


Abstract
Chemical and physical agents can alter the structure of DNA by modifying the bases and the phosphate–sugar backbone, consequently compromising both replication and transcription. During transcription elongation, RNA polymerase complexes can stall at a damaged site in DNA and mark the lesion for repair by a subset of proteins that are utilized to execute nucleotide excision repair, a pathway commonly associated with the removal of bulky DNA damage from the genome. This RNA polymerase-induced repair pathway is called transcription-coupled nucleotide excision repair. Although our understanding of DNA lesion effects on transcription elongation and the associated effects of stalled transcription complexes on DNA repair is broadening, the attainment of critical data is somewhat impeded by labor-intensive, time- consuming processes that are required to prepare damaged DNA templates. Here, we describe an approach for building linear DNA templates that contain a single, site-specific DNA lesion and support transcription by human RNA polymerase II. The method is rapid, making use of biotin–avidin interactions and paramagnetic particles to purify the final product. Data are supplied demonstrating that these templates support transcription, and we emphasize the potential versatility of the protocol and compare it with other published methods.
INTRODUCTION
Environmental, industrial and therapeutic chemical and physical agents can induce changes in DNA, often resulting in DNA lesions that include modified bases, alterations to the phosphate–sugar backbone, and single- and double-strand breaks (1). The ensuing DNA modifications compromise replication and transcription because DNA and RNA polymerases do not always function properly on aberrant templates (2–4). DNA polymerases can generate mutations when they add an incorrect base during lesion bypass, or the replication machinery can actually stall at the damaged site, thus inducing recombination (1,5). RNA polymerases can also misincorporate ribonucleotides into transcripts during elongation past damaged DNA bases, resulting in anomalous transcript function during translation or in altered RNA processing events. In addition, RNA polymerase can stall at damaged sites in DNA, producing prematurely truncated transcripts which might remain in the nucleus or be degraded (2).
DNA repair pathways function to remove DNA damage and preserve genomic integrity in both prokaryotes and eukaryotes. Among these repair pathways is nucleotide excision repair (NER), which clears a wide array of DNA adducts from the genome, including cyclobutane pyrimidine dimers induced by ultraviolet radiation, and bases with large DNA adducts derived from chemicals such as polycyclic aromatic hydrocarbons (PAHs), e.g. benzo[a]pyrene diol epoxide (BPDE) or benzo[c]phenanthrene diol epoxide (BPhDE) (6–10). In eukaryotic cells, over thirty proteins are required to effect NER, including seven proteins called XPA through XPG that are named for the complementation groups associated with the disease xeroderma pigmentosum (XP) (11). The XP proteins are required for DNA damage recognition and incision during NER. Thus, XP complementation groups partially or completely lack the ability to repair bulky DNA damage, and patients with XP are sensitive to sunlight, which produces lesions like cyclobutane pyrimidine dimers which NER normally removes (12,13). In eukaryotes, homologs of the human protein heterodimer XPC/HR23B bind to DNA lesions, marking them for excision and recruiting other XP homologs and additional NER-associated factors that ultimately excise and release the damage as part of a DNA oligomer (14–16). DNA polymerase δ/ε fills the resulting gap, and ligase seals the newly synthesized oligomer to the parental DNA strand (17).
During the past decade, two modes of NER have been identified: global-genomic NER (GG-NER) and transcription-coupled NER (TC-NER) (18–20). In the late 1980s, it was discovered that DNA damage on the transcribed strand of active genes can be cleared faster than that on the non-transcribed strand or the genome overall; this phenomenon was called transcription-coupled DNA repair (21–24). Experiments during the 1990s further demonstrated the existence of significant overlap between transcription-coupled DNA repair and NER, with the principal difference occurring at the level of DNA damage recognition. In GG-NER, XPC/HR23B recognizes lesions throughout the genome, whereas TC-NER involves XPC-independent damage recognition (14). For TC-NER, it has been suggested that the lesion in the transcribed strand blocks RNA polymerase progression, and the resulting stalled elongation complex then acts as a damage recognition marker, obviating the need for XPC/HR23B (9). Although the detailed mechanism of TC-NER is not yet established, the stalled RNA polymerase complex likely recruits additional XP proteins and NER factors to remove the damaged DNA (7).
The importance of TC-NER is evidenced by the clinical phenotype observed when it is aberrant, as is seen in patients with Cockayne syndrome (CS), a disease characterized by severe growth and developmental defects (1,6,25). Cells from CS patients are defective in their ability to execute TC-NER and are hypersensitive to certain DNA-damaging agents. However, the underlying cause of CS appears to be more closely tied to defective transcription than to reduced DNA repair. Thus, the CS phenotype may well result from the consequences associated with the cell’s inability to handle stalled transcription complexes that are formed when a lesion blocks an elongating RNA polymerase (7,9).
There are two CS complementation groups, CSA and CSB, but the underlying role of the CSA and CSB proteins in normal cell function is not clear. CSA has ATPase activity and a series of WD (Trp-Asp) repeats that are characteristic of many proteins involved in signal transduction and, perhaps relevant to TC-NER, RNA processing; CSB has homology to SWI/SNF proteins that are involved in chromatin remodeling (26,27). It has been shown that certain DNA damaging agents like hydrogen peroxide induce the translocation of CSA protein to the nuclear matrix in a process that is dependent on CSB protein (28). It is possible that CSA and CSB attract other essential repair proteins, like the XP homologs, to the site of DNA damage, or they might assist in eliminating stalled transcription complexes from the damaged sites so that repair can occur (14,15).
Although our understanding of DNA lesion effects on transcription elongation is broadening, obtaining critical data is somewhat impeded by labor-intensive, time-consuming processes required in assembling suitable damaged DNA templates (29–31). Here, we describe an approach for building linear DNA templates that contain a single, site-specific DNA lesion and support transcription by human RNA polymerase II. The protocol assumes that a site-specifically modified oligomer is available for synthesizing the transcription templates. The method is relatively rapid, making use of biotin–avidin interactions and paramagnetic particles to purify the final product, a procedure that eliminates some of the protracted steps associated with other methods. Data are supplied demonstrating that these templates support transcription and, in the discussion, we emphasize the potential versatility of the protocol and compare it with other published methods. The template construction and purification method presented here will allow more rapid and straightforward investigation of DNA damage-induced transcription blockage, thus permitting further elucidation of the connection between arrested RNA polymerase elongation and TC-NER.
MATERIALS, METHODS AND RESULTS
Reagents and supplies
T4 polynucleotide kinase, T4 DNA ligase, BbsI and BglII were purchased from New England Biolabs, Inc. (Beverly, MA). [γ-32P]ATP (3000 Ci/mmol), [α-32P]CTP (3000 Ci/mmol) and [α-32P]UTP (3000 Ci/mmol) were purchased from Perkin Elmer Life Sciences, Inc. (Boston, MA). The HeLa Scribe In Vitro Transcription System, EcoRV, I-PpoI, RNasin, Streptavidin Magnesphere Paramagnetic Particles, Magnesphere Technology Magnetic Separation Stand, dephosphorylated φX174 DNA-Hinf I DNA ladder and ribonucleoside triphosphates were obtained from Promega Corp. (Madison, WI). Acrylamide:bis-acrylamide (40%, 19:1) and Micro Bio-spin 6 gel filtration columns were purchased from Bio-Rad, Inc. (Hercules, CA). All other reagents were procured from Fisher Scientific (Pittsburgh, PA).
Construction of a plasmid containing an RNA polymerase II promoter and a G-less cassette
To begin this work, a plasmid was designed that contained the immediate-early enhancer/promoter region of the human cytomegalovirus (CMV) and a G-less cassette. The CMV enhancer/promoter was chosen because it provides a strong, constitutive promoter for human RNA polymerase II. A G-less cassette is a region that is located downstream from the transcription initiation site and contains no cytidine nucleotides on the transcribed strand; hence, transcription initiation can begin and proceed as far as the first cytidine even when there is no GTP present in the reaction. G-less cassettes facilitate single-round transcription studies. Heparin, which binds to free RNA polymerase and prevents it from forming an initiation complex at a promoter, can be added after initiation and before addition of GTP, resulting in the formation of transcription elongation complexes that are stalled at the first cytidine in the template. Once GTP is added, the complex can continue elongation, but subsequent rounds of transcription cannot occur due to the presence of heparin (31).
The pCI-neo (Promega Corp., Madison, WI) vector was used as the source of the CMV enhancer/promoter, and a double-strand G-less cassette was inserted as follows (Fig. 1). pCI-neo was linearized at a SacI restriction site located 24 bp upstream from the +1 start site for transcription off the CMV promoter. The ends of the linearized plasmid were dephosphorylated with calf intestinal phosphatase to minimize subsequent formation of re-ligated vector during the modification procedure. A sequence of double-stranded DNA that contained the G-less cassette, a region to restore the original promoter, SacI overhangs and a unique SacII site to assist with later differentiation of colonies containing the recombinant vector from those with self-ligated vector was phosphorylated at the 5′ ends using 10 U of T4 polynucleotide kinase in 70 mM Tris–HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT and 1 mM ATP in a 10 µl final volume. Competent Escherichia coli strain DH5α was transformed with the recombinant vector using conventional heatshock techniques, and bacteria were selected on Petri dishes containing LB agar and ampicillin. Random selection of 12 colonies was undertaken, plasmid was isolated from each using the technique of alkaline lysis, and each separate preparation of plasmid DNA was digested with SacII to select those that harbored the recombinant vector containing the G-less cassette. Plasmids from the selected colonies were sequenced to identify a vector containing the G-less cassette on the transcribed strand. A colony of E.coli containing the desired vector was propagated, and the recombinant plasmid was isolated and sequenced to confirm the presence of the G-less cassette (data not shown). The recombinant vector containing the CMV immediate-early enhancer/promoter and the G-less cassette will be referred to as pCI-neo-G-less.
Figure 1.

Schematic representation for the insertion of a G-less cassette into pCI-neo to form pCI-neo-G-less. The magnified region shows the transcribed strand in the vicinity of the G-less cassette. Gray, ampicillin resistance gene; yellow, CMV immediate-early enhancer/promoter region; cyan, G-less cassette; red, transcription +1 start site.
RNA polymerase II transcription of pCI-neo-G-less
Once pCI-neo-G-less was prepared, it was necessary to ensure that it could support transcription by human RNA polymerase II. In vitro transcription reactions were carried out using the HeLa Nuclear Extract In Vitro Transcription System from Promega Corp. Each reaction contained 5 mM Tris (pH 7.8, 25°C), 6.4 mM HEPES (pH 7.9, 25°C), 44 mM KCl, 3 mM MgCl2, 88 µM EDTA, 9.4% glycerol, 60 µM PMSF, 220 µM DTT, 400 µM each of ATP, GTP and UTP, 16 µM [α-32P]CTP (25 Ci/mmol), 20 U of RNasin and 0.1 pmol template in a final volume of 25 µl. A total of 8 U of HeLa nuclear extract was added, and the reaction was incubated at 30°C for 60 min. Reactions were terminated by the addition of 175 µl of HeLa extract stop solution [0.3 M Tris–HCl (pH 7.4, 25°C), 0.3 M sodium acetate, 0.5% SDS, 2 mM EDTA, 1 µg/ml tRNA]. RNA was purified by extraction with phenol:chloroform: isoamyl alcohol (25:24:1 by volume) followed by ethanol precipitation. After centrifugation at 14 000 g for 15 min, the samples were washed with 70% ethanol and centrifuged for an additional 2 min. The samples were dried under vacuum without heat for 1 h and the RNA was resuspended in RNase-free water. Loading dye (98% formamide, 10 mM EDTA, 0.1% xylene cyanol, 0.1% bromophenol blue) was added to each sample, each was denatured at 90°C for 10 min, and the products were resolved using 7% PAGE in the presence of 8 M urea. Electrophoresis was carried out at 2000 V for ∼2 h. The gel was then dried and exposed to a Molecular Dynamics PhosphorImager. Intelligent Quantifier (BioImage, Inc., Jackson, MI) software was used to quantify the transcripts.
The results shown in Figure 2 demonstrate that pCI-neo-G-less, linearized by digestion with BbsI, supports transcription by the HeLa nuclear extract. Run-off transcripts produced from pCI-neo-G-less were 256 bases in length. A positive control DNA template from Promega Corp. was used to test for HeLa nuclear extract transcriptional activity.
Figure 2.
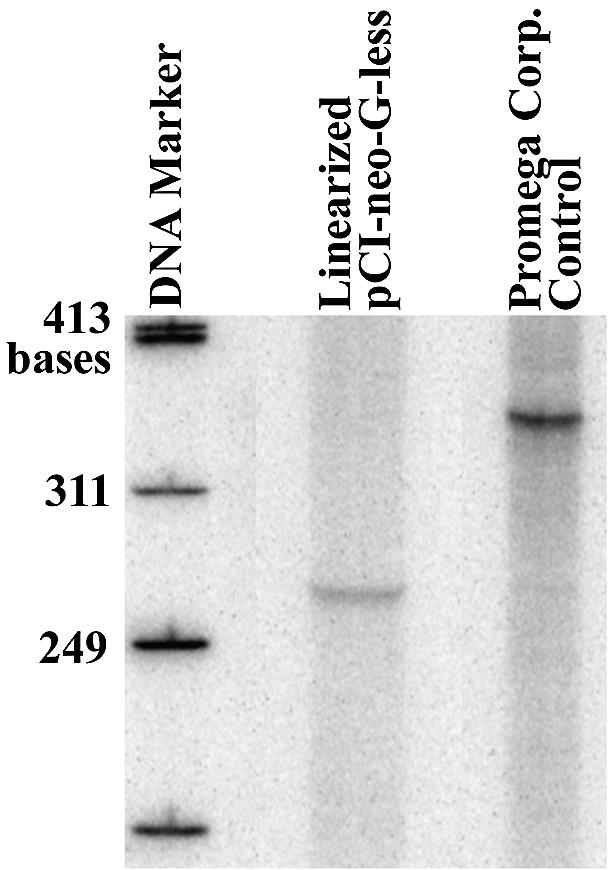
PAGE analysis of run-off transcription of pCI-neo-G-less linearized by BbsI digestion, and the control template from Promega Corp. using HeLa nuclear extract. Contents of each lane are shown above the well, and the DNA marker utilized was the φX174 DNA-Hinf I DNA ladder.
Synthesis of unmodified and site-specifically modified linear DNA templates
The general method for template production is shown in Figure 3. The overall strategy relies on ligating the CMV enhancer/promoter to a tail region that contains the site of damage. pCI-neo-G-less was incised with BbsI, producing a non-palindromic 5′-overhang four bases in length. This restriction site was selected to prevent formation of dimerized vectors. All synthetic oligomers were phosphorylated at the 5′ end using T4 polynucleotide kinase and 1 mM ATP as described earlier. To form the annealed tail region, ∼30 pmol phosphorylated 98mer, 91mer and 12mer were annealed by heating to 80°C and cooling slowly to 4°C. The annealed tail was then ligated to 10 pmol linearized pCI-neo-G-less using T4 DNA ligase. T4 DNA ligase catalyzes the formation of a phosphodiester bond between juxtaposed 3′-hydroxyl and 5′-phosphate groups in nicked, double-stranded DNA. Vector DNA was used in limiting amounts to minimize the presence of free promoter that could interfere during transcription reactions; if it were to act as a template, it would produce run-off transcripts similar in length to those that would result due to transcription complexes stalling at sites of damage. However, the purification procedure can be modified to ligate the vector and tail regions in a 2:1 ratio, respectively, in order to minimize loss of adduct-containing tail region (data not shown).
Figure 3.

Schematic of the general method for template production and purification. Gray, ampicillin resistance gene; yellow, CMV immediate-early enhancer/promoter region; cyan, G-less cassette; green, 98mer; blue, biotinylated 91mer; orange, biotin label; red, 12mer; red hexagon, adduct site.
As illustrated in Figure 3, the 91mer contained a 3′-biotin label to assist in template purification. The biotin label of the ligated template was utilized to purify the full-length, fully ligated product using Streptavidin Paramagnetic Particles (SA-PMPs). The SA-PMPs were stored at 4°C in phosphate-buffered saline, 1 mg/ml BSA and 0.02% sodium azide at a concentration of 1 mg/ml. Approximately 1 mg of SA-PMPs contains 1 nmol of biotin-binding sites. In order to maximize template recovery, 0.6 mg of SA-PMPs was used to isolate 10 pmol of ligated template, which contains 30 pmol biotin-labeled oligodeoxynucleotides. SA-PMPs were pelleted using the Magnesphere Technology Magnetic Separation Stand and washed three times with 500 µl of 0.5× SSC (75 mM NaCl, 7.5 mM sodium citrate). Non-specific DNA binding sites on the SA-PMPs were then blocked with 1 mg/ml salmon sperm DNA for 10 min at room temperature in a total volume of 500 µl, and were again washed three times with 500 µl of 0.5× SSC. The SA-PMPs were resuspended in the 200 µl volume of the template-containing ligation reaction mixture and incubated at room temperature with gentle agitation for 1 h to allow the biotinylated ligated template to associate with the SA-PMPs. The SA-PMPs were pelleted, the supernatant was removed, and the pellet was washed three times with 500 µl of wash buffer [6 mM Tris–HCl, 6 mM MgCl2 and 150 mM NaCl (pH 7.9)]. The SA-PMPs were resuspended in 90 µl of wash buffer and 100 U of BglII was added. The reaction was incubated with gentle agitation at 37°C for 1 h. BglII cuts the template 5 bases upstream from the CMV immediate-early enhancer/promoter, removing all elements of the pCI-neo-G-less that are not necessary for in vitro transcription assays with human RNA polymerase II. Removal of the extraneous vector DNA resulted in cleaner RNA samples from transcription assays (data not shown). Following BglII digestion, the SA-PMPs were pelleted, washed three times with wash buffer, and resuspended in 90 µl of wash buffer. EcoRV (100 U) was added to the suspended SA-PMPs, and the reaction mixture was incubated at 37°C for 2 h with gentle agitation. EcoRV cuts the linear DNA template 28 bases upstream from the biotin label and was utilized to release the template from the SA-PMPs, leaving a blunt end downstream of the adduct site. Hence, the biotin tag on the 91mer facilitated template capture by the SA-PMPs, and the EcoRV site was used to release the ultimate product from the beads.
Verification of DNA template integrity
In order to carry out reliable run-off transcription assays, the DNA template structure needed to be verified, and it was essential to determine that the template did not contain any nicks that could produce truncated transcripts independent of the adduct’s absence or presence. To examine construct integrity, purified templates were digested with I-PpoI, which cuts the DNA at position 900. Approximately 0.5 pmol of template was incubated with 20 U of I-PpoI in 2 mM MgCl2, 1 mM DTT, 25 mM CHES and 25 mM CAPS in a 10 µl final volume at 37°C for 2 h. Digested templates were then end labeled with 32P-phosphate using the T4 polynucleotide kinase exchange reaction. The digested template was incubated with 20 U of T4 polynucleotide kinase, and [γ-32P]ATP in 50 mM imidazole–HCl (pH 6.4), 12 mM MgCl2, 1 mM 2-mercaptoethanol and 0.07 mM ADP in a reaction volume of 20 µl at 37°C for 1 h. Digestion products were visualized on a 7% denaturing polyacrylamide gel containing 8 M urea. Figure 4a schematically illustrates the digestion products.
Figure 4.

(a) Schematic of the digestion of the template by I-PpoI to verify its integrity. Color code is the same as in Figure 3. (b) PAGE analysis of templates constructed and purified using the present protocol. Unmodified templates, as well as templates containing adducts derived from BPDE and BPhDE, are shown. The lanes are marked above each well.
We have used this procedure to produce an unmodified template using 5′-CATGCGTCCTAC-3′ as the 12mer and a modified template containing (+)-trans-anti-N2-BPDE-dG located at G* in 5′-CATGCG*TCCTAC-3′. We also examined (+)-trans-anti-N6-BPhDE-dA at A* in the sequence 5′-CTCTCA*CTTC-3′. In each case, the products were tested for the presence of unligated promoter or single-strand nicks due to incomplete ligation of the promoter. This was done by cutting the template with I-PpoI and analyzing the products by denaturing gel electrophoresis as described previously. Incomplete ligation of the promoter would produce bands 110 and 118 bases in length, whereas complete ligation would produce bands 189 and 193 bases in length. Figure 4b shows the PAGE analysis of the purified products and demonstrates that the linear templates are indeed pure and fully ligated, as evidenced by the absence of products 110 and 118 bases in length.
Human RNA polymerase II transcription of unmodified and site-specifically modified DNA templates
All templates were used in transcription reactions as described earlier. The results are shown in Figure 5. Transcription was supported by templates containing adducts from metabolically activated benzo[a]pyrene and benzo[c]phenanthrene on the transcribed strand, as well as unmodified templates, constructed and purified using our protocol. The unmodified templates produced full-length run-off transcripts, while the modified templates produced truncated transcripts, demonstrating that the bulky DNA lesions block the progression of RNA polymerase II.
Figure 5.

PAGE analysis of transcription of unmodified and damaged DNA templates using HeLa nuclear extract. Lanes are marked above each well. The arrows mark the positions of the full-length and truncated transcripts.
DISCUSSION
Our goal in designing the approach described here, for synthesizing site-specifically damaged DNA templates that are suitable for transcription, was to avoid two separate but related difficulties associated with other methods: (i) the time-consuming nature of protocols that require re-modification of a plasmid each time a new, site-specific modified sequence of DNA is to be examined and (ii) the potential for inducing spurious damage to the template during procedures that expose the DNA to ethidium bromide and/or ultraviolet radiation, both of which can alter the modified site or the DNA itself.
For example, one published method for preparing site-specifically modified transcription templates requires ligating a short, site-specifically modified, double-stranded segment of DNA into a plasmid and using CsCl equilibrium-density gradient centrifugation to purify the closed circular form (30). This technique requires exposing the product to ethidium bromide and ultraviolet light during visualization.
A second method commonly used employs derivatives of M13 DNA that contain a sequence complementary to that of a site-specific, modified oligomer (29). The modified oligomer is annealed to the M13 DNA, and double-stranded plasmid is made by extending the modified oligomer using DNA polymerase. This results in a nicked, circular duplex that is ultimately sealed with DNA ligase. The closed circular DNA is then purified using CsCl equilibrium-density gradient centrifugation, again in the presence of ethidium bromide and ultraviolet light to locate the product. While this method produces high quality DNA, albeit often in low yield, it is time consuming, and the combination of ethidium bromide and ultraviolet radiation can alter the DNA or adduct itself. But the greatest drawback is the limitation afforded by the fact that M13 DNA must be modified with a sequence of DNA complementary to the damaged oligomer, somewhat complicating the study of various modified sequences because a new M13 derivative must be made for each different oligomer.
Previously reported protocols have also employed the biotin–streptavidin interaction to purify different types of DNA templates for use in in vitro transcription assays. One report describes utilizing DNA triplex formation and psoralen crosslinking to produce templates that caused RNA polymerase II to terminate transcription upstream of the crosslinking site (32), another group synthesized immobilized unmodified transcription templates to study transcription initiation (33), and others have used a biotin–streptavidin system to isolate circular templates in order to investigate the effects of torsional constraints on transcription (34). However, our technique is unique in that it exploits this affinity for the purification of linear DNA templates containing a site-specific, stereochemically pure DNA lesion in which the base sequence context of the adduct as well as the downstream template region can easily be adapted for different purposes. Also in contrast to previously reported techniques, our approach yields templates that can be utilized in their free linear form or as an immobilized template system.
The present method for constructing and purifying site-specifically modified DNA templates is efficient, with a yield of ∼30%, relatively rapid, and avoids exposing the templates to ethidium bromide and ultraviolet light; it is also extremely versatile in that it can be adapted to suit a wide array of applications. There are two key requirements for using this system. First, the ligation site between the two elements of the template, which are the promoter and the tail region, must be non-palindromic, so that only the tail attaches downstream of the promoter, and template concatemers are precluded from forming. Secondly, a biotin tag must be added to the terminus of the tail region so that the product can be purified from unligated promoter, and a restriction site must be present so the ultimate product can be released from the biotin– streptavidin–paramagnetic particle complex.
The inclusion of a G-less cassette in the template design allows the investigator to conduct detailed kinetic analyses when only a single round of transcription is permitted to occur. By initiating transcription in the absence of GTP, RNA polymerase II becomes stalled at the end of the cassette, and transcription only proceeds upon the addition of GTP to the reaction, thus synchronizing the transcription machinery. When heparin is added prior to the addition of GTP, only one round of transcription can occur.
The method described here is amenable to making DNA templates for other applications. When the template is not released from the SA-PMPs by omitting digestion with a restriction endonuclease, it remains tethered to the paramagnetic particles, which facilitates capturing transcription complexes that remain associated with the template (T.M.Schinecker, H.Roider, R.A.Perlow, S.J.Kim and D.A.Scicchitano, unpublished results). In addition, the CMV promoter used in this protocol could be replaced by a different RNA polymerase II promoter, or even a promoter that would support transcription initiation by another RNA polymerase. Such a change would merely require situating a non-palindromic restriction site downstream from the new promoter and engineering a complementary site into the tail region.
The method reported here has already been modified to produce a DNA template containing a PAH adduct on the non-transcribed strand. It has also been used to assemble damaged DNA containing a variety of DNA lesions other than those derived from PAHs. We suspect that the protocol can accommodate almost any type of DNA damage as long as the oligomer in which it is placed can be ligated into the system. It is important that the lesion is far enough from the end of the oligomer so it does not hinder the function of DNA ligase. In addition, this system could also be easily adapted to produce site-specifically modified linear templates for experiments not related to transcription, including the preparation of DNA for studies in replication and DNA repair.
In summary, the present protocol provides a relatively rapid, efficient and versatile method for constructing and purifying site-specifically modified DNA templates. Import antly, this protocol avoids exposing the templates to ethidium bromide and ultraviolet light, two agents that are utilized in other methods for obtaining site-specifically modified DNA, but that can alter either the adduct or DNA structure itself (35). The method described here permits easier, more rapid investigation of DNA damage-induced transcription blockage, facilitating the assembly of a database that will catalog the effects of a large number of DNA adducts on transcription. Such data will enhance our understanding of arrested RNA polymerase elongation at DNA adducts, and perhaps shed light on DNA repair, mutagenesis and gene expression when DNA damage is present in actively transcribed genetic loci.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dr Suse Broyde for ongoing helpful discussions. R.A.P. would like to express appreciation for support from the American Cancer Society Postdoctoral Fellowship PF-01-108-01-CNE. This work was supported by grants from the NIH: ES10581 (to D.A.S.) and CA76660 (to N.E.G.).
REFERENCES
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington D.C.
- 2.Scicchitano D.A. and Mellon,I. (1997) Transcription and DNA damage: a link to a kink. Environ. Health Perspect., 105 (Suppl. 1), 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tornaletti S. and Hanawalt,P.C. (1999) Effect of DNA lesions on transcription elongation. Biochimie, 81, 139–146. [DOI] [PubMed] [Google Scholar]
- 4.Kunkel T.A. and Bebenek,K. (2000) DNA replication fidelity. Annu. Rev. Biochem., 69, 497–529. [DOI] [PubMed] [Google Scholar]
- 5.Friedberg E.C., Wagner,R. and Radman,M. (2002) Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science, 296, 1627–1630. [DOI] [PubMed] [Google Scholar]
- 6.Conaway J.W. and Conaway,R.C. (1999) Transcription elongation and human disease. Annu. Rev. Biochem., 68, 301–319. [DOI] [PubMed] [Google Scholar]
- 7.Hanawalt P.C. (2000) DNA repair. The bases for Cockayne syndrome. Nature, 405, 415–416. [DOI] [PubMed] [Google Scholar]
- 8.Bohr V.A., Brosh,R.M.,Jr, von Kobbe,C., Opresko,P. and Karmakar,P. (2002) Pathways defective in the human premature aging disease Werner syndrome. Biogerontology, 3, 89–94. [DOI] [PubMed] [Google Scholar]
- 9.Svejstrup J.Q. (2002) Mechanisms of transcription-coupled DNA repair. Nature Rev. Mol. Cell Biol., 3, 21–29. [DOI] [PubMed] [Google Scholar]
- 10.Batty D.P. and Wood,R.D. (2000) Damage recognition in nucleotide excision repair of DNA. Gene, 241, 193–204. [DOI] [PubMed] [Google Scholar]
- 11.Kraemer K.H. (1994) Nucleotide excision repair genes involved in xeroderma pigmentosum. Jpn J. Cancer Res., 85, inside front cover. [PubMed] [Google Scholar]
- 12.van Steeg H. and Kraemer,K.H. (1999) Xeroderma pigmentosum and the role of UV-induced DNA damage in skin cancer. Mol. Med. Today, 5, 86–94. [DOI] [PubMed] [Google Scholar]
- 13.Rapin I., Lindenbaum,Y., Dickson,D.W., Kraemer,K.H. and Robbins,J.H. (2000) Cockayne syndrome and xeroderma pigmentosum. Neurology, 55, 1442–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugasawa K., Ng,J.M., Masutani,C., Iwai,S., van der Spek,P.J., Eker,A.P., Hanaoka,F., Bootsma,D. and Hoeijmakers,J.H. (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell, 2, 223–232. [DOI] [PubMed] [Google Scholar]
- 15.Sugasawa K., Okamoto,T., Shimizu,Y., Masutani,C., Iwai,S. and Hanaoka,F. (2001) A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev., 15, 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wood R.D., Mitchell,M., Sgouros,J. and Lindahl,T. (2001) Human DNA repair genes. Science, 291, 1284–1289. [DOI] [PubMed] [Google Scholar]
- 17.Araujo S.J., Tirode,F., Coin,F., Pospiech,H., Syvaoja,J.E., Stucki,M., Hubscher,U., Egly,J.M. and Wood,R.D. (2000) Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev., 14, 349–359. [PMC free article] [PubMed] [Google Scholar]
- 18.Balajee A.S. and Bohr,V.A. (2000) Genomic heterogeneity of nucleotide excision repair. Gene, 250, 15–30. [DOI] [PubMed] [Google Scholar]
- 19.Cleaver J.E., Karplus,K., Kashani-Sabet,M. and Limoli,C.L. (2001) Nucleotide excision repair ‘a legacy of creativity’. Mutat. Res., 485, 23–36. [DOI] [PubMed] [Google Scholar]
- 20.Amundson S.A., Patterson,A., Do,K.T. and Fornace,A.J.,Jr (2002) A nucleotide excision repair master-switch: p53 regulated coordinate induction of global genomic repair genes. Cancer Biol. Ther., 1, 145–149. [DOI] [PubMed] [Google Scholar]
- 21.Mellon I., Bohr,V.A., Smith,C.A. and Hanawalt,P.C. (1986) Preferential DNA repair of an active gene in human cells. Proc. Natl Acad. Sci. USA, 83, 8878–8882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bohr V.A. (1987) Preferential DNA repair in active genes. Dan. Med. Bull., 34, 309–320. [PubMed] [Google Scholar]
- 23.Mellon I., Spivak,G. and Hanawalt,P.C. (1987) Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell, 51, 241–249. [DOI] [PubMed] [Google Scholar]
- 24.Mellon I. and Hanawalt,P.C. (1989) Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature, 342, 95–98. [DOI] [PubMed] [Google Scholar]
- 25.Bohr V.A. (2002) DNA damage and its processing. relation to human disease. J. Inherit. Metab. Dis., 25, 215–222. [DOI] [PubMed] [Google Scholar]
- 26.Henning K.A., Li,L., Iyer,N., McDaniel,L.D., Reagan,M.S., Legerski,R., Schultz,R.A., Stefanini,M., Lehmann,A.R. and Mayne,L.V. (1995) The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell, 82, 555–564. [DOI] [PubMed] [Google Scholar]
- 27.Selby C.P. and Sancar,A. (1997) Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem., 272, 1885–1890. [DOI] [PubMed] [Google Scholar]
- 28.Kamiuchi S., Saijo,M., Citterio,E., de Jager,M., Hoeijmakers,J.H. and Tanaka,K. (2002) Translocation of Cockayne syndrome group A protein to the nuclear matrix: possible relevance to transcription-coupled DNA repair. Proc. Natl Acad. Sci. USA, 99, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szymkowski D.E., Lawrence,C.W. and Wood,R.D. (1993) Repair by human cell extracts of single (6-4) and cyclobutane thymine-thymine photoproducts in DNA. Proc. Natl Acad. Sci. USA, 90, 9823–9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donahue B.A., Yin,S., Taylor,J.S., Reines,D. and Hanawalt,P.C. (1994) Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc. Natl Acad. Sci. USA, 91, 8502–8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tornaletti S., Maeda,L.S., Lloyd,D.R., Reines,D. and Hanawalt,P.C. (2001) Effect of thymine glycol on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. J. Biol. Chem., 276, 45367–45371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z. and Rana,T.M. (1997) DNA damage-dependent transcriptional arrest and termination of RNA polymerase II elongation complexes in DNA template containing HIV-1 promoter. Proc. Natl Acad. Sci. USA, 94, 6688–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arias J.A. and Dynan,W.S. (1989) Promoter-dependent transcription by RNA polymerase II using immobilized enzyme complexes. J. Biol. Chem., 264, 3223–3229. [PubMed] [Google Scholar]
- 34.Bentin T. and Nielsen,P.E. (2002) In vitro transcription of a torsionally constrained template. Nucleic Acids Res., 30, 803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida H., Swenberg,C.E. and Geacintov,N.E. (1987) Kinetic flow dichroism study of conformational changes in supercoiled DNA induced by ethidium bromide and noncovalent and covalent binding of benz[a]pyrene diol epoxide. Biochemistry, 26, 1351–1358. [DOI] [PubMed] [Google Scholar]