


Abstract
The Arabidopsis protein AINTEGUMENTA (ANT) is an important regulator of organ growth during flower development. ANT is a member of the AP2 subclass of the AP2/ERF family of plant-specific transcription factors. These proteins contain either one or two copies of a DNA-binding domain called the AP2 domain. Here, it is shown that ANT can act as a transcriptional activator in yeast through binding to a consensus ANT-binding site. This activity was used as the basis for a genetic screen to identify amino acids that are critical for the DNA binding ability of ANT. Mutants that showed reduced or no activation of a reporter gene under the control of ANT-binding sites were identified. The mutations identified in the screen as well as additional site-directed mutations suggest that the mode of DNA recognition by members of the AP2 subfamily is distinct from that of ERF proteins. Surprisingly, it appears that each AP2 domain of ANT uses different amino acids to contact DNA. Identification of several linker mutations argues that this sequence acts in the positioning of each AP2 domain on the DNA or makes direct DNA contacts.
INTRODUCTION
The Arabidopsis protein AINTEGUMENTA (ANT) functions in several aspects of reproductive development. ANT promotes growth within floral meristems and developing organ primordia (1–4). It is required for integument initiation in ovules (1,2,5,6) and plays roles in gynoecium (7) and petal development (8). ANT is a member of a large family of plant transcriptional regulators called the AP2/ERF family (1,2). All members of this family contain at least one copy of a DNA-binding domain called the AP2 domain (9). The 144 AP2/ERF proteins in the Arabidopsis genome (10) can be subdivided into five groups: the AP2 subfamily, RAV subfamily, DREB subfamily, ERF subfamily, and others (11). Members of the AP2 subfamily contain two AP2 domains connected by a 25 amino acid conserved linker. Members of the other subgroups contain a single AP2 domain, which has been referred to as the ERF domain (12) and GCC box-binding domain (13).
Structural information is available for one member of the ERF subfamily, AtERF1. The solution structure of the ERF domain of AtERF1 bound to the GCC box (5′-AGCCGCC-3′) reveals that the protein uses an antiparallel three-stranded β-sheet to make major groove contacts (13). The β-sheet packs against an α-helix that runs approximately parallel to the sheet (13). This structure is stabilized by an extensive number of hydrophobic contacts. The majority of DNA contacts are made by arginine and tryptophan residues located in the β-sheet (13).
Little is known about the interactions between members of the AP2 subfamily of proteins and DNA. Previously, we showed that ANT can bind DNA and that its in vitro consensus binding site is 5′-gCAC(A/G)N(A/T)TcCC(a/g)ANG(c/t)-3′ (14). This sequence is quite distinct from the GCC motif bound by ERF proteins or the DRE/CRT motif (5′-A/GCCGAC-3′) bound by the DREB subfamily (reviewed in 15). Because of these differences in DNA binding specificities and the fact that there is little conservation of amino acid identity between AP2 and ERF proteins at ERF DNA-contacting positions, it appears that these two subfamilies may use distinct DNA binding mechanisms. That the AP2 domain may exhibit flexibility in its mode of DNA recognition is also suggested by the RAV subfamily, in which a single AP2 domain is found in combination with an additional plant-specific DNA-binding domain (the B3 domain).
To test this idea, I have utilized a genetic screen in yeast to identify amino acids that are critical for the DNA binding ability of ANT. This screen combined the inherent infidelity of Taq polymerase and in vivo recombination (16) to generate a large number of mutants containing alterations within the DNA-binding region of ANT. Mutants that were no longer able to activate the expression of a reporter gene under the control of ANT-binding sites were identified. A secondary western blot screen eliminated those mutations resulting in premature termination, frameshifts or grossly altered levels of expression. This screening procedure identified residues in the AP2 repeats and linker region of ANT that appear to play critical roles in DNA binding in vivo. The positions of these mutations provide strong evidence that each AP2 domain of ANT makes unique contacts with DNA and that neither domain utilizes the same mode of DNA recognition as that of the single AP2 domain of ERF proteins.
MATERIALS AND METHODS
Plasmid construction
ANT1–555 was PCR amplified using Pfu polymerase (Stratagene) and primers ANT-13 (5′-TTGCGGATCCATGAAGTCTTTTTGTGATAAT-3′; BamHI site underlined) and ANT-4 (5′-TGAATCTAGATTAAGAATCAGCCCAA GCAGCGAA-3′; XbaI site underlined) using ANT cDNA as template. The PCR product was cloned into the BamHI and XbaI sites of plitmus28 (New England Biolabs) and pGEM3Z (Promega). ANTΔ281–357 was made by replacing the ClaI– HindIII fragment of ANT1–555/plitmus28 with a PCR product generated with ANT-24 (5′-TCAACCCGGGGTTACTAGACATGATACGATC-3′) and ANT-35 (5′-CTATCATCGA TACTTTTGGACAACGAGAAGACATGAAGAACATGA CTAGACAAGAA-3′) primers and digested with ClaI and HindIII. ANTΔ383–451 was made by replacing the NdeI–XbaI fragment of ANT1–555/plitmus28 with a PCR product generated by overlap extension PCR with the primers ANT-12 (5′-GCTGCTCGAGCATATGATCT-3′), ANT-36 (5′-ACTAGACATGATACCCCTAGAGAAACCACTGCT-3′), ANT-37 (5′-TTCTCTAGGGGTATCATGTCTAGTAACACAC TC-3′) and ANT-4. ANT1–451 was made by replacing the HindIII–XbaI fragment of ANT1–555/plitmus28 with a PCR product generated with the primers ANT-10 (5′-TAGGTCTAGATTAACGATCAACATCGTACCTCGT-3′) and ANT-3 (5′-CCAAGAAGAAGCTGCAGAAG-3′). ANTA423T was made by replacing the NdeI–HindIII fragment of ANT1–555/plitmus with a PCR product generated by overlap extension PCR with the primers ANT-6 (5′-TCAAGGATCCACTTT TGGACAACGAACTTCT-3′), ANT-7 (5′-GACAGGATCC GTTACTAGACATGATACGATC-3′), ANT-43 (5′-GTAA GCTTCTGTAGCTTCTTCTTG-3′) and ANT-44 (5′-CAAGAAGAAGCTACAGAAGCTTAC-3′). To create an epitope tagged ANT, an ANT clone that lacked a stop codon was generated in the BamHI site of pGEM3Z. A double hemagglutinin (DHA) tag was added by ligating two annealed oligos DHA-1 (5′-CTAGATACCCATACGACGTTCCAGA CTACGCTGGTTACCCATACGACGTTCCAGACTACGC TTAAT-3′) and DHA-2 (5′-CTAGATTAAGCGTAGTCTGGAACGTCGTATGGGTAACCAGCGTAGTCTGGAACG TCGTATGGGTAT-3′) into the XbaI site of this clone. ANT(sites)DHA was made by replacing the ClaI–BpmI fragment of ANT-DHA/pGEM3Z with a PCR product generated with ANT-50 (5′-AAATCTATCGATACTTTTGGACAACGTACGTCT-3′; introduced BsiWI site underlined) and ANT-51 (5′-GTTGTTGTTCCTTCGCGCGAGCTCTCC-3′; introduced SacI site underlined). Addition of these restriction sites did not alter the amino acid sequence of the protein. Those constructs not made in pGEM3Z were subcloned into pGEM3Z as BamHI–XbaI fragments. The ANT constructs were subsequently subcloned as KpnI–HincII fragments into pGAD424 (Clontech) that had been digested with KpnI and SmaI to remove the GAL4 activation domain. All of these constructs retain the SV40 T-antigen nuclear localization signal. The reporter plasmid was made by cloning three copies of a 29 bp sequence (5′-CTGTAAGCATCGGGATATGTGCACCAAGT-3′) containing the 16 bp ANT consensus binding site (indicated in bold) (14) upstream of the lacZ gene in the vector pLacZi (Clontech). The sequence of each plasmid was confirmed using an ABI 377.
PCR
The PCR product for the yeast transformation was generated by either error-prone PCR (E1–E3) or standard PCR (E4–E8) using Taq polymerase. Standard PCR provided an error rate of 1–2 nt changes per clone. Primers ANT-15 (5′-GAGAC GACGACAATGGCGGCT-3′) and ANT-53 (5′-ACTGACT TCTTTGTTGGAACC-3′) were added to a final concentration of 1.4 µM and the ANT(sites)DHA plasmid (10–20 ng) was used as template for both error-prone and standard PCR. Error-prone PCRs were carried out in 10 mM Tris pH 8.3, 50 mM KCl, 7 mM MgCl2, 0.5 mM MnCl2, 1 mM dCTP, 1 mM dTTP, 0.2 mM dATP and 0.2 mM dGTP. Standard PCRs were carried out in 10 mM Tris pH 9, 50 mM KCl, 1.5 mM MgCl2, 0.1% Triton X-100, 0.2 mM dCTP, 0.2 mM dTTP, 0.2 mM dATP and 0.2 mM dGTP. The reactions were cycled for 12 (E1 and E2), 5 (E3) or 30 rounds (E4–E8) with denaturation at 94°C for 1 min, annealing at 55°C for 1 min, extension at 72°C for 3 min and a final cycle of 72°C for 5 min. For E3, an additional five rounds of standard PCR were performed on the error-prone PCR product. The products were purified using a StrataPrep PCR purification column (Stratagene) and quantitated on a gel. These PCR products contained 109 and 97 bp of homology with either end of the gapped plasmid.
Yeast strains and transformation
The yeast reporter strain was made by integration of the linearized ANT target reporter plasmid into the yeast strain YM4271 (MATa ura3-52, his3-200, lys2-801, ade2-101, trp1-901, leu2-3, 112, tyr1-501, gal4-Δ512, gal80-Δ538, ade5::hisG). This new reporter strain (called BK1) was then transformed with the different ANT constructs or the PCR product (∼200 ng)/gapped plasmid (∼200 ng) combination using a lithium acetate procedure (17). Transformants were selected on plates containing synthetic medium lacking leucine and screened for β-galactosidase activity using a colony-lift filter assay. Plasmid DNA was isolated (18) and sequenced from those mutants showing no (white) or reduced (light blue) β-galactosidase activity but normal levels of protein expression.
β-Galactosidase assays
A colony-lift filter assay was used to screen the yeast transformants. Colonies were lifted onto nitrocellulose membranes (BA85; Schleicher & Schuell) and cells were lysed by freezing in liquid nitrogen. The thawed membranes were laid on filter paper soaked with 100 mM sodium phosphate pH 7, 10 mM KCl, 1 mM MgSO4, 38 mM β-mercaptoethanol and 0.33 mg/ml X-gal and incubated at 30°C overnight. White and light blue colonies were identified by aligning the membranes to the original plates. Quantitative β-galactosidase assays were performed on 1.5 ml liquid cultures grown in synthetic medium lacking leucine. The yeast cells were harvested by centrifugation, resuspended in lysis buffer (100 mM potassium phosphate pH 7.8, 0.2% Triton X-100) and vortexed in the presence of glass beads (425–600 µm diameter; Sigma). An aliquot of 5 µl of the crude yeast extract was added to 100 µl of a 1:100 dilution of the chemiluminescent substrate Galacton (Applied Biosystems). After incubation for 1 h at room temperature, the enzyme activity was terminated and light emission was initiated by the addition of 150 µl of the accelerator. Luminescence was measured in a luminometer. The assays were performed in triplicate. The amount of protein present in each sample was determined by a Bradford assay. The ratio of β-galactosidase levels for each truncated construct compared to full-length ANT was determined.
Western blots
Yeast cultures (5 ml) grown in synthetic medium lacking leucine were harvested after growth for 2 days at 30°C. The cells were washed once in 1 ml of cold sterile water and then lysed in 100 µl lysis buffer (100 mM sodium phosphate pH 7.8, 5 mM EDTA, 0.2% Triton X-100, 2 mM DTT, 80 µg/ml PMSF and 2 µg/ml each of leupeptin, pepstatin A, antipain and aprotinin) by vortexing with glass beads for 8 min at 4°C. The supernatant was collected after centrifugation for 10 min. Total protein concentration was determined by a Bradford assay. An aliquot of 10 µg of protein was separated on a 10% SDS–PAGE gel in Laemmli buffer. The proteins were transferred onto PVDF nylon membranes (Hybond-P; Amersham Pharmacia). The blot was air dried, blocked with 5% non-fat dry milk in PBS containing Tween for 1 h, incubated in a 1:1000 dilution of anti-HA antibody (clone HA-7; Sigma) for 4 h at room temperature, and incubated in a 1:1000 dilution of a secondary antibody coupled to horseradish peroxidase (Bio-Rad) for 2 h at room temperature. The blots were developed with ECL-Plus (Amersham Pharmacia) and scanned on a Molecular Dynamics Storm 860 phosphorimager.
Protein expression
The DNA-binding domain (amino acids 276–456) of each mutant was cloned into the BamHI site of pQE12 (Qiagen) using PCR and the ANT-6 and ANT-7 primers. Plasmid sequences were confirmed by sequencing on an ABI 377. Proteins were expressed in XL1-Blue MRF′ Tet Escherichia coli cells (Stratagene) by induction with 1 mM IPTG. The cells were grown at 30°C and harvested 8 h post-induction. The 6-His tagged proteins were purified under denaturing conditions on Ni-NTA-agarose resin (Qiagen) as described by the manufacturer.
Gel mobility shift assays
Gel mobility shift assays were performed as described previously (14) except the binding reactions were left at 4°C overnight. DNA-binding site probes were prepared as described previously (14). Quantitation of bound and free DNA was performed using a Molecular Dynamics Storm 860 phosphorimager.
RESULTS
ANT activates transcription in yeast through binding to its consensus site
Recently, we isolated DNA sequences to which ANT binds in vitro (14). I utilized a yeast system to determine whether ANT can activate transcription when bound to these sites in vivo. A yeast reporter strain (BK1) containing the lacZ reporter gene under the control of a multimerized ANT consensus binding site and the TATA portion of the CYC1 gene (Fig. 1) was utilized for these studies. Full-length ANT (ANT1–555) expressed in BK1 under the control of the ADH1 promoter produced high β-galactosidase activity, indicating that the ANT-binding site determined in vitro does function to control gene expression in vivo. DNA binding experiments previously indicated that both AP2 repeats are required for high affinity in vitro binding to this site (14). To determine whether both AP2 repeats were required for DNA binding in vivo, β-galactosidase activity was measured in yeast expressing ANT proteins lacking either the first (ANTΔ281–357) or second (ANTΔ383–451) AP2 repeat (Fig. 1). Neither ANTΔ383–451 nor ANTΔ281–357 was able to activate expression of the reporter gene (Fig. 1) while a construct lacking the C-terminal 104 amino acids (ANT1–451) conferred high levels of β-galactosidase activity. Since the transcriptional activation domain of ANT maps to the N-terminal half of the protein (C.Sulli and B.A.Krizek, unpublished observations), the loss of transcriptional activation when either AP2 repeat is missing suggests that binding to this site in vivo requires both AP2 repeats.
Figure 1.

Transcriptional activation by ANT when bound to the consensus binding site in yeast. Schematics of full-length (ANT1–555), internally deleted (ANTΔ281–357, ANTΔ383–451), truncated (ANT1–451) or mutated (ANTA423T) ANT proteins are shown on the left. ANTΔ281–357 lacks the first AP2 domain (AP2-R1), ANTΔ383–451 lacks the second AP2 domain (AP2-R2), ANT1–451 lacks the C-terminal 104 amino acids and ANTA423T contains a replacement of A423 by T. Each of these effector plasmids was tested for their ability to activate expression of the reporter plasmid. The reporter plasmid contained lacZ under the control of three ANT-binding sites and the TATA region of the CYC1 promoter. Relative transcriptional activation conferred by the different ANT proteins are indicated on the right. All values are expressed relative to full-length ANT (1.00). Error bars show standard deviations.
Genetic screen to isolate mutations in ANT that disrupt DNA binding
From these preliminary data, it appeared that the BK1 yeast strain might be a useful tool to further probe ANT–DNA interactions. I developed a genetic screen utilizing this yeast strain to rapidly identify amino acids that are critical to the DNA binding ability of ANT. To determine whether the screen would work (i.e. whether decreased β-galactosidase activity in BK1 is observed with single amino acid mutants that show decreased DNA binding ability in vitro), I tested ANTA423T. This mutation, which corresponds to that present in the weak ant-8 allele (3), results in a dramatically reduced DNA binding ability in vitro (data not shown). BK1 cells expressing ANTA423T showed no β-galactosidase activity (Fig. 1 and Table 1). Thus, it appeared that the BK1 strain would be an appropriate system for identifying mutants with reduced or no DNA binding ability.
Table 1. Mutants isolated in the yeast genetic screen or constructed by PCR.
| Clone | Mutation(s) | Phenotype | Predicted location | Relative β-gal activity | DNA binding |
|---|---|---|---|---|---|
| ANT | Blue | 1.00 | +++ | ||
| R285G | Blue | R1 | 0.18 ± 0.10 | ||
| E5-15 | V287D | White | R1-β1 | 0.00062 ± 0.00015 | |
| E5-52 | V287A, N303D | White | R1-β1, R1 | ||
| E5-41 | T288A, L319P | White | R1-β1, R1-β3 | ||
| T288A | Blue | R1-β1 | 0.62 ± 0.037 | ||
| H290A | Light blue | R1-β1 | 0.017 ± 0.0042 | ||
| E4-11 | W301R, E421G | Light blue | R1-β2, R2-α | ||
| W301R | Light blue | R1-β2 | 0.016 ± 0.0057 | ++ | |
| E5-52 | N303D, V287A | White | R1, R1-β1 | ||
| E5-5 | G314R, Y353C | White | R1, R1 | ||
| E1-2 | V317D | Light blue | R1-β3 | 0.0065 ± 0.00055 | +++ |
| E5-33 | Y318C | White | R1-β3 | 0.00069 ± 0.00031 | + |
| E5-43 | Y318C | White | R1-β3 | 0.00069 ± 0.00031 | + |
| E3-5 | L319Q | White | R1-β3 | 0.00034 ± 0.000068 | + |
| E5-2 | L319P | White | R1-β3 | ||
| E5-26 | L319P | White | R1-β3 | ||
| E8-9 | L319P | White | R1-β3 | ||
| E8-5 | L319P, E356G | White | R1-β3, R1 | ||
| E5-41 | L319P, T288A | White | R1-β3, R1-β1 | ||
| E6-1 | Y322F, Y386D | White | R1-β3, R2 | ||
| E1-1 | D333E, I452T | White | R1-α, C-term | ||
| E1-4 | D333G | White | R1-α | 0.00067 ± 0.00014 | ++ |
| E5-4 | L337P | White | R1-α | 0.00043 ± 0.000067 | + |
| E4-6 | L337P, R387G | White | R1-α, R2 | ||
| E5-5 | Y353C, G314R | White | R1, R1 | ||
| E8-5 | E356G, L319P | White | R1, R1-β3 | ||
| E5-44 | E358G | White | L | ||
| E5-50 | H371P, V389A | White | L, R2-β1 | ||
| E2-9 | F379S | White | L | 0.00092 ± 0.00021 | |
| E5-18 | F379S | White | L | 0.00092 ± 0.00021 | |
| E5-37 | F379S | White | L | 0.00092 ± 0.00021 | |
| E4-13 | S380P | Light blue | L | 0.0092 ± 0.0025 | ++ |
| G382D | White | L | 0.00095 ± 0.000064 | ||
| E5-27 | S384P | White | R2 | ||
| E5-34 | S384P | White | R2 | ||
| E6-1 | Y386D, Y322F | White | R2, R1-β3 | ||
| E4-6 | R387G, L337P | White | R2, R1-α | ||
| R387G | White | R2 | 0.00053 ± 0.000042 | – | |
| E5-50 | V389A, H371P | White | R2-β1, L | ||
| V389D | Blue | R2-β1 | 0.41 ± 0.038 | ||
| H392A | Blue | R2-β1 | 0.22 ± 0.065 | ||
| H393A | Blue | R2 | 0.50 ± 0.13 | ||
| Y412C | Blue | R2-β3 | 0.20 ± 0.037 | ||
| L413Q | Light blue | R2-β3 | 0.041 ± 0.015 | ||
| E4-11 | E421G, W301R | Light blue | R2-α, R1-β2 | ||
| A423T | White | R2-α | 0.00091 ± 0.00033 | + | |
| E5-42 | D427G | White | R2-α | 0.00041 ± 0.00010 | + |
| E6-39 | I431S | Light blue | R2-α | 0.0031 ± 0.0010 | ++ |
| E1-1 | I452T, D333E | White | C-term, R1-α |
To generate mutations in just the DNA-binding domain of ANT, I utilized a method (16) involving recombination between Taq-derived PCR products corresponding to the AP2 repeat region of ANT and a gapped ANT plasmid lacking the AP2 repeat region (Fig. 2). Homology between the ends of the PCR product and the gapped plasmid allowed in vivo recombination between these DNA pieces. Yeast transformants were initially screened for β-galactosidase activity. To eliminate mutations resulting in premature truncations or grossly altered stability of the protein, a secondary western blot screen was performed on all mutants showing reduced or no β-galactosidase activity (Fig. 3A). Each of the β-galactosidase phenotypes was confirmed by re-transformation of the BK1 yeast strain with DNA from the individual clone (Fig. 3B).
Figure 2.

Method used to generate mutations in the DNA-binding domain of ANT. BsiWI and SacI restriction sites engineered into the ANT sequence allowed the production of a gapped ANT plasmid lacking the DNA-binding domain. In vivo recombination occurred between PCR products generated with Taq polymerase and the gapped plasmid. Yeast transformants containing the repaired ANT plasmid were screened for β-galactosidase activity using a colony-lift filter assay. Clones producing reduced (light blue) or no (white) β-galactosidase activity were then analyzed using western blots and an antibody against the DHA epitope present at the C-terminus of ANT.
Figure 3.
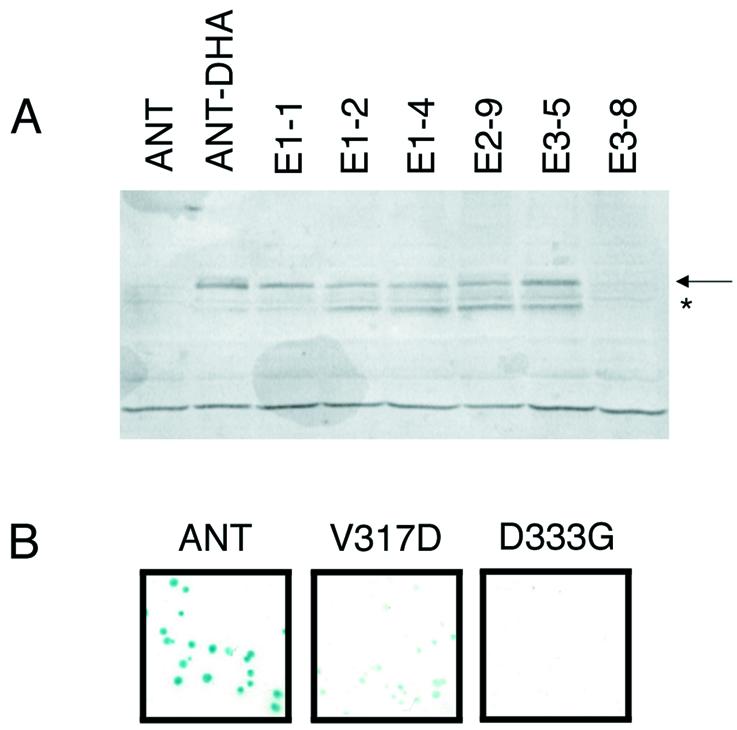
Western blots and colony-lift filter assays of some mutants identified in the yeast screen. (A) Western blot of individual yeast clones. ANT (indicated with the arrow) is detected using the anti-HA antibody. No protein is detected in protein extracts from yeast expressing an ANT protein lacking the DHA tag (ANT) or the mutant E3-8. Full-length ANT protein is detected in extracts from ANT-DHA and the mutants E1-1, E1-2, E1-4, E2-9 and E3-5. A smaller ANT fragment (marked with an asterisk) is also detected in some of the mutants. (B) Colony-lift filter β-galactosidase assays for yeast expressing ANT (blue phenotype), V317D (light blue phenotype) and D333G (white phenotype).
Characterization of mutants identified by the screen
Using this two-step screening procedure, 28 individual colonies were identified that expressed a full-length ANT protein but showed either reduced (four colonies) or no (24 colonies) β-galactosidase activity. Each clone had either one (19 clones) or two (nine clones) amino acid changes within the DNA-binding region of ANT (Table 1). Mutations in seven positions (V287, Y318, L319, D333, L337, F379 and S384) were represented multiple times within the collection. Quantitative β-galactosidase assays were performed for 10 of the mutants. There is a good correlation between the results from the quantitative β-galactosidase assay and those from the colony-lift assay. White colonies produced β-galactosidase levels at least 1000-fold lower than ANT while light blue colonies produced β-galactosidase levels at least 100-fold lower than ANT (Table 1).
The 37 mutations correspond to 24 different amino acid positions within ANT. These include 13 positions within the first AP2 repeat (ANT-AP2R1), four positions within the linker and seven positions within the second AP2 repeat (ANT-AP2R2). The positions of these mutations are shown in Figure 4A. For the most part, the mutations mapped to different positions within the first and second AP2 repeats.
Figure 4.

Mapping of mutants onto the ANT sequence. Each of the two AP2 repeats of ANT is shown aligned with the single ERF domain of AtERF1. Secondary structural elements as determined in the AtERF1 structure (13) are indicated above the AtERF1 sequence. The first and last amino acids are numbered at the left and right of each sequence. Hydrophobic residues stabilizing the packing of these secondary structural elements are shaded with gray. DNA-contacting amino acids of AtERF1 are shown in bold (base-contacting) and/or underlined (phosphate backbone-contacting). (A) Summary of mutants identified in the yeast genetic screen. Amino acids shown in red correspond to mutations conferring a white phenotype in the colony-lift filter assay while amino acids shown in blue correspond to mutations conferring a light blue phenotype in the colony-lift filter assay. Residues shown in green were present in a double mutant and thus their individual effects in the yeast system could not be determined. (B) Summary of all mutants with confirmed white (shown in red), light blue (shown in blue) or blue (shown in green) phenotypes in the colony-lift filter assay.
The single ERF domain from AtERF1 folds into a compact structure consisting of a three-stranded β-sheet that packs against an α-helix (13). Amino acids from each of the three β-strands as well as two residues N-terminal to the first β-strand contribute to DNA binding (13). These DNA-contacting positions include five arginines (R147, R150, R152, R162 and R170) and two tryptophans (W154 and W172). Three of the arginines and the two tryptophan residues contact both the nucleotide base and the sugar–phosphate backbone. The secondary structural elements and DNA-contacting residues of AtERF1 are shown in Figure 4.
A large number of hydrophobic residues (Y146, V149, F157, A159, I161, V171, L173, F176, A179, A182, A183, A185, Y186, A189, A190, A198, L200 and F202 in AtERF1) stabilize the packing of these secondary structural elements against one another (13). The positions of these hydrophobic residues are well conserved in the sequences of ANT-R1 and ANT-R2, suggesting that each AP2 repeat of ANT might fold into a similar three-dimensional structure as the ERF domain of AtERF1. Homology modeling using Swiss-Model predicts an α-helix in each AP2 domain similar in length and location to the α-helix in AtERF1 (Fig. 5) (19–21). These models differed from AtERF1 however in the β-sheet region. No β-sheet was predicted in ANT-AP2R2 while that proposed for ANT-AP2R1 consisted of two strands at somewhat different positions than the β-strands in AtERF1 (Fig. 5).
Figure 5.

Structure of AtERF1 and homology models of ANT-AP2R1 and ANT-AP2R2. α-Helices are indicated in red and β-strands in yellow. D187 and F191 of AtERF1 lie on the face of the helix pointing away from the β-sheet. The corresponding positions in ANT-AP2R1 (D333 and L337) and ANT-AP2R2 (D427 and I431) lie on this same face of the predicted helices.
In the absence of any structural information on the AP2 domain, I compared the positions identified in the screen to positions within AtERF1 as determined by aligning the sequences using Clustal X. This alignment is shown in Figure 4. Approximately half of the mutations (13 of 24) map to regions predicted to fold into secondary structural elements based on the alignment with AtERF1. Although the α-helix makes no contacts with DNA in the AtERF1 structure, mutation of two positions within the putative helix of both ANT-AP2R1 and ANT-AP2R2 results in loss of function in the yeast assay. These two positions are predicted to lie on the face of the helix that points away from the DNA-contacting β-sheet (Fig. 5). Outside of the α-helix, similar mutations are not present in ANT-AP2R1 and ANT-AP2R2. For example, mutations in at least three amino acids (V317, Y318 and L319) of ANT-AP2R1 that align with the third β-strand (β3) of AtERF1 result in decreased ANT activity, yet no mutations in the corresponding positions of ANT-AP2R2 were isolated in the screen. Similarly, three residues (S384, Y386 and R387) located near the N-terminus of ANT-AP2R2 were identified in the screen but no similar positions in ANT-AP2R1 were isolated.
Few mutations map to predicted DNA-contacting residues
Few of the identified mutations map to the DNA-contacting positions of AtERF1 that are within and adjacent to the β-strands. One single mutant (Y318C) corresponds to the base and backbone-contacting W172 residue of AtERF1. Three double mutants (T288A L319P, W301R E421G and L227P R387G) potentially have mutations corresponding to AtERF1 DNA-contacting residues. T288, W301 and R387 correspond to R150, R162 and R147, respectively. The single mutants T288A, W301R and R387G were constructed to determine the effects of these individual mutations on β-galactosidase activity. T288A mutants exhibited high β-galactosidase activity, W301R mutants produced low β-galactosidase activity and R387G mutants showed significantly reduced β-galactosidase activity (Table 1). Thus, three residues in ANT (W301, Y318 and R387) aligning to DNA-contacting positions within AtERF1 alter the ability of ANT to activate a reporter gene. Mutations in two of these positions, Y318 in ANT-AP2R1 and R387 in ANT-AP2R2, cause dramatic reductions in function as measured in the yeast assay.
Analyses of site-directed mutants
Even though there is high sequence similarity between the first and second AP2 repeats of ANT, few corresponding mutations in each repeat were identified in the screen. It is possible that such mutations would have been isolated with additional screening. To address further whether the two AP2 repeats of ANT might interact differently with DNA, additional amino acid substitutions were made in ANT and assayed in the BK1 system. For several mutants (V287D, Y318C, L319Q and R387G) that exhibited no β-galactosidase activity in the screen, I made the corresponding amino acid substitution in the other AP2 repeat (V389D, Y412C, L413Q and R285G, respectively). V389D, Y412C and R285G mutants exhibited high levels of β-galactosidase activity ranging between 18 and 40% that of wild-type ANT and produced a blue color in the colony-lift assay (Table 1). The L413Q mutant displayed a partial loss of function in the yeast assay giving a light blue color in the colony-lift filter assay and β-galactosidase activity that was ∼4% that of wild-type ANT (Table 1).
The importance of one additional DNA-contacting position (R152) in AtERF1 was investigated in each of the AP2 repeats of ANT. Histidines are found at the corresponding locations in ANT-AP2R1 and ANT-AP2R2 (H290 and H392, respectively). In ANT-AP2R2, a second histidine (H393) is found next to H392. All three positions were mutated to alanine and assayed in BK1. The H290A mutation produced a light blue color in the colony-lift assay and exhibited significantly lower β-galactosidase activity compared to ANT (Table 1). The H392A and H393A mutations confer high levels of β-galactosidase activity (Table 1). Thus, in only one (L319Q/L413Q) of the five pairs tested did identical amino acid changes in the two AP2 repeats of ANT produce similar β-galactosidase phenotypes in yeast.
One additional mutant was assayed in the BK1 system. The G382D mutation, corresponding to the molecular defect present in the strong ant-2 mutation, contains a replacement of a highly conserved glycine at the last position of the linker (2). Yeast expressing the G382D mutant gave a white phenotype in the colony-lift filter assay (Table 1).
Characterization of the in vitro DNA binding ability of several mutants
The ability of several of these mutants to bind DNA in vitro was examined using gel mobility shift assays. Mutations in ANT-AP2R1 (W301R, V317D, Y318C, L319Q, D333G and L337P), ANT-AP2R2 (R387G, D427G and I431S) and the linker (S380P) were chosen for these studies. This group of mutants corresponds to both partial (W301R, V317D, S380P and I431S) and complete (Y318C, L319Q, D333G, L337P, R387G and D427G) loss of yeast activity mutants. In addition, some members of the group correspond to positions that may contact DNA (W301R, V317D, Y318C, L319Q, S380P and R387G), while others correspond to positions that appear unlikely to contact DNA (D333G, L337P, D427G and I431S). The mutations were created and assayed in the context of the two AP2 repeats of ANT, as this was the ANT construct previously used in DNA binding characterization (called ANT-AP2R1R2) (14). All tested proteins from mutants that gave a light blue β-galactosidase phenotype were able to bind the ANT consensus site in vitro with fairly high affinity. The V317D mutant had a DNA binding ability comparable to that of ANT while W301R, S380P and I431S bound with decreased ability (Fig. 6 and Table 1). Five of the six proteins from mutants that gave a white colony-lift β-galactosidase phenotype showed significantly decreased DNA binding (Y318C, L319Q, L337P and D427G) or absolutely no DNA binding (R387G). The lone exception was D333G; this protein exhibited relatively high DNA binding ability but had no activity in the yeast β-galactosidase assay. Thus, the in vitro DNA binding results are generally in good agreement with the yeast in vivo results.
Figure 6.

Gel mobility shift assay of ANT-AP2R1R2 and mutants. The experiment used radiolabeled ANT consensus binding site 15 (14). Equal amounts of protein (∼100 ng), as estimated from the intensity of Coomassie Brilliant Blue stained gels, were loaded in each lane. B and F denote the positions of bound and free probe, respectively. The fractional saturation of bound probe to total probe is shown below the gel. Weak DNA binding was observed for D427G at higher protein concentration.
DISCUSSION
Different positions within each AP2 domain of ANT appear to play important roles in DNA recognition
Using a genetic screen in yeast, I identified a number of amino acids that disrupt the ability of ANT to activate expression of a reporter gene under the control of ANT-binding sites. Most of these mutations do not map to positions corresponding to AtERF DNA-contacting positions and thus their importance could not have been predicted from the AtERF structure. To test whether these mutations did indeed affect DNA binding, several were subsequently assayed using gel mobility shift assays. For almost all the mutants tested, the decreased activity in vivo correlated well with the decreased DNA binding activity measured in vitro, confirming the validity of the approach. The different activities of the D333G mutant in the two assays may result from the different protein contexts utilized in the yeast and gel mobility shift experiments. This mutation displayed a more severe phenotype when assayed in the context of the full-length protein, perhaps due to an effect on a region of the protein not present in the shortened version used in the in vitro DNA binding experiments. Most mutants still possessed some DNA binding ability, suggesting that the majority of the mutations did not significantly affect the overall folding of the protein but had more specific alterations that affected DNA binding affinity.
This genetic screen and subsequent targeted mutagenesis have identified 19 residues that are likely to play important roles in DNA binding and at least six residues that are dispensable for DNA binding (summarized in Fig. 4B). Several residues were isolated at least twice, suggesting that the screen was approaching saturation. Based on the number and severity of the mutations, the amino acids most critical to DNA binding by ANT correspond to those within ANT-AP2R1 that align with β3 of AtERF1, those at the C-terminal part of the linker and those at the N-terminus of ANT-AP2R2. These results suggest that the two AP2 repeats of ANT differ in their interactions with this DNA-binding site. The two AP2 repeats might use different positions to contact the DNA. Alternatively, similar positions might be used but contribute differently to the overall affinity for DNA. The former appears more likely as the ANT consensus site does not consist of two similar half-sites, even though ANT-AP2R1 and ANT-AP2R2 contain identical amino acids at most of the positions identified in this study.
No in vivo targets of ANT regulation have been identified. Thus, the level of similarity between the in vitro determined ANT consensus binding site (used in this study) and natural promoter elements bound by ANT in vivo is not known. Once such sites have been identified, it will be important to determine whether the residues identified in this study mediate critical DNA interactions in the context of these other sites. Such studies will be necessary to determine if the same amino acids are used to contact native ANT target promoters and to determine whether differential DNA interactions by each AP2 repeat is a natural feature of the molecular recognition of DNA by ANT.
Importance of the linker in ANT–DNA interactions
The linker connecting the two AP2 repeats of ANT appears to be an important component for DNA recognition as demonstrated by the conservation of the linker in members of the AP2 subfamily (reviewed in 9) and the fact that mutations in several positions within the linker negatively affect DNA binding. Several families of transcription factors use tandem repeats of structurally independent DNA-binding domains to contact DNA. The linkers connecting these domains appear to play several different roles in these proteins. In zinc finger proteins and Myb proteins, the DNA-binding domains (zinc fingers or helix–turn–helix related motifs) are connected by well conserved linkers (5 or 13 amino acids) that allow the independent structural units to follow the turn of the DNA helix and make continuous contacts in the major groove. The importance of these linkers thus appears to derive primarily from a role in positioning of the DNA-binding domains (22,23).
Two other families of proteins that contain two DNA-binding domains are the paired and POU domain proteins. In the paired domain, a conserved linker of 16 amino acids makes extended contacts along the minor groove while connecting two helix–turn–helix type domains that contact the major groove (24). These linkers thus directly contribute to DNA binding affinity and specificity. In POU domain proteins, the linker sequence is not well conserved, of variable length (15–56 amino acids) and seems to primarily serve a tethering role linking the two helix–turn–helix motifs of the POU-specific (POUS) and POU-homeodomain (POUH) domains. This flexible linker allows POU domain proteins to recognize diverse DNA sequences due to alterations in the positioning, spacing and orientation of the POUS and POUH domains (reviewed in 25). The conserved nature of the linker in AP2-like proteins and the identification of several linker residues in ANT that are indispensable for DNA binding suggests that this region directly contacts DNA or functions in positioning the two AP2 repeats on DNA.
Possible effects of mutations in the predicted α-helical regions of ANT
Since the α-helix does not make any DNA contacts in AtERF1, few residues in this region would be expected to affect DNA binding. Only six of the 24 mutations from the screen were located in this region. However, five of the six mutations map to the same two positions (D333/D427 and L337/I431) within the predicted α-helix of both AP2 repeats. Of particular surprise is that these two positions are predicted to lie on the exposed face of the helix (Fig. 5). Thus, their effect on DNA binding does not appear to derive from simply decreased stabilization of the packing between the predicted α-helix and the rest of the AP2 domain as might be predicted for the A423T mutation.
These residues could be involved in contacting other regions of ANT, although the D333G, L337P, D427G and I431S mutants all showed reduced DNA binding activity in vitro when they are assayed in the context of an ANT protein consisting of just the two AP2 repeats and the linker. These amino acids seem unlikely to be involved in interactions with components of the general transcriptional machinery as the transcriptional activation domain of ANT is located in the N-terminal half (C.Sulli and B.A.Krizek, unpublished observations). A third possibility is that the mutations more indirectly affect DNA binding by altering the structure of the AP2 domain. Proline and glycine tend not to be found in α-helices as they are helix-destabilizing residues. Thus, it is possible that these mutations prematurely terminate the helix and decrease the stability of the protein–DNA complex. Such an idea could be tested by mutating the amino acids in these positions to residues compatible with an α-helical structure and testing them in the yeast BK1 assay.
Members of the AP2 and ERF subfamilies may use distinct modes for DNA binding
One surprising result from the screen was that few residues in ANT predicted to be DNA-contacting residues based on an alignment with AtERF1 were identified as conferring a loss of function phenotype in yeast. Of the three positions identified (W301R, Y318C and R387G), R387G showed the most dramatic losses of activity in vivo and in vitro. Site-directed mutations of additional positions in ANT (R285, T288, H290, H392 and Y412) that correspond to DNA-contacting positions in AtERF1 confirmed that most of these alterations do not disrupt ANT function in the yeast assay. Of these mutants, only H290A showed any loss of activity in the yeast assay. This suggests that the mode of DNA binding by ANT is distinct from that of AtERF1. However, it is also possible that these amino acids have critical roles in DNA binding when ANT is bound to a natural binding site. Such investigations await the identification of in vivo target sites of ANT.
Any differences in DNA binding between ANT and AtERF1 could arise from a different three-dimensional structure of the AP2 domains of ANT and consequently different interactions with DNA. Alternatively, each of the AP2 domains could fold into a structure that resembles the ERF domain but which has a distinct mode of DNA recognition due to the covalent linkage of two domains. Structural studies are needed to better address the three-dimensional structure of the AP2 domain and the role of the linker in this class of transcription factors.
Acknowledgments
ACKNOWLEDGEMENTS
I thank Sandra Snyder for performing gel shift analyses of the A423T mutant and Staci Nole-Wilson, Erin Connolly and Cliff Robinson for comments on the manuscript. This work was supported by Department of Energy grant 98ER20312.
REFERENCES
- 1.Elliott R.C., Betzner,A.S., Huttner,E., Oakes,M.P., Tucker,W.Q.J., Gerentes,D., Perez,P. and Smyth,D.R. (1996) AINTEGUMENTA, an APETALA2-like gene of Arabidopsis with pleiotropic roles in ovule development and floral organ growth. Plant Cell, 8, 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klucher K.M., Chow,H., Reiser,L. and Fischer,R.L. (1996) The AINTEGUMENTA gene of Arabidopsis required for ovule and female gametophyte development is related to the floral homeotic gene APETALA2. Plant Cell, 8, 137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krizek B.A. (1999) Ectopic expression of AINTEGUMENTA in Arabidopsis plants results in increased growth of floral organs. Dev. Genet., 25, 224–236. [DOI] [PubMed] [Google Scholar]
- 4.Mizukami Y. and Fischer,R.L. (2000) Plant organ size control: AINTEGUMENTA regulates growth and cell numbers during organogenesis. Proc. Natl Acad. Sci. USA, 97, 942–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker S.C., Robinson-Beers,K., Villanueva,J.M., Gaiser,J.C. and Gasser,C.S. (1997) Interactions among genes regulating ovule development in Arabidopsis thaliana. Genetics, 145, 1109–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneitz K., Hulskamp,M., Kopczak,S.D. and Pruitt,R.E. (1997) Dissection of sexual organ ontogenesis: a genetic analysis of ovule development in Arabidopsis thaliana. Development, 124, 1367–1376. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z., Franks,R.G. and Klink,V.P. (2000) Regulation of gynoecium marginal tissue formation by LEUNIG and AINTEGUMENTA. Plant Cell, 12, 1879–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krizek B.A., Prost,V. and Macias,A. (2000) AINTEGUMENTA promotes petal identity and acts as a negative regulator of AGAMOUS. Plant Cell, 12, 1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riechmann J.L. and Meyerowitz,E.M. (1998) The AP2/EREBP family of plant transcription factors. Biol. Chem., 379, 633–646. [DOI] [PubMed] [Google Scholar]
- 10.Riechmann J.L., Heard,J., Martin,G., Reuber,L., Jiang,C.-Z., Keddie,J., Adam,L., Pineda,O., Ratcliffe,O.J., Samaha,R.R., Creelman,R., Pilgrim,M., Broun,P., Zhang,J.Z., Ghandehari,D., Sherman,B.K. and Yu,G.-L. (2000) Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science, 290, 2105–2110. [DOI] [PubMed] [Google Scholar]
- 11.Sakuma Y., Liu,Q., Dubouzet,J.G., Abe,H., Shinozaki,K. and Yamaguchi-Shinozaki,K. (2002) DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem. Biophys. Res. Commun., 290, 998–1009. [DOI] [PubMed] [Google Scholar]
- 12.Hao D., Ohme-Takagi,M. and Sarai,A. (1998) Unique mode of GCC box recognition by the DNA-binding domain of ethylene-responsive element-binding factor (ERF domain) in plant. J. Biol. Chem., 273, 26857–26861. [DOI] [PubMed] [Google Scholar]
- 13.Allen M.D., Yamasaki,K., Ohme-Takagi,M., Tateno,M. and Suzuki,M. (1998) A novel mode of DNA recognition by a β-sheet revealed by the solution structure of the GCC-box binding domain in complex with DNA. EMBO J., 17, 5484–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nole-Wilson S. and Krizek,B.A. (2000) DNA binding properties of the Arabidopsis floral development protein AINTEGUMENTA. Nucleic Acids Res., 28, 4076–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shinozaki K. and Yamaguchi-Shinozaki,K. (2000) Molecular responses to dehydration and low temperature: differences and cross-talk between two stress signaling pathways. Curr. Opin. Plant Biol., 3, 217–223. [PubMed] [Google Scholar]
- 16.Muhlrad D., Hunter,R. and Parker,R. (1992) A rapid method for localized mutagenesis of yeast genes. Yeast, 8, 79–82. [DOI] [PubMed] [Google Scholar]
- 17.Gietz D., St Jean,A., Woods,R.A. and Schiestl,R.H. (1992) Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res., 20, 1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffman C.S. and Winston,F. (1987) A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene, 57, 267–272. [DOI] [PubMed] [Google Scholar]
- 19.Peitsch M.C. (1995) Protein modeling by e-mail. Biotechnology, 13, 658–660. [Google Scholar]
- 20.Peitsch M.C. (1996) ProMod and Swiss-Model: internet-based tools for automated comparative protein modeling. Biochem. Soc. Trans, 24, 274–279. [DOI] [PubMed] [Google Scholar]
- 21.Guex N. and Peitsch,M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 22.Hegvold A.B. and Gabrielsen,O.S. (1996) The importance of the linker connecting the repeats of the c-Myb oncoprotein may be due to a positioning function. Nucleic Acids Res., 24, 3990–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolfe S.A., Nekludova,L. and Pabo,C.O. (2000) DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct., 29, 183–212. [DOI] [PubMed] [Google Scholar]
- 24.Xu H.E., Rould,M.A., Xu,W., Epstein,J.A., Maas,R.L. and Pabo,C.O. (1999) Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev., 13, 1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herr W. and Cleary,M.A. (1995) The POU domain: versatility in transcriptional regulation by a flexible two-in-one DNA-binding domain. Genes Dev., 9, 1679–1693. [DOI] [PubMed] [Google Scholar]