use the candidate genes identified in these lines to return to natural populations and use linkage disequilibrium mapping determine the nature and frequency of molecular polymorphisms affecting increased longevity (DeLuca et al. 2003). Molecular population genetic analyses of DNA sequences will also reveal whether there is evidence for historical balancing selection, as expected from antagonistic pleiotropy, or other evidence for departure from neutrality. Given the conservation of key biological processes across diverse taxa, it is not impossible that the same genes may be responsible for variation in life span in human populations.
Acknowledgments
We thank Philip Service for the generous gift of the fly stocks used in this study and Stefanie Heinsohn for technical assistance. This work was supported by National Institutes of Health grant GM 45344 to T.F.C.M. This is a publication of the W. M. Keck Center for Behavioral Biology.
References
- Bellen, H. J., R. W. Levis, G. Liao, Y. He, J. W. Carlson et al., 2004. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 167: 761–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., 1980. Evolution in Age-Structured Populations. Cambridge University Press, London.
- Chippendale, A. K., D. T. Hoang, P. M. Service and M. R. Rose, 1994. The evolution of development in Drosophila melanogaster selected from postponed senescence. Evolution 48: 1880–1899. [DOI] [PubMed] [Google Scholar]
- Curtsinger, J. W., and A. A. Khazaeli, 2002. Lifespan, age-specificity, and pleiotropy in Drosophila. Mech. Ageing Dev. 123: 82–93. [DOI] [PubMed] [Google Scholar]
- De Luca, M., N. V. Roshina, G. L. Geiger-Thornsberry, R. F. Lyman, E. G. Pasyukova et al., 2003. Dopa-decarboxylase affects variation in Drosophila longevity. Nat. Genet. 34: 429–433. [DOI] [PubMed] [Google Scholar]
- Dillin, A., A. L. Hsu, N. Arantes-Oliveira, J. Lehrer-Graiwer, H. Hsin et al., 2002. Rates of behavior and aging specified by mitochondrial function during development. Science 298: 2398–2401. [DOI] [PubMed] [Google Scholar]
- Finch, C. E., and G. Ruvkun, 2001. The genetics of aging. Annu. Rev. Genomics Hum. Genet. 2: 435–462. [DOI] [PubMed] [Google Scholar]
- Finch, C. E., and R. E. Tanzi, 1997. Genetics of aging. Science 278: 407–411. [DOI] [PubMed] [Google Scholar]
- Forbes, S. N., R. K. Valenzuela, P. Keim and P. M. Service, 2004. Quantitative trait loci affecting life span in replicated populations of Drosophila melanogaster. I. Composite interval mapping. Genetics 168: 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger-Thornsberry, G. L., and T. F. C. Mackay, 2004. Quantitative trait loci affecting natural variation in Drosophila longevity. Mech. Ageing Dev. 125: 179–189. [DOI] [PubMed] [Google Scholar]
- Graves, J. L., E. C. Toolson, C. Jeong, L. N. Vu and M. R. Rose, 1992. Desiccation, flight, glycogen, and postponed senescence in Drosophila melanogaster. Physiol. Zool. 65: 268–286. [Google Scholar]
- Griswold, C. M., A. L. Matthews, K. E. Bewley and J. W. Mahaffey, 1993. Molecular characterization and rescue of acatalasemic mutants of Drosophila melanogaster. Genetics 134: 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente, L., and C. Kenyon, 2000. Genetic pathways that regulate ageing in model organisms. Nature 408: 255–262. [DOI] [PubMed] [Google Scholar]
- Houle, D., B. Morikawa and M. Lynch, 1996. Comparing mutational variabilities. Genetics 143: 1467–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii, N., M. Fujii, P. S. Hartman, M. Tsuda, K. Yasuda et al., 1998. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394: 694–697. [DOI] [PubMed] [Google Scholar]
- Kirkwood, T. B. L., and S. N. Austad, 2000. Why do we age? Nature 408: 233–238. [DOI] [PubMed] [Google Scholar]
- Kirkwood, T. B. L., and M. R. Rose, 1991. Evolution of senescence: late survival sacrificed for reproduction. Philos. Trans. R. Soc. Lond. B 332: 15–24. [DOI] [PubMed] [Google Scholar]
- Lee, C. K., R. G. Klopp, R. Weindruch and T. A. Prolla, 1999. Gene expression profile of aging and its retardation by caloric restriction. Science 285: 1390–1393. [DOI] [PubMed] [Google Scholar]
- Lee, S. S., R. Y. N. Lee, A. G. Fraser, R. S. Kamath, J. Ahringer et al., 2003. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33: 40–48. [DOI] [PubMed] [Google Scholar]
- Leips, J., and T. F. C. Mackay, 2000. Quantitative trait loci for life span in Drosophila melanogaster: interactions with genetic background and larval density. Genetics 155: 1773–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leips, J., and T. F. C. Mackay, 2002. The complex genetic architecture of Drosophila life span. Exp. Aging Res. 28: 361–390. [DOI] [PubMed] [Google Scholar]
- Long, A. D., S. L. Mullaney, T. F. C. Mackay and C. H. Langley, 1996. Genetic interactions between naturally occurring alleles at quantitative trait loci and mutant alleles at candidate loci affecting bristle number in Drosophila melanogaster. Genetics 144: 1497–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckinbill, L. S., and M. J. Clare, 1985. Selection for life span in Drosophila melanogaster. Heredity 55: 9–18. [DOI] [PubMed] [Google Scholar]
- Luckinbill, L. S., R. Arking, M. J. Clare, W. C. Cirocco and S. A. Buck, 1984. Selection of delayed senescence in Drosophila melanogaster. Evolution 38: 996–1003. [DOI] [PubMed] [Google Scholar]
- Mackay, T. F. C., 2001. The genetic architecture of quantitative traits. Annu. Rev. Genet. 35: 303–339. [DOI] [PubMed] [Google Scholar]
- Mackay, T. F. C., R. F. Lyman and F. Lawrence, 2005. a Polygenic mutation in Drosophila melanogaster: mapping spontaneous mutations affecting sensory bristle number. Genetics 170: 1723–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, T. F. C., N. V. Roshina, J. W. Leips and E. G. Pasyukova, 2005. b Complex genetic architecture of Drosophila longevity, pp. 181–216 in Handbook of the Biology of Aging, Ed. 6, edited by E. J. Masaro and S. N. Austad.
- Martin, L. G., and S. H. Preston, 1994. The Demography of Aging. National Academy Press, Washington, DC. [PubMed]
- Medawar, P. B., 1952. An Unsolved Problem of Biology. H. K. Lewis, London.
- Melov, S., J. Ravenscroft, S. Malik, M. S. Gill, D. W. Walker et al., 2000. Extension of life-span with superoxide dismutase/catalase mimetics. Science 289: 1567–1569. [DOI] [PubMed] [Google Scholar]
- Nuzhdin, S. V., E. G. Pasyukova, C. L. Dilda, Z.-B. Zeng and T. F. C. Mackay, 1997. Sex-specific quantitative trait loci affecting longevity in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 94: 9734–9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes, T. L., A. J. Elia, D. Dickinson, A. J. Hilliker, J. P. Phillips et al., 1998. Extension of Drosophila life span by over-expression of human SOD1 in motorneurons. Nat. Genet. 19: 171–174. [DOI] [PubMed] [Google Scholar]
- Parks, A. L., K. R. Cook, M. Belvin, N. A. Dompe, R. Fawcett et al., 2004. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 36: 288–292. [DOI] [PubMed] [Google Scholar]
- Partridge, L., and K. Fowler, 1992. Direct and correlated responses to selection on age of reproduction in Drosophila melanogaster. Evolution 46: 76–91. [DOI] [PubMed] [Google Scholar]
- Partridge, L., and D. Gems, 2002. Mechanisms of ageing: public or private? Nat. Rev. Genet. 3: 165–175. [DOI] [PubMed] [Google Scholar]
- Pasyukova, E. G., C. Vieira and T. F. C. Mackay, 2000. Deficiency mapping of quantitative trait loci affecting longevity in Drosophila melanogaster. Genetics 156: 1129–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher, S. D., S. J. Macdonald, R. Marguerie, U. Certa, S. C. Stearns et al., 2002. Genome-wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr. Biol. 12: 712–723. [DOI] [PubMed] [Google Scholar]
- Rose, M. R., 1984. Laboratory evolution of postponed senescence in Drosophila melanogaster. Evolution 38: 1004–1010. [DOI] [PubMed] [Google Scholar]
- Rose, M. R., and B. Charlesworth, 1980. A test of evolutionary theories of senescence. Nature 287: 141–142. [DOI] [PubMed] [Google Scholar]
- Rowe, J. W., 1997. The new gerontology. Science 278: 367.. [DOI] [PubMed] [Google Scholar]
- SAS Institute, 1998. SAS/STAT User's Guide, Release 6.03 Edition. SAS Institute, Cary, NC.
- Service, P. M., 1987. Physiological mechanisms of increased stress resistance in Drosophila melanogaster selected for postponed senescence. Physiol. Zool. 60: 321–326. [Google Scholar]
- Service, P. M., 1993. Laboratory evolution of longevity and reproductive fitness components in male fruit flies: mating ability. Evolution 47: 387–399. [DOI] [PubMed] [Google Scholar]
- Service, P. M., and R. E. Vossbrink, 1996. Genetic variation in “first” male effects on egg laying and remating by female Drosophila melanogaster. Behav. Genet. 26: 39–48. [DOI] [PubMed] [Google Scholar]
- Service, P. M., E. W. Hutchinson, M. D. MacKinley and M. R. Rose, 1985. Resistance to environmental stress in Drosophila melanogaster selected for postponed senescence. Physiol. Zool. 58: 380–389. [Google Scholar]
- Service, P. M., R. Ochoa, R. Valenzuela and C. A. Michieli, 1998. Cohort size and maximum likelihood estimation of mortality parameters. Exp. Gerontol. 33(4): 331–342. [DOI] [PubMed] [Google Scholar]
- Sgro, C. M., and L. Partridge, 1999. A delayed wave of death from reproduction in Drosophila. Science. 286(5449): 2521–2524. [DOI] [PubMed] [Google Scholar]
- Sun, J., D. Folk, T. J. Bradley and J. Tower, 2002. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics 161: 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar, M., A. Bartke and A. Antebi, 2003. The endocrine regulation of aging by insulin-like signals. Science 299: 1346–1351. [DOI] [PubMed] [Google Scholar]
- Thibault, S. T., M. A. Singer, W. Y. Miyazaki, B. Milash, N. A. Dompe et al., 2004. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat. Genet. 36: 283–287. [DOI] [PubMed] [Google Scholar]
- Tower, J., 1996. Aging mechanisms in fruit flies. BioEssays 18: 799–807. [DOI] [PubMed] [Google Scholar]
- Vieira, C., E. G. Pasyukova, Z.-B. Zeng, J. B. Hackett, R. F. Lyman et al., 2000. Genotype-environment interaction for quantitative trait loci affecting life span in Drosophila melanogaster. Genetics 154: 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Williams, G. C., 1957. Pleiotropy, natural selection, and the evolution of senescence. Evolution 11: 398–411. [Google Scholar]
- Wong, P. C., J. D. Rothstein and D. L. Price, 1998. The genetic and molecular mechanisms of motor neuron disease. Curr. Opin. Neurobiol. 8: 791–799. [DOI] [PubMed] [Google Scholar]
- Zeng, Z.-B., 1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, S., S. Meadows, L. Sharp, L. Y. Jan and Y. N. Jan, 2000. Genome-wide study of aging and oxidative stress response in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 97: 13726–13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaan, B. J., R. Bijlsma and R. F. Hoekstra, 1995. Direct selection on lifespan in Drosophila melanogaster. Evolution 49: 649–659. [DOI] [PubMed] [Google Scholar]