

Abstract
A modified version (mBIC) of the Bayesian Information Criterion (BIC) has been previously proposed for backcross designs to locate multiple interacting quantitative trait loci. In this article, we extend the method to intercross designs. We also propose two modifications of the mBIC. First we investigate a two-stage procedure in the spirit of empirical Bayes methods involving an adaptive (i.e., data-based) choice of the penalty. The purpose of the second modification is to increase the power of detecting epistasis effects at loci where main effects have already been detected. We investigate the proposed methods by computer simulations under a wide range of realistic genetic models, with nonequidistant marker spacings and missing data. In the case of large intermarker distances we use imputations according to Haley and Knott regression to reduce the distance between searched positions to not more than 10 cM. Haley and Knott regression is also used to handle missing data. The simulation study as well as real data analyses demonstrates good properties of the proposed method of QTL detection.
CONSIDER a situation where we have a fairly densely spaced molecular marker map and our goal is to locate multiple interacting quantitative trait loci (QTL) influencing the trait of interest. We assume that marker genotype and quantitative trait value data are obtained by carrying out an intercross experiment using two inbred lines.
Classical methods of QTL mapping based on one-dimensional genome searches like interval mapping (Lander and Botstein 1989), composite-interval mapping (CIM) (Zeng 1993, 1994), and multiple-QTL mapping (MQM) (Jansen 1993; Jansen and Stam 1994) aim at locating QTL with significant main effects. The common practice of estimating epistatic effects only at those positions for which main effects have been found causes an underestimation of the frequency and importance of epistasis (Wolf et al. 2000) and fails to identify interesting regions of the genome. Epistatic effects are a common phenomenon, however, which is supposed to play an important role in the genetic determination of complex traits (see, e.g., Doerge 2002; Carlborg and Haley 2004 and references therein) as well as in the evolution process (see, e.g., Wolf et al. 2000). Neglecting these effects may lead to oversimplified models describing the inheritance of complex traits and as noted by Carlborg and Haley (2004) may result in a relatively low economic gain if such models are used for marker-assisted selection. Also, “in cases in which the epistasis is ignored . . . the estimated effects of detected QTLs could be severely biased” (Carlborg and Haley 2004, p. 624). The growing awareness of the importance of epistasis led to the development of new methods of QTL mapping that allow us to detect epistatic QTL (see, e.g., Kao et al. 1999; Carlborg et al. 2000; Jannink and Jansen 2001; Boer et al. 2002; Carlborg and Andersson 2002; Yi and Xu 2002; Yi et al. 2003; Bogdan et al. 2004; Narita and Sasaki 2004; Zhang and Xu 2005; Yi et al. 2005).
Taking into account epistatic effects usually relies on using more complicated ANOVA or regression models, allowing to take into account the joint influence of many genes. In the context of multiple regression, the identification of QTL requires the identification of nonzero coefficients. In the context of classical statistics on the other hand, one usually performs a sequence of statistical tests to estimate the number and positions of QTL as suggested, e.g., in Doerge and Churchill (1996), Kao et al. (1999), and Carlborg and Andersson (2002). This approach, however, allows us to compare only nested models. It is also unclear how to choose significance levels for each consecutive test. Another possibility is to choose variables for multiple regression or ANOVA, using one of many model selection criteria, like the Bayesian information criterion (BIC) (Schwarz 1978) or the Akaike information criterion (Akaike 1974). In the context of QTL mapping, these criteria were used or discussed, e.g., by Jansen (1993), Jansen and Stam (1994), Broman (1997), Ball (2001), Nakamichi et al. (2001), Piepho and Gauch (2001), Broman and Speed (2002), Silanpää and Corander (2002), and Bogdan et al. (2004). In particular, Broman (1997) and Broman and Speed (2002) report that the BIC, which is considered to be one of the most conservative model selection criteria, has a strong tendency to overestimate QTL number. These findings strongly undermine the possibility of using other, more liberal, criteria in the context of QTL mapping. Bogdan et al. (2004) explain the phenomenon of overestimation and propose a new, modified version of the BIC (mBIC), with an extra penalty depending on the number of markers used in the study and prior knowledge on the QTL number. They investigate the performance of the mBIC in the simple setting of a backcross design with equally spaced markers.
In this article we extend the method and propose a modified version of the Bayesian information criterion for the intercross design. Due to the increased number of genotypes for the intercross design, the corresponding number of potential regressor variables describing additive and epistatic QTL effects is much larger than that for the backcross design. We thus adapt the approach of Bogdan et al. (2004) and construct a modified version mBIC of the BIC for the intercross design. Additionally, we propose two new modifications of the BIC. The first of them is in the spirit of empirical Bayes approaches and is based on a two-step procedure. In the first step, the proposed mBIC is used for an initial estimation of the number of QTL and interactions. In the second step, QTL are located using the mBIC with the penalty modified according to the estimates obtained in step one. The second modification relies on extending the search procedure and is aimed at increasing the power of detection of interaction effects. We propose to consider an additional search for interactions that are related to at least one of the additive effects found in the original scan based on the mBIC. Restricting our attention to a limited set of interactions reduces the multiplicity of the testing problem and allows us to use a smaller penalty for including interactions.
We perform an extensive simulation study verifying the performance of our method. To account for the more complicated model structure in the intercross design, the range of models considered in the simulations is substantially larger than that in Bogdan et al. (2004). We also include models with nonequidistant and missing marker data. In situations when the distance between markers is large, we use imputations according to Haley and Knott regression to keep the distance between searched positions ≤10 cM. We also investigate the use of Haley and Knott regression to handle missing data. Additionally, we apply our procedure to real data sets and compare the results to standard QTL mapping techniques. Our simulations as well as the analysis of real data suggest good properties of the proposed method and demonstrate that the proposed modifications of the mBIC may help to increase the power of QTL detection while keeping the proportion of false discoveries at a relatively low level.
METHODS
Statistical model:
To model the dependence between QTL genotypes and trait values, we use a multiple-regression model with regressors coded as described in Kao and Zeng (2002). This method of coding effects is known as Cockerham's model and involves an additive and a dominance effect for each QTL locus as well as effects modeling epistasis between two loci. With r QTL this leads to the linear model
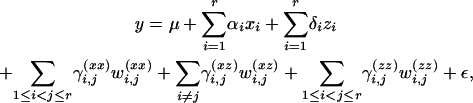 |
(1) |
where y is the trait value, and ɛ ∼ N(0, σ) summarizes environmental effects. The variables are coded as specified below:
Additive effects:
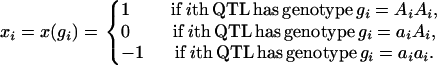 |
Dominance effects:
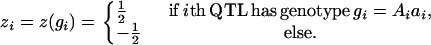 |
Epistatic effects:
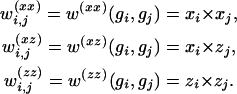 |
The advantage of the Cockerham parameterization is that under linkage equilibrium, the effects are orthogonal and the coefficients αi, δi, and γi,j have a natural ge netic interpretation (see Kao and Zeng 2002). The formulation of the model allows some of the coefficients to be zero to accommodate cases when there are either QTL that are not involved in epistatic effects or QTL that do not have their own main effects yet influence the quantitative trait by interacting with other genes, i.e., epistatic effects.
If the experiment is based on a relatively dense set of markers, the first step in QTL localization could rely on identifying markers that are closest to a QTL. Thus our task reduces to choosing the best model of the form
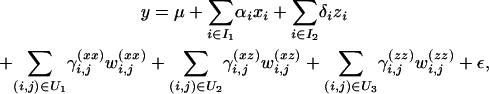 |
(2) |
where I1 and I2 are certain subsets of the set 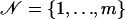 ; m is the number of available markers; and U1, U2, and U3 are certain subsets of
; m is the number of available markers; and U1, U2, and U3 are certain subsets of 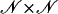 . Analogous to the formulas given above, the values of the regressor variables xi, zi,
. Analogous to the formulas given above, the values of the regressor variables xi, zi, 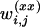 ,
, 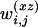 , and
, and 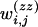 are defined according to the genotypes of the ith and jth markers. Similarly to Bogdan et al. (2004), we allow interaction terms to appear in our model even when the related main effects are not included.
are defined according to the genotypes of the ith and jth markers. Similarly to Bogdan et al. (2004), we allow interaction terms to appear in our model even when the related main effects are not included.
A modified version of the BIC:
Many variable selection techniques are available in the statistical literature. One of the most popular tools for choosing important regressor variables is the BIC. The BIC belongs to the class of penalized likelihood methods and recommends choosing the simplest model for which
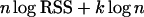 |
obtains a minimal value. Here RSS is the residual sum of squares from regression, k is the number of regressor variables, and n is the sample size (number of considered individuals).
While generally considered to be conservative, the BIC has a strong tendency to overestimate the QTL number when used in the context of QTL mapping (see, e.g., Broman 1997; Broman and Speed 2002). Bogdan et al. (2004) explain the phenomenon of overfitting and propose a new, modified version of the BIC (mBIC), with an extra penalty depending on the number of markers used in the study as well as on the prior knowledge on the QTL number. This proposed criterion recommends choosing the model that minimizes the quantity
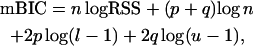 |
where p is the number of main effects in the model and q is the number of epistatic terms. The additional penalty coefficients l := mv/ENv and u := me/ENe depend on the number of possible regressors mv and me (corresponding to main and epistatic effects, respectively), as well as the expected number of main and epistatic QTL effects ENv and ENe, with the expectation chosen according to some prior.
Note that for the backcross design mv = m and me = m(m−1)/2. In the absence of prior information, Bogdan et al. (2004) propose to use ENv = ENe = 2.2. In the context of the backcross design, this choice guarantees that for sample sizes n ≥ 200 and a moderate number of markers (M > 30), the probability of the type I error of the resulting procedure (i.e., probability of detecting at least one QTL when there are none) is <7%.
In this article, we construct a version of mBIC suitable for intercross design. Note that in this design mv = 2m (m possible additive and m possible dominance terms), and me = 2m(m − 1). Choosing again ENv = ENe = 2.2, the resulting modified version of the BIC recommends the model for which
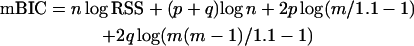 |
(3) |
obtains a minimum. Here p is equal to the sum of the number of additive and dominance effects present in the model and q is the number of epistatic terms.
Observe that the proposed penalty for including individual terms is larger in the intercross design than in the backcross design. This is a result of a larger number of possible terms in the regression model, which forces us to increase the threshold for adding an additional term to keep control of the overall type I error. An upper bound for the type I error of the search procedure is derived using the Bonferroni inequality (see appendix for details). Simulations show that the upper bound is close to the observed type I error for markers that are not closer than 5 cM.
Figure 1 compares the upper bound on the type I error of the mBIC when the penalty is adjusted for intercross designs (see Equation 3) with the related type I error when the penalty designed for backcross (2p log(m/2.2 − 1) + 2q log(m(m − 1)/2.2 − 1)) is used. The results are for m = 132 markers. The graph clearly shows that for common sample sizes adjusting the penalty is necessary to control the type I error at a 5% level.
Figure 1.—

Comparison of the Bonferroni type I error bounds under the null model (no effects) for the intercross design when the same penalty as in backcross is used and when the penalty is adjusted accordingly.
Apart from using mBIC in its standard form (Equation 3), we developed adaptive strategies to modify the size of the penalty on the basis of the data. In general, available prior information on the number of main and epistatic effects may be used to adjust the criterion in the following way,
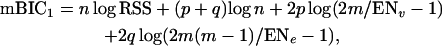 |
(4) |
where ENe and ENv denote the expected values of Ne and Nv under the prior distribution. If we have no knowledge on the number of QTL, an obvious option is to use the data to obtain an initial estimation of Ne and Nv. Such estimates for Ne and Nv could in principle be obtained using standard methods for QTL localization, e.g., interval mapping. However, due to the known problems related to interval mapping (many local maxima between markers, difficulties with separating linked QTL, and “ghost” effects) we recommend the application of the standard version of mBIC (3) for an initial search. We denote the number of additive and epistatic effects found in this initial search by 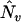 and
and 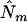 . In the second step, the final localization of QTL is based on version (4) of the criterion, with ENv replaced by
. In the second step, the final localization of QTL is based on version (4) of the criterion, with ENv replaced by 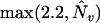 and ENe replaced by
and ENe replaced by 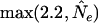 .
.
In the case of a large number of underlying QTL, the reduced penalty in the second search step increases the power of QTL detection. If in the first search step two or fewer main and epistatic effects are found, the penalty is not decreased. Thus in particular under the null model of no effects, the type I error will still be close to 5%.
We also consider a second extension to the search strategy to increase the power of detecting epistatic effects. The described application of the mBIC takes into account epistatic effects regardless of whether the corresponding main effects were included in the model or not. Therefore, epistasis can be detected in cases where main effects are weak or not present at all. Wolf et al. (2000) list the common practice of fitting epistatic terms after main effects have been included in the model as a main reason why in many QTL studies, epistasis has not been detected. However, the price for the possibility of detecting epistasis even if main effects are not detectable is a relatively large penalty for interaction terms. In particular for small sample sizes, this results in low detection rates (see Figure 2). This observation confirms the statement of Carlborg and Haley (2004, p. 620) that epistatic studies “are at their most powerful if they use good quality data for 500 or more F2 individuals.”
Figure 2.—

The solid curves show the percentage of correctly identified additive, dominance, and epistatic effects depending on the heritability. The shaded curves display the expected number of incorrectly selected (linked and unlinked) markers (n = 200).
For the above reasons, we deploy a third search step that increases the power of detection for epistatic terms by considering a restricted set of potential terms on the basis of prior analysis. Specifically, we restrict our attention to those epistatic effects related to at least one of the main effects detected by an initial search based on (4). Thus the set of epistatic effects to be searched through in this third step consists of not more than 4p(m − 1) elements, where p is the number of main effects detected in the two-step procedure. This allows us to decrease the penalty for interactions accordingly. The mBIC version used in this last step chooses the model that minimizes the quantity
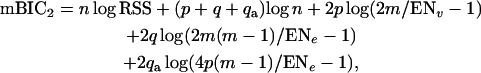 |
(5) |
where q is the number of epistatic effects found in the two-step procedure and qa is the number of extra epistatic terms considered in the additional search for epistasis.
The penalty for the extra interaction terms in (5) is now of the same order as the penalty for additive terms and thus the power for detecting such epistatic effects should be comparable to the power of detecting main effects with the same heritability.
The identification of the model minimizing (3), (4), or (5) within the huge class of potential models is by no means trivial. Our approach is to use a forward selection procedure with the following stopping rule: if a local minimum of the modified BIC is reached, we still proceed with forward selection, trying to include (one-by-one) five additional terms. If, at some point, this leads to a new minimum, we temporarily accept this “best” model and continue again with forward selection. Otherwise none of the additional five effects are added. This approach helps to avoid premature stopping of the search algorithm at a local minimum. This can be the case when including two additional regressors improves the model even if each single one of them does not. The maximum number of additional regressors is set to five because it is very unlikely that five additional regressors improve the model while each of them alone does not or only marginally improves it.
Finally, backward elimination is tried; i.e., it is checked whether mBIC can still be improved by deleting some of the previously added variables.
SIMULATIONS
Simulations are carried out to investigate the performance of our proposed method of QTL detection in the intercross design under a variety of parameter settings. We consider several scenarios involving equidistant markers that are relatively easy to analyze and three realistic scenarios designed according to an actual QTL experiment described in Huttunen et al. (2004).
In our equidistant scenarios, we simulate QTL and marker genotypes on 12 chromosomes, each of length 100 cM. Markers are equally spaced at a distance of 10 cM with the first marker at position 0 and the 11th marker at position 100 of each chromosome. This leads to a total number of available markers m of 132 and the standard version of mBIC (3) becomes
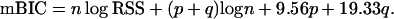 |
Genome length and marker density are kept constant in all simulations and are in accordance with previous simulation studies (Piepho and Gauch 2001; Bogdan et al. 2004) to increase comparability.
Further details for the equidistant (both simple and more complex) scenarios and the realistic scenarios are provided below. We simulated the trait data under different models of the form (1). In all simulations the overall mean μ and the standard deviation of the error term σ were set to be 0 and 1, respectively. For each scenario and parameter setting, the simulation results are based on 500 replications.
Among the simulation results we include are the average number of correctly identified effects, which we denote by cadd, cdom, and cepi for additive, dominance, and epistatic effects, respectively. In the case of simple models with just one effect, these quantities are estimates of the power. A main effect is classified to be correctly identified if the regression model chosen by mBIC includes the corresponding effect related to a marker within 15 cM of QTL. An epistatic effect is classified as correctly identified when the mBIC finds a corresponding effect with both markers falling within 15 cM of the corresponding QTL. If more than one effect is detected in such a window, only one of them is classified as true positive. All the other effects are considered to be false positives.
In our simulation study of more complex equidistant scenarios, we simulated many QTL with weak effects. In this situation, the confidence intervals for the estimates of QTL location are often much wider than 30 cM (see, e.g., Bogdan and Doerge 2005). Thus each of such weak effects will bring a certain proportion of “false” positives related to a weak precision of QTL localization, while still providing an approximation to the best regression model. As a result of this phenomenon, the total number of false positives typically increases with the size of the model used in the simulation. Therefore, additionally to the average number of false positives fp, we report the estimated proportion of false positives within the total number of identified effects, pfp = fp/(cadd + cdom + cepi + fp).
Simple equidistant scenarios:
We first consider the null model, i.e., the situation where there are no QTL at all. As shown in Figure 1, the probability that at least one effect is incorrectly selected should be <0.05 when the sample size is at least 200. Our simulations lead to a percentage of 0.038 of such (familywise) type I errors when n = 200, thus confirming the theoretical results. The percentage of errors should decrease with increasing sample size, and indeed for a sample size of n = 500, the number goes down to 0.02.
Next we consider two experiments to investigate the detectability of QTL effects depending on their strength, their effect type (additive, dominance, or epistatic), and the total number of QTL. In these experiments we use a sample size of n = 200.
For the first experiment, we generate the data according to three simple models of the form (1). In the first two models (scenarios 1 and 2), one QTL is located at the fifth marker on the first chromosome. In scenario 1 the QTL has only an additive effect with the effect size α ranging from 0.2 to 0.6. In scenario 2, the additive effect is constant (α = 0.7) and a dominance effect δ with values in the interval between 0.4 and 1.2 is added. For scenario 3 only one epistatic effect (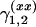 ) between marker numbers five of chromosome 5 and six, respectively, is considered. The effect size of (
) between marker numbers five of chromosome 5 and six, respectively, is considered. The effect size of (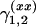 ) ranges between 0.4 and 1.6.
) ranges between 0.4 and 1.6.
In the context of scenarios 1 and 3, we investigate the power of detection in dependence on the classical heritability
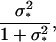 |
(6) |
with 1 being the environmental variance, and 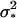 denoting the variance due to the single genetic effect present (i.e.,
denoting the variance due to the single genetic effect present (i.e., 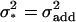 in the case of an additive effect, and
in the case of an additive effect, and 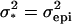 in the case of epistasis between two loci).
in the case of epistasis between two loci).
In scenario 2, the power of detection of the dominance effect should also depend on whether the corresponding additive effect can be detected, since the error variance gets smaller, if the additive effect is included into the regression model. In our experiment the additive effect was almost always detected (power 99%) and we observed that a good indicator for the power of detection of the dominance term is its heritability in the model without the additive term:
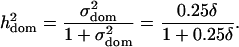 |
(7) |
A comparison of detection rates of additive, dominance, and epistatic effects in dependence on the heritability [as defined in (6) for additive and epistatic effects, respectively, and in (7) for the dominance effect] is given in Figure 2. The relationship can be seen to be S-shaped and nearly identical for additive and dominance effects. Although dominance and additive effects are detected with the same power at a fixed heritability, the actual size of the dominance effects has to be larger (by 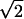 ) than the additive effects (
) than the additive effects (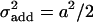 and
and 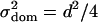 for an additive effect of size a and dominance effect of size d. Hence if
for an additive effect of size a and dominance effect of size d. Hence if 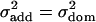 , d has to be
, d has to be 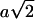 ). For epistatic effects, the power of detection is lower. This can be explained by the increased penalty of the model selection criterion.
). For epistatic effects, the power of detection is lower. This can be explained by the increased penalty of the model selection criterion.
The shaded curves in Figure 2 display the average number of falsely detected effects, which can be used as an estimate of the expected number of false positives. This quantity is an upper bound to the probability of having at least one incorrect effect in the model. The displayed error rates are fairly constant over the range of heritabilities considered. They vary between 0.05 (the value achieved by the model with no effects) and 0.11.
The purpose of the second experiment is to investigate to what extent the power of detection of individual signals is affected by the amount of QTL influencing the trait. The number of QTL varies between 1 and 10, and all QTL are on different chromosomes and therefore unlinked and have only additive effects with αi = 0.5.
Figure 3 shows that the probability of detection using the standard version of mBIC (3) decreases with the number of effects present. This can be explained by the fact that criterion (3) is based on the assumption that the expected number of effects is 2.2. If the correct (but in practice unknown) number of effects were used instead of 2.2, the percentage of correctly identified additive terms would increase from 0.543 to 0.761 for 10 underlying effects, from 0.672 to 0.781 for 7, and from 0.763 to 0.7995 for 4 underlying effects. We can obtain a comparable improvement by applying the two-step procedure defined in Equation 4 that involves an estimation of the number of expected effects in the first search step. The dotted line in Figure 3 shows that the power of detection increases while the proportion of false positives remains stable.
Figure 3.—

Percentage of correctly identified additive effects vs. number of additive effects. The QTL are unlinked, i.e., located on different chromosomes, and have effect sizes of 0.5. The solid line is based on simulations where no prior information is used to derive the penalty terms of the modified BIC. The dashed line represents simulations with the correct number of underlying effects (1, 2, 4, 7, and 10) assumed known. The dotted line corresponds to the two-step search procedure based on formula (4).
Complex equidistant scenarios:
Here, we consider nine more complex models that involve several effects of different size and type.
For all models, the overall broad sense heritability 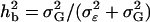 is kept at 0.7; i.e., 70% of the phenotypic variance is explained by genotypic variation
is kept at 0.7; i.e., 70% of the phenotypic variance is explained by genotypic variation 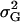 . Fixing the variance caused by environmental effects
. Fixing the variance caused by environmental effects 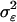 to 1 leads to a genotypic variation of
to 1 leads to a genotypic variation of 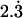 , which is then distributed among additive effects (45%), dominance effects (25%), and epistatic effects (30%). The resulting narrow sense heritability has an expected value of 0.315. All simulations are done both with sample sizes 200 and 500.
, which is then distributed among additive effects (45%), dominance effects (25%), and epistatic effects (30%). The resulting narrow sense heritability has an expected value of 0.315. All simulations are done both with sample sizes 200 and 500.
We consider all combinations of situations involving two, four, and eight additive and epistatic effects. Dominance effects are assigned to half of the loci where additive effects occur. The epistatic QTL are taken both from the additive effect positions and from other genome locations. If p additive effects are present, the relative size of effect i is chosen to yield 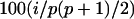 % of the additive heritability. For dominance and epistatic effects the relative strengths are chosen analogously. We consider the worst-case situation where the QTL positions are always exactly in the middle of two markers. Table 1 contains a brief summary of the resulting nine scenarios. A detailed description of all effect positions and strengths can be found on our web page, http://homepage.univie.ac.at/andreas.baierl/pub.html.
% of the additive heritability. For dominance and epistatic effects the relative strengths are chosen analogously. We consider the worst-case situation where the QTL positions are always exactly in the middle of two markers. Table 1 contains a brief summary of the resulting nine scenarios. A detailed description of all effect positions and strengths can be found on our web page, http://homepage.univie.ac.at/andreas.baierl/pub.html.
TABLE 1.
Description of scenarios 1–9
| Scenario | nadd | ndom | 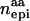 |
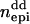 |
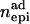 |
|---|---|---|---|---|---|
| 1 | 2 | 1 | 1 | 1 | 0 |
| 2 | 2 | 1 | 3 | 0 | 1 |
| 3 | 2 | 1 | 7 | 1 | 0 |
| 4 | 4 | 2 | 1 | 1 | 0 |
| 5 | 4 | 2 | 3 | 0 | 1 |
| 6 | 4 | 2 | 7 | 1 | 0 |
| 7 | 8 | 4 | 1 | 1 | 0 |
| 8 | 8 | 4 | 3 | 0 | 1 |
| 9 | 8 | 4 | 7 | 1 | 0 |
Columns contain numbers of additive (nadd), dominance (ndom), and epistatic QTL for each scenario. Epistatic effects can be of additive–additive (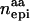 ), dominance–dominance (
), dominance–dominance (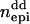 ) or additive–dominance (
) or additive–dominance (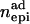 ) type.
) type.
Results for simulations with sample sizes of 200 and 500 are described in the following. Table 2 summarizes the average number of correctly identified effects as well as the average number of false positives and the proportion of false positives for the standard version of the mBIC (3). Table 3 gives the corresponding statistics for modifications based on the two-step procedure (see Equation 4) as well as for the additional search for epistatic terms with reduced penalty based on Equation 5. Table 3 demonstrates that the two-step procedure has the potential to increase the detection power while keeping the observed proportion of false positives at a level similar to the standard version of the mBIC. The increase in detection rates gained by this procedure is apparent for models with a larger number of underlying QTL (scenarios 7–9). The performance of the additional search for epistasis based on (5) depends on the actual model. In some cases the corresponding increase in the number of false positives is larger than the increase in the average number of correctly identified effects. We observed this situation to occur under scenarios 1, 4, and 7 (one relatively weak epistatic effect related to one of the main effects) and the sample size n = 200. Note, however, that under all these scenarios the gain in the detection rate was decisively larger than the increase in false positives when the sample size was n = 500. The additional search for epistatic effects is especially successful for scenario 9 with a large number of underlying main and epistatic effects.
TABLE 2.
Simulation results for sample sizes 200 and 500 (initial penalty)
| Scenario
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| cadda | 1.818 | 1.712 | 1.668 | 2.424 | 2.334 | 2.182 | 2.950 | 2.676 | 2.478 |
| caddb | 1.994 | 1.988 | 1.982 | 3.180 | 3.166 | 3.158 | 4.982 | 4.976 | 4.874 |
| cdoma | 0.966 | 0.968 | 0.978 | 1.296 | 1.204 | 1.064 | 1.348 | 1.164 | 0.972 |
| cdomb | 0.932 | 0.962 | 0.958 | 1.814 | 1.870 | 1.840 | 2.626 | 2.260 | 2.530 |
| cepia | 1.040 | 0.860 | 0.470 | 1.020 | 0.830 | 0.390 | 0.900 | 0.650 | 0.250 |
| cepib | 1.730 | 2.350 | 3.690 | 1.730 | 2.260 | 3.280 | 1.630 | 1.990 | 3.110 |
| pfpa | 0.040 | 0.053 | 0.063 | 0.050 | 0.044 | 0.059 | 0.061 | 0.065 | 0.080 |
| pfpb | 0.027 | 0.024 | 0.024 | 0.020 | 0.023 | 0.026 | 0.023 | 0.019 | 0.029 |
| fpa | 0.160 | 0.200 | 0.210 | 0.250 | 0.200 | 0.230 | 0.340 | 0.310 | 0.320 |
| fpb | 0.130 | 0.130 | 0.160 | 0.140 | 0.170 | 0.220 | 0.220 | 0.180 | 0.310 |
Columns contain the average numbers of correctly identified additive (cadd), dominance (cdom), and epistatic (cepi) effects. “pfp” denotes the proportion of false positives and “fp” the average number of falsely detected effects, respectively.
Results are for a sample size of 200.
Results for a sample of size 500.
TABLE 3.
Simulation results for sample sizes 200 and 500 (adjusted penalty)
| Scenario
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| cadda | 1.822 | 1.700 | 1.694 | 2.494 | 2.456 | 2.353 | 3.086 | 3.014 | 2.600 |
| caddb | 1.993 | 1.990 | 1.995 | 3.280 | 3.238 | 3.218 | 5.200 | 5.163 | 5.140 |
| cdoma | 0.978 | 0.987 | 0.966 | 1.302 | 1.240 | 1.180 | 1.504 | 1.280 | 1.032 |
| cdomb | 0.938 | 0.945 | 0.943 | 1.865 | 1.848 | 1.865 | 2.833 | 2.390 | 2.755 |
| cepia | 1.064 | 0.940 | 0.482 | 1.016 | 0.806 | 0.380 | 0.962 | 0.630 | 0.252 |
| cepib | 1.745 | 2.365 | 3.780 | 1.685 | 2.220 | 3.410 | 1.625 | 1.963 | 3.278 |
| fpa | 0.156 | 0.200 | 0.228 | 0.260 | 0.262 | 0.280 | 0.370 | 0.336 | 0.346 |
| fpb | 0.125 | 0.150 | 0.230 | 0.205 | 0.205 | 0.260 | 0.233 | 0.218 | 0.270 |
| pfpa | 0.039 | 0.052 | 0.068 | 0.051 | 0.055 | 0.067 | 0.062 | 0.064 | 0.082 |
| pfpb | 0.026 | 0.028 | 0.033 | 0.029 | 0.027 | 0.030 | 0.024 | 0.022 | 0.024 |
| Δca.epia | 0.018 | 0.130 | 0.158 | 0.046 | 0.112 | 0.100 | 0.058 | 0.128 | 0.100 |
| Δca.epib | 0.150 | 0.040 | 0.070 | 0.185 | 0.035 | 0.085 | 0.145 | 0.110 | 0.395 |
| Δfpaa | 0.07 | 0.11 | 0.104 | 0.082 | 0.09 | 0.08 | 0.066 | 0.086 | 0.084 |
| Δfpab | 0.065 | 0.04 | 0.065 | 0.035 | 0.025 | 0.055 | 0.05 | 0.027 | 0.048 |
| pfpaa | 0.055 | 0.074 | 0.088 | 0.065 | 0.069 | 0.080 | 0.071 | 0.075 | 0.095 |
| pfpab | 0.038 | 0.034 | 0.042 | 0.033 | 0.030 | 0.035 | 0.028 | 0.025 | 0.027 |
Simulation results for the two-step procedure based on formula (4) and the three-step procedure based on formula (5) are shown. Columns contain the average numbers of correctly identified additive (cadd), dominance (cdom), and epistatic (cepi) effects as well as the average number of falsely detected effects (fp) and the proportion of false positives (pfp) for the two-step procedure. Δca.epi and Δfpa display average numbers of correctly identified and false-positive epistatic effects that were detected additionally in the third search step on the basis of formula (5); “pfpa” on the other hand denotes the total proportion of false positives based on the finally selected model at the end of the third search step.
Results are for a sample size of 200.
Results for a sample of size 500.
Figures 4 and 5 are based on the final search results described in Table 3. They indicate that the ability to detect an effect of a given size depends mainly on the individual effect heritability 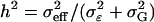 .
.
Figure 4.—

Percentage of correctly identified additive, dominance, and epistatic effects vs. individual effect heritabilities 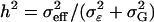 . Detection rates are taken from simulations of scenarios 1–9 (see Table 3) for n = 200.
. Detection rates are taken from simulations of scenarios 1–9 (see Table 3) for n = 200.
Figure 5.—

Percentage of correctly identified additive, dominance, and epistatic effects vs. individual effect heritabilities 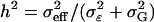 . Detection rates are taken from simulations of scenarios 1–9 (see Table 3) for n = 500.
. Detection rates are taken from simulations of scenarios 1–9 (see Table 3) for n = 500.
For the sample size n = 200, the majority of large additive effects (h2 > 0.07) is detected with a high power (>0.8). While only some fraction of moderate effects (h2 ∈ (0.04, 0.07)) is detected for n = 200, moderate additive and dominance effects are almost always detected when n = 500. Epistasis effects are somewhat harder to detect; the type of epistasis, however, does not influence the detectability. The observed proportion of false positives never exceeds 9% for n = 200 and 4% for n = 500.
Realistic scenarios:
As an alternative model, we take the marker setup from a Drosophila experiment by Huttunen et al. (2004) and also include missing data. To obtain a more densely spaced set of genome locations, genotype values were imputed at 35 positions chosen equidistantly between adjacent markers, keeping the maximum distance between the considered genome locations at not more than 10 cM. Haley–Knott regression (Haley and Knott 1992) was used to impute values. See Figure 6 for the marker locations.
Figure 6.—

Genetic map for the D. virilis experiment by Huttunen et al. (2004). Solid horizontal lines indicate observed marker positions, and dotted lines show imputed positions. QTL localized by our proposed method are symbolized by diamonds. Intervals with significant additive and/or dominance effects found by Huttunen et al. (2004) applying composite interval mapping are indicated by solid vertical lines.
Our three scenarios permit for different expected proportions (0, 5, and 10%, resp.) of marker locations per chromosome where the genotype information is missing. To permit for comparison, both heritabilities and QTL characteristics are chosen as in the above-mentioned complex equidistant scenario 4 involving four additive, two dominance, and two epistatic effects, and furthermore the QTL effects have again been positioned in a distance of 5 cM to the closest marker. For this experiment we use the sample size n = 200.
According to Table 4, the obtained results are similar to those obtained for the complex equidistant scenario 4 that has the same number and relative strength of effects. This suggests that our approach does not rely on the somewhat unrealistic assumption of equidistant markers and no missing data.
TABLE 4.
Simulation results for different percentages of missing data
| % missing | cadd | cdom | cepi | pfp |
|---|---|---|---|---|
| 0 | 2.486 | 1.302 | 1.290 | 0.043 |
| 5 | 2.362 | 1.264 | 1.140 | 0.054 |
| 10 | 2.262 | 1.160 | 0.980 | 0.070 |
Results of simulations for the realistic scenario with 0, 5, and 10 of missing marker data. Columns contain the average number of correctly identified additive (cadd), dominance (cdom), and epistatic effects (cepi) and the proportion of false positives (pfp) derived with the two-step search strategy on the basis of formula (4).
Not surprisingly, the average number of correctly identified markers decreases slightly when the proportion of missing data increases. The proportion of false positives on the other hand somewhat increases. This results from a loss of power as well as a loss of precision in localizing QTL.
ILLUSTRATIONS
We apply our proposed method to data sets from QTL experiments on Drosophila virilis and mice, respectively. Huttunen et al. (2004) analyzed the variation in male courtship song characters in D. virilis. We considered their intercross data set obtained from 520 males and the quantitative trait PN (number of pulses in a pulse train). Figure 6 shows the positions of the markers used in this experiment (solid lines). Depending on the chromosome, between 2 and 5% of the marker data were missing. We used the same imputation strategy as for our considered realistic scenarios, both for the missing data and for the additional genome positions.
Huttunen et al. (2004) used single-marker analysis as well as composite-interval mapping. They found one QTL on chromosome 2, five QTL on chromosome 3, and another QTL on chromosome 4. As they note, four of the five positions found on chromosome 3 are close together and may well correspond to only one single underlying QTL.
With our approach and the penalization based on 59 search positions, we found the same QTL positions on chromosomes 2 (at 53.7 cM) and 4 (at 100.2 cM), but only two positions (at 25.4 and 108.25 cM) on chromosome 3. All QTL found were classified as additive. The QTL locations pointed out by our method as well as intervals suggested by Huttunen et al. (2004) are presented in Figure 6. In the results of the additional regression analysis we observed that none of the putative QTL suggested by Huttunen et al. (2004) on chromosome 3 that were not found by our method significantly improves our model (corresponding P-values for adding these QTL were 0.85, 0.34, 0.06, and 0.32). Given these results and the above remark by Huttunen et al. (2004), our method might have led to a more precise localization of the respective QTL on chromosome 3.
Shimomura et al. (2001) investigated the circadian rhythm amplitude in mice on 192 F2 individuals. Genotypes were observed on 121 markers spread across the 19 autosomal chromosomes with 0–5% of the data missing for most markers. Again, the same imputation strategy as described in the section on realistic scenarios was used.
The analysis presented in Shimomura et al. (2001) consists of single and pairwise marker genome scans with permutation tests for assessing statistical significance. They identified one main effect on chromosome 4 at 42.5 cM and one epistatic term between the previous position and a marker on chromosome 1 at 81.6 cM.
Both QTL were detected by our method, one additive main effect on chromosome 4 and one dominant × additive epistatic effect between the QTL on chromosomes 4 and 1. The epistatic term was found in an additional search step based on the mBIC described in Equation 5.
DISCUSSION
In this article we use a modification of the Bayesian information criterion (mBIC) to locate multiple interacting quantitative trait loci in intercross designs. The proposed procedure allows us to detect multiple interacting QTL while controlling the probability of the type I error at a level close to 0.05 for sample sizes n ≥ 200. The main advantages of this procedure include that it is straightforward to apply and computationally efficient, which makes an extensive search for epistatic QTL practically feasible.
We presented results from simulations with single effects (additive, dominance, and epistatic) of different magnitude and for complex scenarios to investigate detection thresholds. We applied our proposed procedure to realistic parameter settings including nonequidistant marker positions and different proportions of missing values. To demonstrate the applicability of our proposed method when applied to real data, we analyzed two sets of QTL experiments from the genetic literature, namely one dealing with D. virilis and another one with mice. Both our simulation results and our real data analysis confirm good properties of the proposed modifications to the BIC.
Compared to the original BIC (see Schwarz 1978), the mBIC contains an extra penalty term that accounts for the large number of markers included in typical genome scans and the resulting multiple-testing problem. While a modification of the BIC is already required when only main effects are considered (see, e.g., Broman and Speed 2002 and theoretical calculations in Bogdan et al. 2004), the multiple-testing problem becomes even more important when epistatic effects can enter the model regardless of the related main effects, as in our approach.
The simulations reported in this article show that the mBIC appropriately separates additive, dominant, and epistatic effects. In the case of closely linked markers, however, our approach sometimes leads to a misclassification of the effect type, while still correctly identifying the presence of an effect. Hence, we suggest using the mBIC rather to locate QTL than to identify the specific effect type. The procedure should also not be extended to estimate the magnitude of QTL effects or heritabilities. Estimating parameters after model selection leads to upward-biased estimators of the effect sizes. This is true for any method leading to the choice of a single set of regressors, i.e., also in the case of the widespread methods based on multiple tests or interval mapping (see, e.g., Bogdan and Doerge 2005).
The prior for the number of main and epistatic effects in the standard version of the mBIC (with expected values ENv = 2.2 and ENe = 2.2) allows us to control the probability of the type I error. This is suggested for an initial search in the case of no prior knowledge on the number of effects. When reliable information on the number of effects is available, we strongly recommend using it for defining ENv and ENe. Our simulations also show that modifying the prior choices of ENv and ENe in Equation 4 using estimates of the QTL number from an initial search based on the standard version of the mBIC allows for some increase of power of QTL detection while preserving the observed proportion of false positives at a level similar to the standard version of the mBIC. The same holds for the additional search for epistatic terms with modified penalties according to Equation 5.
In this article we apply the mBIC to locate multiple interacting QTL by choosing the best of competing regression models. Our simulations as well as results reported in Broman (1997), Broman and Speed (2002), and Bogdan et al. (2004) show that the proposed forward selection strategy performs very well in this context. However, the mBIC has also great potential to be used in a stricter Bayesian context. The majority of currently used Bayesian Markov chain Monte Carlo methods for QTL mapping require multiple generation of all regression parameters and multiple visits of a given model to estimate its posterior probability by the frequency of such visits (see, e.g., Yi et al. 2005 and references therein). As a result, the proposed methods are computationally intensive and are very rarely verified by thorough simulation studies, which could provide insight into the influence of the prior distributions. The influence of the choice of priors on the outcome of Bayesian model selection methods is discussed, e.g., by Clyde (1999). Note that the mBIC provides a method to estimate the posterior probability of a given model by visiting this model just once. This is because exp(−mBIC/2) is an asymptotic approximation for P(Y | M) × P(M), where P(Y | M) stands for the likelihood of the data given model M (see Schwarz 1978) and P(M) is the prior probability of a given model. Thus the posterior probability of a given model Mi could be estimated by
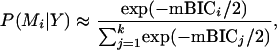 |
given that the k visited models contain all plausible models. To reach all sufficiently plausible models, a suitable search strategy needs to be designed. The construction of such an efficient search strategy is difficult due to the huge number of possible models (for 200 markers we potentially have 2638,800 models). However, we believe that a numerically feasible procedure permitting us to use mBIC in a Bayesian context might be found by exploiting the specific structure of QTL mapping problems, restricting the search space, and applying a proper adaptation of an efficient Markov chain Monte Carlo sampler (see, e.g., Broman and Speed 2002) or a heuristic search strategy like genetic algorithms (see, e.g., Goldberg 1989), simulated annealing (Kirkpatrick et al. 1983), tabu search (Glover 1989a,b), or ant colony optimization (see, e.g., Dorigo and Di Caro 1999). This would allow us to estimate posterior probabilities of different plausible models as well as to use model averaging to estimate parameters like effect sizes and heritabilities.
Acknowledgments
We thank Susanna Huttunen, Jouni Aspi, Anneli Hoikkala, Christian Schlötterer, and Joseph S. Takahashi for providing us with data sets from their QTL studies as well as Jayanta K. Ghosh and R. W. Doerge for helpful discussions. We thank furthermore the associate editor and two anonymous reviewers for comments and suggestions that led to a substantial improvement of the manuscript.
APPENDIX
The difference between the mBIC of the null model (mBIC0) and the mBIC of any one-dimensional model Mi (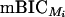 ) is log n + 2 log(l − 1) or log(u − 1), depending on whether the effect included in the one-dimensional model is a main or an epistatic effect. The number of possible one-dimensional models Mi for intercross designs is 2m + 4m(m − 1)/2.
) is log n + 2 log(l − 1) or log(u − 1), depending on whether the effect included in the one-dimensional model is a main or an epistatic effect. The number of possible one-dimensional models Mi for intercross designs is 2m + 4m(m − 1)/2.
To derive a bound for the type I error under the null model, we note that two times the difference of the likelihoods of a one-dimensional model and the null model is approximately χ2-distributed with 1 d.f.
Applying the Bonferroni inequality gives
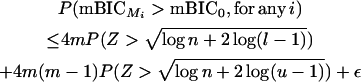 |
(A1) |
for the probability of choosing any one-dimensional model, if the null model is true.
The curves shown in Figure 1 are derived by evaluating the right-hand side of equation A1 for values of n between 100 and 500 and m = 132. For the backcross penalty, the parameters l and u are set to m/2.2 and m(m − 1)/4.4, respectively, whereas for the intercross penalty u = m/1.1 and l = m(m − 1)/1.1.
References
- Akaike, H., 1974. A new look at the statistical model identification. IEEE Trans. Automat. Control AC-19: 716–723. [Google Scholar]
- Ball, R., 2001. Bayesian methods for quantitative trait loci mapping based on model selection: approximate analysis using the Bayesian information criterion. Genetics 159: 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer, M. P., C. J. F. ter Braak and R. C. Jansen, 2002. A penalized likelihood method for mapping epistatic quantitative trait loci with one-dimensional genome searches. Genetics 162: 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan, M., and R. W. Doerge, 2005. Biased estimators of quantitative trait locus heritability and location in interval mapping. Heredity 95: 476–484. [DOI] [PubMed] [Google Scholar]
- Bogdan, M., J. K. Ghosh and R. W. Doerge, 2004. Modifying the Schwarz Bayesian information criterion to locate multiple interacting quantitative trait loci. Genetics 167: 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., 1997. Identifying quantitative trait loci in experimental crosses. Ph.D. Dissertation, University of California, Berkeley, CA.
- Broman, K. W., and T. P. Speed, 2002. A model selection approach for the identification of quantitative trait loci in experimental crosses. J. R. Stat. Soc. B 64: 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlborg, Ö., and L. Andersson, 2002. The use of randomisation testing for detection of multiple epistatic QTL. Genet. Res. 79: 175–184. [DOI] [PubMed] [Google Scholar]
- Carlborg, Ö., and C. S. Haley, 2004. Epistasis: Too often neglected in complex trait studies? Nat. Rev. Genet. 5: 618–625. [DOI] [PubMed] [Google Scholar]
- Carlborg, Ö., L. Andersson and B. Kinghorn, 2000. The use of a genetic algorithm for simultaneous mapping of multiple interacting quantitative trait loci. Genetics 155: 2003–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyde, M., 1999. Comment for “Bayesian model averaging: a tutorial” by Hoeting et al. Stat. Sci. 14: 401–404. [Google Scholar]
- Doerge, R. W., 2002. Mapping and analysis of quantitative trait loci in experimental populations. Nat. Rev. Genet. 3: 43–52. [DOI] [PubMed] [Google Scholar]
- Doerge, R. W., and G. A. Churchill, 1996. Permutation tests for multiple loci affecting a quantitative character. Genetics 142: 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorigo, M., and G. Di Caro, 1999. The ant colony optimization meta-heuristic, pp. 11–32 in New Ideas in Optimization, edited by D. Corne, M. Dorigo and F. Glover. McGraw–Hill, London.
- Glover, F., 1989. a Tabu search–part 1. ORSA J. Comput. 1: 190–206. [Google Scholar]
- Glover, F., 1989. b Tabu search–part 2. ORSA J. Comput. 2: 4–32. [Google Scholar]
- Goldberg, D. E., 1989. Genetic Algorithms in Search, Optimization and Machine Learning. Addison-Wesley, Reading, MA.
- Haley, C. S., and S. A. Knott, 1992. A simple method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69: 315–324. [DOI] [PubMed] [Google Scholar]
- Huttunen, S., J. Aspi, A. Hoikkala and C. Schlötterer, 2004. QTL analysis of variation in male courtship song characters in Drosophila virilis. Heredity 92: 263–269. [DOI] [PubMed] [Google Scholar]
- Jannink, J. L., and R. C. Jansen, 2001. Mapping epistatic quantitative trait loci with one-dimensional genome searches. Genetics 157: 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, R. C., 1993. Interval mapping of multiple quantitative trait loci. Genetics 135: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, R. C., and P. Stam, 1994. High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136: 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C.-H., and Z-B. Zeng, 2002. Modeling epistasis of quantitative trait loci using Cockerham's model. Genetics 160: 1243–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C-H., Z-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152: 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, S., C. D. Gelatt and M. P. Vecchi, 1983. Optimization by simulated annealing. Science 220: 671–680. [DOI] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamichi, R., Y. Ukai and H. Kishino, 2001. Detection of closely linked multiple quantitative trait loci using genetic algorithm. Genetics 158: 463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita, A., and Y. Sasaki, 2004. Detection of multiple QTL with epistatic effects under a mixed inheritance model in an outbred population. Genet. Sel. Evol. 36: 415–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepho, H.-P., and H. G. Gauch, Jr., 2001. Marker pair selection for mapping quantitative trait loci. Genetics 157: 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, G., 1978. Estimating the dimension of a model. Ann. Stat. 6: 461–464. [Google Scholar]
- Shimomura, K., S. S. Low-Zeddies, D. P. King, T. D. L. Steeves, A. Whitely et al., 2001. Genome-wide epistatic interaction analysis reveals complex genetic determinants of circadian behavior in mice. Genome Res. 11: 959–980. [DOI] [PubMed] [Google Scholar]
- Silanpää, M. J., and J. Corander, 2002. Model choice in gene mapping: what and why. Trends Genet. 18: 301–307. [DOI] [PubMed] [Google Scholar]
- Wolf, J. B., E. D. Brodie, III and M. J. Wade (Editors), 2000. Epistasis and the Evolutionary Process. Oxford University Press, New York.
- Yi, N., and S. Xu, 2002. Mapping quantitative trait loci with epistatic effects. Genet. Res. 79: 185–198. [DOI] [PubMed] [Google Scholar]
- Yi, N., S. Xu and D. B. Allison, 2003. Bayesian model choice and search strategies for mapping interacting quantitative trait loci. Genetics 165: 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., B. S. Yandell, G. A. Churchill, D. B. Allison, E. J. Eisen et al., 2005. Bayesian model selection for genome-wide epistatic QTL analysis. Genetics 170: 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z-B., 1993. Theoretical basis of separation of multiple linked gene effects on mapping quantitative trait loci. Proc. Natl. Acad. Sci. USA 90: 10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z-B., 1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.-M., and S. Xu, 2005. A penalized maximum likelihood method for estimating epistatic effects of QTL. Heredity 95: 96–104. [DOI] [PubMed] [Google Scholar]