


Abstract
Oligonucleotide microarrays are based on the hybridization of labeled mRNA molecules to short length oligonucleotide probes on a glass surface. Two effects have been shown to affect the raw data: the sequence dependence of the probe hybridization properties and the chemical saturation resulting from surface adsorption processes. We address both issues simultaneously using a physically motivated hybridization model. Based on publicly available calibration data sets, we show that Langmuir adsorption accurately describes GeneChip hybridization, with model parameters that we predict from the sequence composition of the probes. Because these parameters have physical units, we are able to estimate absolute mRNA concentrations in picomolar. Additionally, by accounting for chemical saturation, we substantially reduce the compressive bias of differential expression estimates that normally occurs toward high concentrations.
INTRODUCTION
Hybridization of complementary oligonucleotide sequences lies at the heart of microarray technology. The detailed understanding of this process is crucial for perfecting both the design of arrays and analyses of experiments. Yet, few studies have addressed the sequence specificity in the binding of oligonucleotides to DNA probes near a glass surface. Several practically relevant consequences of sequence specificity have been reported in the case of high-density oligonucleotide arrays, also known as GeneChips (1). For instance, non-linearities in the probe responses and differences in the onset of saturation between exactly complementary probes and probes with a single mismatch were discussed in (2,3). Additionally, the sequence-specificity in the behavior of mismatched probes was mentioned in Naef et al. (4). In a recent article (5), the difference in hybridization kinetics between specific and non-specific targets is described in the context of spotted oligonucleotide arrays, and it is shown how such differences can be exploited to reduce contaminating non-specific contributions.
Here, we show how most of these issues can be understood within a simple model of surface adsorption, and how the sequence composition of the probes can be used to calibrate GeneChips. We proceed in several steps: we first show how GeneChip data beautifully follows Langmuir isotherms (Fig. 1). Next, we fit the three model parameters to the sequence composition of each probe. Finally, we explain how to construct estimators of absolute concentration and expression ratio and test their predictions.
Figure 1.

Langmuir isotherms provide a very accurate description of GeneChip hybridization. After each probe has been fitted to the form I = ax / (b + x) + d, the rescaled variables X = x / b and Y = (I – d) / a collapse onto the form Y = X / (1 + X). Notice the range on the x-axis covers six orders of magnitude. The significant density of points near the shoulders indicates that saturation is not a marginal effect. Specifically, 69% of all PM probes have b < 512 pM. For these, at least 2 out of the 14 measurements lie above X = 1. The total fraction of measurements above X = 1 (respectively X = 0.5) is 20% (respectively 28%). The MM case is only slightly noisier. All probes with a, b, Y > 0 were plotted representing 94% of all probes for the PM (5472 out of 5824 measurements with positive target RNA concentration), and 87% in the MM case.
Our procedure offers several advantages among which the estimation of absolute concentration, and a strong reduction in bias of differential expression measures that occurs when a linear relationship between measured fluorescence and target RNA concentration is assumed. We emphasize that extant methods, either similar to MAS 5.0 or model-based (6), are designed around the notion that predicted concentrations can be compared for the same transcript measured in different experiments, but not for different transcripts. The reason is that sequence specificity is not taken into account at all (MAS 5.0) or only partially (6). In contrast, the approach described below yields estimates that permit the comparison of, say, α-tubulin versus β-tubulin within the same experiment.
MATERIALS AND METHODS
The GeneChip technology is based on a photolithographic oligonucleotide deposition process: individual probes consist of 25 base DNA sequences. As such short length hybridization should not be expected to be specific enough, labeled mRNA transcripts are probed by 22–40 of those probes (depending on chip models), introducing redundancy. Additionally, the probes come in two varieties: half are perfect matches (PM) identical to templates found in databases, and the other half single mismatches (MM), carrying a single base substitution at the middle (13th) base position. MM probes were introduced as non-specific hybridization controls, with the idea that the true signal (proportional to the target’s mRNA concentration) would be proportional to the difference of match versus mismatch (PM – MM) signal.
Data sets
The Human HG-U95A Latin Square (LS) experiment is a calibration data set produced by Affymetrix (available at http://www.netaffx.com), in which 14 genes are spiked onto 14 different arrays at concentrations corresponding to all cyclic permutations of the series (0, 0.25, 0.5, 1, 2, …, 1024) pM. Each gene is therefore probed at 14 different concentrations one of which is zero. The remaining are logarithmically spaced by a factor 2, ranging from 0.25 to 1024 pM. In addition to the spiked-in target cRNAs, a complex RNA background extracted from human pancreas was added to the sample. Each experiment was hybridized twice, leading to two groups of 14 arrays named Groups 1521 and 1532 (an additional Group 2353 was not used because it is incomplete). The probe sequences of all transcript are also available at the above website.
Normalization
In this article we compare the default MAS 5.0 algorithm with the method described below. In particular, we are interested in how chemical saturation affects the sensitivity of differential expression scores. For fair comparison, we used a single normalization method throughout the paper: all arrays were normalized to the first array in Group 1521 using the default (global) normalization provided by MAS 5.0.
Background subtraction
We like to distinguish between two background sources: the physical background, e.g. reflection from the glass surface or photo-multiplier dark current, and the biological background resulting from the hybridization of non-specific RNA molecules. The physical background ε was estimated as explained in Naef et al. (3) and subtracted from all raw PM and MM intensities. We will exclusively discuss the quantity I = IF – ε, where IF is the raw fluorescence intensity. We found that estimating ε separately, instead of including it into parameter d in equation 1, slightly increases sensitivity.
RESULTS
Langmuir adsorption model
The most elementary model of surface adsorption is the Langmuir adsorption isotherm (7). Let x be the specific target RNA concentration. Then, the fraction of occupied probe sites θ is given by
![]()
where x0 is the concentration at which half of the surface sites is occupied. This model assumes that the molecules in solution are in large excess compared to the number of adsorption sites. Assuming the measured fluorescence intensity to be linearly dependent on the amount of complementary RNA bound to a probe leads to the following model for the intensity I:
![]()
where a, b and d are probe specific parameters. Both a and d have units of intensity; b can be interpreted as the concentration at which the complementary RNA saturates half of the probes if there were no non-specific hybridization. The background term d contains contributions from non-specific hybridization. Other sources of fluorescence have been subtracted already (see Materials and Methods). At high intensity, the model predicts the saturation of I at a value of a + d. We emphasize that this effect describes chemical saturation, which is different from optical saturation that would result from a high photo-multiplier gain. Recently, the relevance of this model to microarrays was also suggested in Dai et al. (5) and Kepler et al. (8).
We proceed to show that competitive cross-hybridization by non-specific RNAs in the target solution does not change the functional dependence on concentration of equation 1, but only affects the parameter values. To see this, examine an extension of the Langmuir model for two competing species. Let z be the concentration of a competing non-specific RNA, with z0 being its half-saturation concentration; aS and aNS denote the dependence of fluorescence signal on the fractions of specific and non-specific hybridizing molecules. Then, the fluorescence reads

Inclusion of multiple non-specific compounds is straightforward and does not affect the conclusion that the functional dependence on the specific concentration x is preserved. The effective parameters (a, b, d) in equation 1 can easily be read off equation 2. The magnitude of the non-specific background can be estimated from the ratios d / a = z / z0. It turns out that non-specific background is small (z / z0 < 1%) in 66.5% of the probes (see Supplementary Material).
The Langmuir form provides a nearly perfect description of the calibration data. To illustrate this, a, b and d were determined for all probes (PM and MMs) separately by weighted least-squares fits of equation 1 to the fluorescence measurements Ii, where i is the concentration index. We minimized the sum S of weighted squared errors:

where the weights wi = 1 / Ii are consistent with a noise model in which the uncertainties in Ii are proportional to ![]() . Subsequently, we rescaled the data for each probe according to
. Subsequently, we rescaled the data for each probe according to
![]()
using the fitted hybridization parameters. According to the model, all measurements should then satisfy a single relationship:
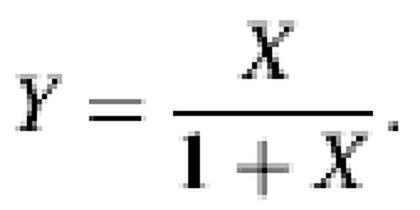
The resulting collapsed data are shown in Figure 1, providing a striking demonstration that the Langmuir model thoroughly captures the physical chemistry of GeneChip hybridization. We emphasize the high density of points in the non-linear regime, proving that chemical saturation is not a marginal effect (see Fig. 5A for the consequences of saturation).
Figure 5.

Differential expression scores for expected ratios of 2 and 1 (no change). Results for ratios of 2 are shown in (A) and (B); control of false positive rates in (C) and (D). (A) and (C) were obtained from MAS 5.0; (B) and (D) from our own estimates using only the PM probes. The compressive bias is clearly visible in (A) as the median ratio lies systematically below the expected value indicated by the upper red line. (B) shows how much our method is capable of reducing bias; sensitivity is also improved despite increased noise levels (Table 3). Low intensity results in (C) and (D) suggest that the normalization is not ideal. For the results in (B), more than half the probes were kept in 85.4% of the comparisons, and more than 12 probes (out of 16) were retained in 333 out of 336 cases. Full box plots are shown in the Supplementary Material.
Comparison of perfect match and mismatch hybridization parameters
A comparison of the values of the hybridization parameters a, b and d between PM probes and their MM partners is shown in Figure 2. In essence, we observe systematically larger as and smaller bs in the PM probe, on the other hand, d is on average equal in the PM and MM cases. The results for b and d can be interpreted in terms of our hybridization model.
Figure 2.

Comparison of the Langmuir parameters a (A), b (B) and d (C) for the PM and MM probes. The line in (B) corresponds to bMM = 3.13 bPM.
First, b is of the form b = x0(1 + z / z0). Considering that non-specific background level is found to be generally low (see the discussion above), the factor (1 + z / z0) is close to 1, and we expect:

In the Langmuir model x0 can be interpreted as an inverse equilibrium constant, and so the difference in binding free energies EB between PM and MM probes is given by:

where kB is Boltzmann’s constant and T is the temperature at which hybridization was performed (45°C). Figure 2 shows that this difference is negative for almost all probes. As a guide to the eye, the line in Figure 2B represents 3.13 bPM = bMM, which corresponds to a difference in binding energy of 1.15 kBT = 3.0 kJ/mol at T = 45°C (318 K).
Turning to the non-specific background d, equation 2 implies that
![]()
when z / z0 is small. As shown in Figure 2C, d has comparable magnitude for PM and MM probes, which is expected for non-specific contributions. We show in the Supplementary Material that the middle base largely determines whether d is larger for the PM or MM. Specifically, we obseverve that dPM > dMM when the PM middle base is a C or a T, while the opposite holds for G or A. This purine–pyrimidine effect could be related to the cRNA labeling protocol, as C and Us are the biotinylated bases (see our preprint at http://xxx.lanl.gov/abs/physics/0208095).
On the contrary, the origin of the result for the as is more difficult to understand. a describes the dependence of the fluorescence on the amount of complementary RNA bound. From equation 2 we identify
![]()
when z / z0 is small. Since aS is the expected fluorescence when the complementary RNA fully saturates the probe, we would not expect this quantity to differ between PM and MM probes; however, we almost exclusively see aPM > aMM. One plausible explanation invokes the washing of the arrays before the scan, to which MM probes are likely more susceptible than PM probes [see Dai et al. (5) and the discussion of b above].
Prediction of probe hybridization parameters from sequence
It is natural to expect that a large fraction of the variability in the probe parameters has a sequence specific origin. We therefore undertook to predict the parameters a, b and d from the sequences of the probes. A cursory inspection of the hybridization parameters suggested a linear model for the logarithms of the hybridization parameters:

which for the case of b is compatible with a model where each base would have an additive contribution to the free energy of binding. Here, nL is the number of letters L = A, C or G in the sequence of a probe, γs are letter specific susceptibilities, Cs are intercepts, and ![]() is an error term. Because the total number of letters must add up to 25, this representation is equivalent to one without intercept but with one additional term γT nT. In the above form, the intercepts C correspond to the estimates for ln a (or, b or d) when the probe sequence would be composed of Ts only. For example, γbC should be understood as the change in ln b when a C base is substituted for a T.
is an error term. Because the total number of letters must add up to 25, this representation is equivalent to one without intercept but with one additional term γT nT. In the above form, the intercepts C correspond to the estimates for ln a (or, b or d) when the probe sequence would be composed of Ts only. For example, γbC should be understood as the change in ln b when a C base is substituted for a T.
The linear model in equation 3 was fit to the hybridization parameters a, b and d from the previous section. The results of the parameters γ are shown in Table 1 for PMs, Table 2 for the MMs, and in Figure 3. The small errors in the fitted parameters indicate that the simple linear model does capture sequence specific effects (see Supplementary Material for a comparison of fitted versus original parameters). We find it hard to assign a physical basis to the results but we point out the following features: (i) PM and MM parameters are very similar (within the errors of one another); (ii) surprisingly, only a exhibits the symmetry between A and T or G and C bases; (iii) letter A has a large negative contribution to ln d. It is unclear to what extent the labeling protocol, only the pyrimidines C and U on the cRNA strand are labeled, contributes to the A-T or C-G asymmetry.
Table 1. Linear regression parameters for the model in equation 3 for the PM data.
| PM | Intercept | γA | γC | γG | R2 |
|---|---|---|---|---|---|
| ln a | 6.617 ± 0.167 | 0.008 ± 0.014 | 0.219 ± 0.014 | 0.195 ± 0.013 | 0.56 |
| ln b | 0.768 ± 0.324 | 0.154 ± 0.022 | 0.206 ± 0.028 | 0.377 ± 0.026 | 0.44 |
| ln d | 2.533 ± 0.416 | –0.305 ± 0.028 | 0.354 ± 0.035 | 0.168 ± 0.033 | 0.48 |
Most parameters have small standard errors compared to their values, indicating that the fits truly capture sequence specificity. Probabilities p(γ = 0) < 10–6 under the hypothesis of no sequence-specificity, except for γaA. Probes were excluded from the fit according to the following criteria: (i) (a, b, d) had to be strictly positive because of the logarithms; (ii) an upper limit on b < 10 000 excluded probes in which no saturation effects were observed and hence a and b could not be determined independently; (iii) d < a / 5 excluded probes that were probably subject to significant cross-hybridization; and (iv) the calibration curves had to follow good Langmuir isotherms: the correlation coefficient ρ(ln Iobs, ln Ifit) between the observed and fitted intensities had to be >0.99. In total, this procedure removed 29.7% of the probes.
Table 2. Linear regression parameters for the model in equation 3 for the MM data.
| MM | Intercept | γA | γC | γG | R2 |
|---|---|---|---|---|---|
| ln a | 5.526 ± 0.256 | 0.012 ± 0.017 | 0.277 ± 0.023 | 0.219 ± 0.018 | 0.57 |
| ln b | 1.066 ± 0.489 | 0.108 ± 0.032 | 0.268 ± 0.043 | 0.418 ± 0.035 | 0.46 |
| ln d | 2.200 ± 0.564 | –0.213 ± 0.036 | 0.322 ± 0.050 | 0.178 ± 0.040 | 0.37 |
Probabilities p(γ = 0) < 10–3 under the hypothesis of no sequence-specificity, except for γaA.
Figure 3.

Data from Tables 1 and 2. The sign flip in the contribution from letter A to ln(d) as compared to ln(a) and ln(b) is particularly obvious.
The small size of the calibration set (14 genes × 16 probes per gene = 224 probes) could only support a model using the overall base composition of each probe. Nevertheless, we show below that even this crude level of modeling is useful in practice.
Prediction of absolute RNA concentration
We now turn to the practically relevant aspects. First, we show how the predicted probe specific hybridization parameters can be exploited to construct an estimator of absolute mRNA concentration. We really mean absolute here, in the sense that RNA levels for different genes can be compared. This adds an interesting new feature to GeneChips.
The Langmuir model relates fluorescence intensity to absolute mRNA concentration. We proceed by inverting equation 1 in which we substitute the predicted parameters from equation 3 (denoted with hats). Each probe p (PM or MM) then yields an estimate of concentration:

which has a vertical asymptote at I = â + d̂. Occasionally, measured intensities will fall above the asymptote or below background, resulting in unphysical values for x̂p. We therefore exclude probes with I > â + d̂ or I < d̂. The values x̂p are then combined to obtain an estimate of probe set concentration:

where the prime (′) indicates exclusion of probes for which I < d̂ or I > â + d̂, and n′ is the number of probes included in the sum. For the analysis presented in the Results section, we included only the PM probes, as inclusion of the MMs appeared to increase the noise in the estimates without improvement in the sensitivity.
A comparison of the real versus estimated concentrations is shown in Figure 4. It is important to note that no scale adjustment was made, and hence the different probe sets can be compared on the same plot. Figure 4A shows three transcripts, which were themselves excluded from the training set determining the parameters γ (the training set consists of the remaining 11 transcripts). Two of them show remarkable linearity throughout the range, while one is not very precise below 16 pM. The average behavior in Figure 4B shows overall good linear behavior in the range from 2 to 256 pM, although residual bias at both ends of the scale can be observed. In the linear range, we observe that the predicted concentrations are systematically too low by a factor <1.5. One contributing factor to this bias is the imperfect prediction of the hybridization parameters (â, b̂, d̂), which have smaller dynamic range than the original parameters (see Fig. S3 in Supplementary Material).
Figure 4.

Absolute concentration estimates: no scale adjustments were made. (A) We tested generalization by using 11 out of 14 transcripts for fitting the parameters γ, then used these parameters to predict the concentrations of the other three. Here, we picked the first three transcripts (according to alphabetically sorted Affymetrix labels) and show predicted versus real concentrations in pM for the two duplicated experiments 1521 and 1532. (B) We tested all transcripts; no probe sets were excluded for determining the γs. The dots represent first quartile, median and third quartile of the 28 measurements (14 transcripts in duplicate). Full box plots are shown in the Supplementary Material.
We found the above way of estimating concentrations to be the most favorable among many we have tried. For instance, we tried more robust estimators (instead of the mean in equation 4) like the median or M-estimators, but we found that these do not offer any obvious advantage for this data set. The result for the median (shown in the Supplementary Material), have slightly lower noise but larger bias, but were on average very close to those obtained using the mean. Alternatively, we tried estimators based on the minimization of functions like

where wi are weights that can depend on (I, a, b, d). Unfortunately, we were unable to achieve similar results as those from equation 5.
Estimates of differential expression
Each probe p in a probe set provides a differential expression estimate f̂p between two conditions 1 and 2. From equation 4, we obtain

where I1 and I2 are the measured fluorescence intensities of probe p in conditions 1 and 2. Notice that the parameter b drops out of the equation. We have factorized the expression as the naïve linear estimate (I2 – d̂) / (I1 – d̂) times a saturation-correcting factor. The ratio for the full probe set is then calculated as the geometric mean of a restricted set of probes:

Here, n′′ is the number of probes included in the sum. The restrictions are the following: we exclude any probe if I1 < d̂ or I2 < d̂, or I1 > (â + d̂) or I2 > (â + d̂), as in the previous section. Because the saturation-correcting factor becomes very large or small when I1 or I2 is close to â + d̂, we also exclude probes for which the saturation-correcting factor was larger than 4 or smaller than one-quarter.
To test the sensitivity of this method, we estimated the relative changes in mRNA levels between measurements taken at subsequent concentrations in the calibration set, i.e. we compared concentrations 0.5 versus 0.25 pM, 1 versus 0.5, …, 1024 versus 512 pM for each probe set. In this way, ratios of 2 are expected in all cases. Results are shown in Figure 5, for comparison, scores from MAS 5.0 are also shown. We also estimated the false positive rate (Fig. 5C and D) by comparing measurements from the replicated Groups (see Materials and Methods). In this case, we expect ratios of 1 (or 0 in log coordinates). Figure 5A clearly shows that the MAS 5.0 ratios are biased throughout the range, most severely at large RNA concentration [see Naef et al. (3) for similar results on a yeast data set]. Notice that the inter-quartiles indicated by the boundary of the boxes lie entirely under the expected line. We emphasize that this qualitative behavior is a feature of all current analysis methods, not just MAS 5.0. Our method (Fig. 5B) clearly reduces the bias in the whole range above 1 pM, with nearly perfect medians in the concentration window spanning 1–128 pM. It is not surprising that these improvements come at the cost of slightly larger variability; however, gain in signal detection overcomes the increase in noise as indicated by the paired t-statistics reported in Table 3.
Table 3. Sensitivity for detection of changes.
| Baseline concentration (pM) | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Langmuir | 1.04 | 2.47 | 8.42 | 10.85 | 10.98 | 13.60 | 12.41 | 21.27 | 21.62 | 11.93 | 12.36 | 7.14 |
| MAS 5.0 | 2.28 | 2.70 | 5.74 | 9.51 | 13.14 | 18.99 | 16.87 | 27.09 | 12.96 | 11.81 | 5.62 | 4.43 |
A paired t-statistic between ratio estimates of 2 and 1 (no change). According to the test, the Langmuir method has higher sensitivity above baseline concentrations of 32 pM.
DISCUSSION
We demonstrated that the assumption of a linear relation between measured intensity and concentration is inaccurate in the case of GeneChips. Instead, we have proven that the calibration curves saturate exactly as one would expect from Langmuir isotherms. In practice, this saturation induces a marked compressive bias in differential expression estimates, most severely at high concentrations. It is likely that similar effects are affecting other versions of microarrays, e.g. cDNA slides or spotted oligonucleotide arrays. We proceeded to show how the three parameters in the Langmuir model could be estimated from the sequence composition of the probes. Despite the small size of the training set, we obtained good results for the prediction of absolute concentration. Additionally, we were able to provide estimates of differential expression with a significant reduction in bias without decrease in signal-to-noise ratio.
One attractive feature of the technique is that it naturally lends itself to fine-tuning as more extensive calibration data are produced. The main improvements should result from more detailed modeling of the Langmuir parameters as a function of probe sequence. Here, only the crudest linear model was used, and it is likely that larger data sets would support models incorporating base position information or nearest-neighbor interactions. We also expect that refinements in the estimator for combining the information from the redundant probes will be possible. So far, our results show that geometric means (equations 5 and 7) lead to similar results as more outlier-robust estimators like the median, suggesting that outliers do not play a crucial role here.
We also observed that inclusion of the MMs generally resulted in increased noise levels, no matter whether we subtracted them from the PM, or pooled them with the PMs. This suggests that this technology would benefit from the replacement of MM probes by additional PMs with non-redundant sequences.
In practice, an effective implementation of our scheme will require some modifications in the current protocols. First, its wide applicability will depend on advances in standardization, but there is general consensus that this is imperative (9). Secondly, it will be crucial to test to what extent the estimated parameters can be transferred across different experiments and/or chip series. After normalization, we expect little variability in the parameters a and b. On the other hand, the parameter d could be dependent on sample particularities. However, the incorporation of a set of non-genomic (random) probes on each array should permit determination of the level of non-specific hybridization and hence calibration of the parameters d.
We believe that using the sequence composition of probes to calibrate arrays will be the key to perfecting microarray-based transcriptional studies. This work provides a step in this direction.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Erik van Nimwegen and Nicolas Socci for helpful discussions.
REFERENCES
- 1.Chee M., Yang,R., Hubbell,E., Berno,A., Huang,X.C., Stern,D., Winkler,J., Lockhart,D.J., Morris,M.S. and Fodor,S.P. (1996) Accessing genetic information with high-density DNA arrays. Science, 274, 610–614. [DOI] [PubMed] [Google Scholar]
- 2.Chudin E., Walker,R., Kosaka,A., Wu,S.X., Rabert,D., Chang,T.K. and Kreder,D.E. (2002) Assessment of the relationship between signal intensities and transcript concentration for Affymetrix GeneChip arrays. Genome Biol., 3, RESEARCH0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naef F., Socci,N. and Magnasco,M. (2002) A study of accuracy and precision in oligonucleotide arrays: extracting more signal at large concentrations. Bioinformatics, 19, 178–184. [DOI] [PubMed] [Google Scholar]
- 4.Naef F., Lim,D.A., Patil,N. and Magnasco,M. (2002) DNA hybridization to mismatched templates: a chip study. Phys. Rev. E Stat. Nonlin. Soft Matter Phys., 65, 040902. [DOI] [PubMed] [Google Scholar]
- 5.Dai H., Meyer,M., Stepaniants,S., Ziman,M. and Stoughton,R. (2002) Use of hybridization kinetics for differentiating specific from non-specific binding to oligonucleotide microarrays. Nucleic Acids Res., 30, e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li C. and Wong,W.H. (2001) Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc. Natl Acad. Sci. USA, 98, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atkins P.W. (1994) Physical Chemistry, 5th Edn. Oxford University Press, Oxford, UK.
- 8.Kepler T.B., Crosby,L. and Morgan,K.T. (2002) Normalization and analysis of DNA microarray data by self-consistency and local regression. Genome Biol., 3, RESEARCH0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brazma A., Hingamp,P., Quackenbush,J., Sherlock,G., Spellman,P., Stoeckert,C., Aach,J., Ansorge,W., Ball,C.A., Causton,H.C., Gaasterland,T., Glenisson,P., Holstege,F.C., Kim,I.F., Markowitz,V., Matese,J.C., Parkinson,H., Robinson,A., Sarkans,U., Schulze-Kremer,S., Stewart,J., Taylor,R., Vilo,J. and Vingron,M. (2001) Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nature Genet., 29, 365–371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.