


Abstract
The current model for influenza virus mRNA transcription involves the sequential interaction of the viral polymerase with the 5′- and 3′-ends of vRNA, with each RNA–protein interaction triggering a polymerase function necessary for cap-primed transcription. Here we show that the order in which this ternary complex is assembled is in fact important. Polymerase bound simultaneously to a pre-annealed duplex of the 5′- and 3′-ends of vRNA had greatly increased levels of primer binding and endonuclease activities compared to a sequentially assembled complex. Increased primer binding was due to the activation of a high affinity binding site with a preference for primer length RNAs. This correlated with enhanced levels of cap-primed transcription. Polymerase that was bound initially to just 5′ vRNA had low primer binding activity, but was endonucleolytically active. Neither activity was significantly increased by the subsequent addition of 3′ vRNA, and this sequentially assembled complex had correspondingly low mRNA transcription activity. Nevertheless, both routes of assembly led to complexes that were highly competent for dinucleotide ApG-primed transcription. Therefore, polymerase complexes assembled on pre-annealed 5′ and 3′ terminal viral RNA sequences have distinct properties from those assembled by sequential loading of polymerase onto the 5′-end followed by the 3′-end. This suggests a mechanism by which the virus couples transcription initiation and termination during mRNA transcription.
INTRODUCTION
Transcription of influenza virus mRNA is initiated using a capped 10–15 nt RNA primer endonucleolytically scavenged from the 5′-end of host hnRNA and is terminated prematurely 15–18 nt from the 5′-terminus of the template vRNA. The mRNA is also polyadenylated in a template-directed reaction catalysed by influenza polymerase. In contrast, synthesis of the complementary replication intermediate (cRNA) is initiated without a primer, is not prematurely terminated and lacks a 3′ poly(A) tail (1). The polymerase complex is composed of three subunits (PB1, PB2 and PA). PB1 binds specifically to the conserved terminal sequences of the vRNA and cRNA and performs the active polymerase and RNA endonuclease activities (2–4). PB2 binds to the 5′ caps of host cell mRNAs (5,6). PA stimulates the template binding activity of PB1 and is essential for polymerase activity (7–9). PA mutants show selective defects in both vRNA and mRNA synthesis (10,11). A recent report showed that recombinant sub-complexes composed of PB1 and PB2 or PB1 and PA have distinct transcriptase and replicase activities, respectively (12). However, previous attempts to demonstrate this have concluded that all three P proteins were required for significant levels of polymerase activity (8,9,13).
The viral genome consists of eight segments of single-strand negative polarity vRNA with highly conserved partially complementary termini. The termini of vRNA modulate the transcriptional activities of the influenza virus polymerase. The polymerase binds tightly to the terminal 11 nt of the 5′-end of vRNA (5′ vRNA) (14,15). This stimulates the cap binding activity of PB2 (16,17) and increases the affinity of the polymerase for the 3′-end of vRNA (3′ vRNA). Binding to both the 5′- and 3′-ends of vRNA is required for full activation of the viral endonuclease activity. Binding to 5′ cRNA also stimulates cap binding, but binding to both ends of cRNA fails to activate the endonuclease (17,18). The 5′- and 3′-ends of vRNA can base pair to form a panhandle structure (19), but the functional significance of this is uncertain. An NMR structure of a short vRNA analogue indicates an extensively base paired bulged stem (20). However, mutagenesis studies suggest that both conserved terminal sequences must be able to adopt a very different secondary structure known as the ‘corkscrew’ configuration for full polymerase activity (21–23). Synthetic vRNA less than 100 nt long forms the panhandle structure spontaneously in solution. However, longer vRNA molecules and isolated vRNPs require the presence of the polymerase to maintain this structure (24).
The current model of influenza virus mRNA transcription proposes that the polymerase binds to the 5′- and 3′-ends of the same vRNA template (cis binding). The polymerase migrates away from the 3′-end, but remains tightly bound to the 5′-end of vRNA. When it reaches a stretch of 6–8 uridine residues, adjacent to the 5′-end binding site still occupied by the polymerase, it encounters a steric block and further elongation is prevented. Non-processive copying of the U stretch results in reiterative incorporation of adenosine residues, thus explaining premature termination and polyadenylation (1,25). It has been suggested replication might be performed by polymerase bound to linear forms of template vRNAs in trans (15,25), which would not be subject to this steric blockade. It is unclear from this model how the mode of transcription initiation and termination would be coordinated to prevent the excessive formation of nonsense transcripts such as uncapped, polyadenylated or capped, non-polyadenylated RNA.
Previously, we reported the development of a versatile in vitro assay for studying the transcriptional activity of the influenza virus RNA polymerase (8). The assay exploits the high affinity of recombinant vaccinia virus-expressed influenza polymerase for a synthetic RNA oligonucleotide corresponding to the 5′-end of vRNA. This RNA is used as a ligand to immobilise the polymerase onto paramagnetic beads. Binding is essentially irreversible over the duration of the assay and, importantly, the bead-immobilized polymerase is active when supplemented with a synthetic 3′ vRNA template. We have now used this assay to show that complexes assembled on pre-annealed 5′ and 3′ terminal viral RNA sequences have distinct properties from those assembled by sequential loading of polymerase onto the 5′-end and then the 3′-end.
MATERIALS AND METHODS
The preparation of the influenza virus polymerase using recombinant vaccinia virus expression vectors (Vac3P) and the control extract (VacT7) have been described previously (8).
RNAs specific to this study
RNAs were produced by run-off transcription using MboII-linearised templates and gel purified as described previously (8). The RNA oligonucleotide, 5′v16B, was synthesised by Cruachem. Sequences of the individual RNAs were as follows, where the underlined letters correspond to the conserved 5′ and/or 3′ terminal vRNA sequences: 5′v16B, 5′-AGUAGAAACAAGGGUG(CH2)12-biotin; PHV, 5′-AGUAGAAACAAGGGUGUUUUUUCCCGGGAATTCGGATCCACACCCUGCUUUUGCU-3′; v34, 5′-GGAGACGAAUUCGGAUCCACACCCUGCUUUUGCU-3′; v41, 5′-AGGAGACGCCCGGGAAUUCGGAUCCACACCCUGCUUUUGCU-3′. Radiolabelled G20 and G11 RNAs were prepared by treatment of synthetic RNA oligonucleotides (pppm2GAAUACUCAAGOH and pppm2GAAUACUCAAGCUAUGCAUCOH, respectively) with vaccinia virus guanylyltransferase/m7-transferase (26) and [α-32P]GTP (3000 Ci/mM; Amersham). This results in the formation of 5′ terminally labelled RNA with a m7Gpppm2G (cap 1) structure that is recognised efficiently by the influenza virus RNA polymerase. G20 is a substrate for the viral endonuclease and is cleaved predominantly after the indicated G residue to produce a product identical to G11. A minor cleavage occurs at the highlighted G residue 5 bases further away from the cap to produce what is referred to as the G16 product. Synthesis and characterisation of capped RNA molecules were as described previously (27). 5′-Biotinylated PHV RNA was synthesized as follows. An aliquot of 5 nmol (∼100 µg) of purified PHV RNA was dephosphorylated using calf intestinal phosphatase and then treated with T4 polynucleotide kinase and ATPγS, which resulted in the transfer of a phosphorothioate group to the 5′-end. This RNA was then incubated with 2 mM EZ-Link iodoacetyl-LC-biotin (Pierce) in 90 mM potassium phosphate buffer, pH 8.0, for 1 h at 50°C. Unincorporated biotinylation reagent was removed by three passes through a 0.5 ml Vivaspin concentrator (3000 Da cut-off) (Sartorius Group).
Immobilised polymerase cap binding and in vitro transcription assays
A detailed description of the bead assay protocol is reported in Lee et al. (8). Briefly, streptavidin-coated paramagnetic beads were coated in 1 pmol of the biotinylated RNA oligonucleotide (5′v16B) per reaction. Pre-annealing of the 5′- and 3′-ends was achieved by heating 5′v16B with a 5-fold molar excess of 3′ vRNA (v34) in 10 mM Tris, pH 7.6, 50 mM NaCl, allowing it to cool slowly to room temperature and then binding it to the beads. The charged beads were washed thoroughly and then incubated with 5 µg of nuclear extract containing the polymerase, followed by a final wash. Western blot samples were subjected to 3%/7.5% discontinuous SDS–PAGE, electroblotted onto PVDF membranes and analysed using polyclonal rabbit antisera specific for the PB1 and PB2 proteins (8). Transcription reactions were resuspended in 10 µl transcription buffer containing the appropriate primer, nucleotides and template [typically, 50 mM Tris, pH 8.2, 100 mM KCl, 2 mM MgCl2, 100 ng tRNA, 10 mM DTT, 10 pmol 3′-end template (referred to as the 3′v ligand), 5 µCi [α-32P]GTP (400 Ci/mmol; Amersham Pharmacia Biotech), 0.8 mM ATP, 0.4 mM CTP, 0.2 mM UTP, 1 µM GTP and 0.5 mM ApG (Sigma-Aldrich) or 20 ng globin mRNA (Gibco BRL)]. Reactions were incubated at 30°C for 30 min then analysed by 18% (19:1 acrylamide:bis-acrylamide), 7 M urea PAGE. Primer binding assay samples were incubated in 50 mM Tris, pH 7.6, 100 mM KCl, 2 mM MgCl2, 0.25% Triton N101, 100 ng tRNA, 10 mM DTT (or for Mg2+-free reactions substitute 3.75 mM EDTA for MgCl2) supplemented with 1 × 104 c.p.m. (∼100–200 fmol) of radiolabelled G20 or G11 RNA. Reactions were incubated at room temperature for 10 min unless otherwise indicated. After the completion of the binding reaction, the beads were washed to remove unbound probe and analysed by 7 M urea PAGE. For the dissociation studies, the purified polymerase was incubated for 15 min with cap-labelled G20 RNA as above. The beads were then washed and diluted 2-fold into fresh, capped RNA-free buffer, supplemented with 1 mM m7GpppG cap analogue (New England Bioloabs) to prevent rebinding of dissociated RNA. Samples were removed from the reaction, separated into bound and free fractions and analysed by scintillation counting. Sigma Plot 2001 was used for data analysis. The data were fitted to equations 1 and 2
B = c + Afast × exp(–kfast × t) + Aslow × exp(–kslow × t)1
R = c + Afast × [1 – exp(–kfast × t)] + Aslow × [1 – exp(–kslow × t)]2
where B is the relative amount of bound RNA, R is the relative amount of released RNA, c is the relative amount of RNA remaining bound at infinite time, Afast and Aslow are the amplitudes of the fast and slow dissociation phases, kfast and kslow are the relative rate constants and t is the incubation time after dilution.
RESULTS
Activation of capped primer binding and endonuclease by 5′ and 3′ vRNA sequences
The bead assay that we have developed is particularly suited to study the process of polymerase activation because of the ease with which the reaction components can be manipulated in a sequential manner. Beads were ‘charged’ by coating them in three different ways: (i) with the 5′-end alone (referred to as the 5′v ligand) using 5′v16B; (ii) with a 5′-biotinylated RNA molecule containing both the 5′ and 3′ vRNA sequences that are predicted to form a partially double-stranded panhandle structure in solution (PHV ligand); (iii) with separate RNAs for the 5′- and 3′-ends that had been pre-annealed together before binding to the bead and washed thoroughly such that any 3′-end that remained associated with the beads did so through base pairing with the biotinylated 5′v16B RNA (5′v3′v ligand). The charged beads were incubated with the polymerase extract under conditions optimised for specific binding of the polymerase and then washed to remove any unbound proteins. To study the interaction with capped RNA, a 20 nt, 32P-cap-labelled RNA molecule (G20, Fig. 1A, lane 2) was synthesised in vitro. The polymerase-coated beads were incubated with the G20 RNA, washed and then analysed by denaturing PAGE to determine the G20 binding and endonuclease activities of the bound polymerase. G20 RNA is cleaved rapidly by influenza virus endonuclease to generate a major 11 nt and a minor 16 nt product (27,28), which will be referred to as ‘primer length’ products. Thus, the level of primer binding when the endonuclease is active is indicated by the sum of the G20, G11 and G16 bands. The polymerase that was bound to the 5′v ligand alone (5′v/Pol, Fig. 1A, lane 3) bound very little of the labelled capped primer RNA, whereas the polymerase that was bound to the PHV ligand containing both 5′ and 3′ vRNA sequences (PHV/Pol, Fig. 1A, lane 7) bound substantially more capped primer RNA. We then assessed how the 3′ vRNA sequences contributed to this effect. Supplementing the reaction with 3′ vRNA (v34) at the same time as the G20 RNA had very little effect on the level of capped primer binding (Fig. 1A, lane 5). Incubating the v34 RNA with the 5′v/Pol for 10 min prior to adding G20 RNA led to a small increase in capped primer binding (Fig. 1A, lane 4). Full capped primer binding by polymerase was only obtained by pre-annealing 5′ and 3′ vRNAs prior to polymerase binding (Fig. 1A, lane 6). This would be expected to closely mimic the conformation of the termini of the PHV ligand. Notably, in all cases, the majority of the G20 RNA was cleaved into the G11 and G16 products. The trivial explanation that the different ligands bound different amounts of polymerase was discounted after quantitative western blot analysis comparing the amount of PB1 and PB2 bound by the beads showed that the amounts of polymerase bound by the 5′v and 5′v3′v ligands were identical (Fig. 1B, lanes 6 and 7). Addition of capped primer RNA had no detectable effect on the level or composition of the polymerase bound to the 5′v or 5′v3′v ligands (data not shown). The signals on the blots were within the quantitative range of the assay, as shown by the serial dilution of total nuclear extract (Fig. 1B, lanes 1–5). No cross-reactive bands were evident in the negative control sample composed of 5′v16B charged beads incubated with a nuclear extract prepared from VacT7-infected cells (Fig. 1B, lane 8). These results demonstrated that using a duplex RNA ligand at the time of polymerase binding had a substantial influence on the capped primer binding activity of the resultant complex that was not off-set by supplying the 3′-end sequences at a later stage.
Figure 1.
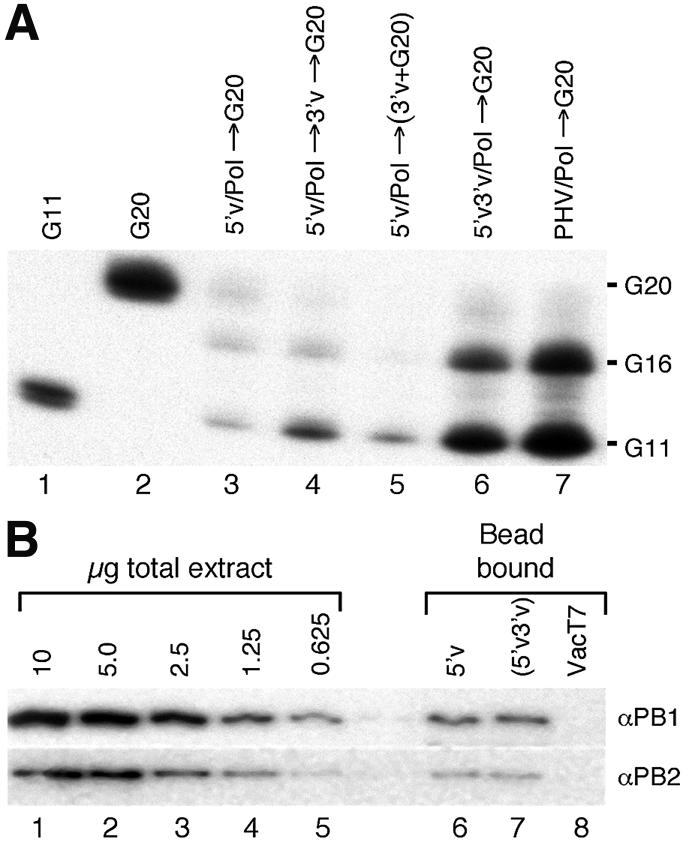
Primer binding by different polymerase/template combinations. (A) 5′v/Pol (lanes 3–5), 5′v3′v/Pol (lane 6) or polymerase bound to 5′- biotinylated PHV RNA (lane 7) were incubated with a mRNA analogue (G20 RNA, lane 2). The beads were washed and the bound G20 was analysed by PAGE. Lane 1, G11 marker; lane 3, 5′v/Pol in the absence of 3′ vRNA; lane 4, effect of incubating 5′v/Pol for 10 min with 3′ vRNA and washing prior to incubating with G20; lane 5, effect of including 3′ vRNA during the G20 binding reaction. Exposure was for 40 h. (B) Western blot to determine the relative amounts of PB1 (upper panel) and PB2 (lower panel) bound to magnetic beads coated with 5′v (lane 6) or with pre- annealed 5′v3′v ligand (lane 7). Lanes 1–5, 2-fold dilutions of nuclear extract from cells infected with Vac3P: lane 8, negative control using nuclear extract from cells expressing T7 RNA polymerase (VacT7) instead of influenza polymerase.
Previously, capped RNA binding has mostly been determined in the absence of Mg2+ (16). In order to characterise further the relationship between capped RNA binding and endonuclease activities of influenza polymerase and the influence of genomic RNA secondary structure on these activities, the capped primer RNA binding assay was performed in the presence or absence of Mg2+. Figure 2A shows the influence of Mg2+ on the stability of capped primer binding by the polymerase bound to 5′v3′v (upper panel) or 5′v alone (lower panel). Comparing the levels of capped primer RNA bound by the polymerase showed that in the presence of Mg2+, the 5′v3′v/Pol had at least 6-fold higher capped primer binding activity than 5′v/Pol (Fig. 2A, lane 1, compare top and bottom panels; see Fig. 2B for replicate data). Greater than 95% of the G20 RNA bound by 5′v3′v/Pol was cleaved to the G11 and G16 products during the 10 min binding reaction. The interaction of the cleaved products with polymerase was stable, as they remained associated with the polymerase bound fraction (B) during a 30 min incubation in fresh primer-free buffer at 30°C (Fig. 2A, lanes 3 and 4, upper panel). Approximately 75% of the G20 RNA bound by 5′v/Pol was also cleaved, despite the absence of the 3′ vRNA, and remained stably bound to the polymerase. The remaining 25% of uncleaved G20 was not stably bound and eluted in the supernatant (F) during the chase period (Fig. 2A, lanes 1, 3 and 4, lower panel). The majority of this uncleaved RNA was later confirmed to be non-specifically associated with the beads (see Fig. 3A and B, lanes 6).
Figure 2.

Cap binding and endonuclease activity of influenza polymerase bound to 5′v or 5′v3′v ligand. (A) Immobilised polymerase (5′3′v/Pol, upper panel; 5′v/Pol, lower panel) was incubated with G20 or G11 RNA. Lane 1, G20 RNA bound by polymerase; lane 2, as for lane 1, but in the absence of Mg2+; lanes 3 and 4, binding reaction was transferred to binding buffer lacking labelled RNA and incubated for 30 min, followed by separation into polymerase-bound (B) and free (F) fractions; lanes 5 and 6, as for lanes 3 and 4, but in the absence of Mg2+; lanes 7–12 are equivalent to lanes 1–6, except that G11 RNA was used as capped RNA ligand. The band intensities on the gel were measured using NIH Image and the relative fold increase in cap binding for 5′v3′v/Pol compared to 5′v/Pol is indicated in the box beneath. Exposure was for 40 h. (B) Fold increase in capped RNA binding displayed by 5′v3′v/Pol relative to 5′v/Pol. The graph shows the means and standard deviations from six separate experiments using G20 and G11 capped RNAs.
Figure 3.

Association and dissociation kinetics of the polymerase/cap binding reaction. Time course of G20 RNA binding by 5′v3′v/Pol (A) and 5′v/Pol (B). Lanes 1–5 correspond to samples removed from the RNA binding reaction at the times indicated above the lanes; lane 6, non-specific binding by beads incubated with a VacT7 control extract; lane 7, G20 marker lane. (C) Time course of G20 (squares) and G11 (triangles) binding by 5′v3′v/Pol (open shapes) or 5′v/Pol (filled shapes). Values are for percentage of total input probe bound by the polymerase. (D) Dissociation of radiolabelled G20 capped RNA from influenza polymerase immobilised on 5′v (squares) or 5′v3′v ligand (circles). Filled black and grey symbols show the proportion of capped RNA remaining bound to the polymerase at various time points after transfer. White symbols show the relative amount of capped RNA released into the free fraction during the time course. For clarity, only the fit to 5′v3′v-bound polymerase data is shown. The calculated rates are shown in Table 1.
In the absence of Mg2+ (Fig. 2A, lane 2) the overall level of capped primer binding was reduced for both complexes (compare total band densities in Fig. 2A, lanes 1 and 2). No cleavage of G20 RNA was observed for these reactions because the viral endonuclease is dependent upon Mg2+ for activity. The levels of capped primer binding by 5′v3′v/Pol and 5′v/Pol converged to within 2-fold of each other because of a disproportionately large decrease in the level of capped primer binding by the 5′v3′v/Pol complex compared to a small decrease for the 5′v/Pol complex. Approximately 70% of the G20 RNA dissociated from the polymerase during the 30 min chase period (Fig. 2A, lanes 5 and 6). These results demonstrate that the association of the polymerase with capped primer RNA was significantly less stable in the absence of Mg2+. This could be interpreted to mean that capped RNA must be cleaved to primer length to allow stable interaction with polymerase or it could be interpreted as showing a more direct dependence upon Mg2+ for stable primer binding.
In order to clarify the situation, the same binding experiment was performed using G11 RNA, which is analogous to the 11 nt endonuclease cleavage product. This experiment is therefore measuring binding that occurs independent of endonuclease activity. In the presence of Mg2+, the binding of G11 to 5′v3′v/Pol was ∼7-fold higher than for 5′v/Pol (Fig. 2A, lane 7), very similar to the situation with G20 RNA (Fig. 2A, lane 1, and B). The G11 RNA remained associated with the polymerase during the 30 min incubation in primer-free buffer (Fig. 2A, lanes 9 and 10). Initial binding of G11 RNA was increased in the absence of Mg2+ for both polymerase forms (Fig. 2A, lane 8). A fraction of this RNA dissociated in the 30 min follow-up period (Fig. 2A, lanes 11 and 12). Importantly, the amount of G11 RNA that remained bound in the absence of Mg2+ was similar to the amount of stably bound RNA in the presence of Mg2+ (Fig. 2A, lanes 9 and 11). This was different from the situation with G20 RNA, where significantly less bound to polymerase in the absence of Mg2+ as compared to the presence of Mg2+ (Fig. 2A, lanes 3 and 5). This suggested that the affinity of the polymerase was greater for primer length G11 RNA than it was for the substrate length G20 RNA. Under all conditions tested, the binding to G11 RNA was significantly higher with 5′v3′v bound polymerase as compared to 5′v bound polymerase (Fig. 2A, compare upper and lower panels).
Capped RNA binding kinetics of the 5′v and 5′v3′v bound polymerase complexes
Under equilibrium binding conditions, an increase in primer binding activity could be achieved through an increased association rate, a decreased dissociation rate or by a shift in the equilibrium between a non-binding- and a binding-competent conformation of the polymerase complex, triggered by the simultaneous interaction with both 5′ and 3′ vRNA. To differentiate between these possibilities, we first evaluated the kinetics of G20 RNA binding. Figure 3A and B shows that the amount of bound, capped RNA reached a plateau after 30 min for both 5′v3′v/Pol and 5′v/Pol, but the level of binding was much lower for 5′v/Pol. Between 70 and 90% of the bound RNA was cleaved to primer length by both complexes. The small amount of G20 that was not cleaved was essentially constant for all reactions, and from comparison with the amount of G20 RNA bound in the absence of the influenza polymerase (Fig. 3A and B, lane 6) it is likely that a proportion of this uncleaved RNA is non-specifically associated with the beads. Thus, neither the association rates nor RNA cleavage rates were sufficiently different between the two forms of polymerase to explain the difference in capped RNA binding at equilibrium. Figure 3C shows graphically the binding of G20 RNA (squares) and G11 RNA (triangles) to the 5′v/Pol and 5′v3′v/Pol complexes over time. Both probes were bound to a significantly greater extent by the 5′v3′v/Pol complex (open symbols) compared to the 5′v/Pol complex (filled symbols). The majority of the binding occurred within the first 5 min of the reaction and reached a plateau by 15–25 min. Consistent with the rapid endonucleolytic cleavage shown in Figure 3A, no significant difference in binding profiles was seen between the G20 and G11 probes.
Figure 3D shows a graphical representation of data for the dissociation of capped RNA from 5′v/Pol and 5′v3′v/Pol. Dissociation was measured either as the loss of polymerase-associated radioactivity or as the increase in released radioactivity with identical results. The data show a biphasic dissociation pattern and were fitted to a double exponential binding model, i.e. a combination of fast and slow phases of RNA dissociation. Very similar time courses and dissociation rates for each of the two phases were obtained for both forms of polymerase (Table 1). The amplitudes for both phases were not significantly different, and the majority of the RNA dissociated during the slow phase. The virtually identical dissociation patterns suggest that the capped RNA that was bound by the 5′v or 5′v3′v selected polymerase dissociated through the same mechanism. This implies that the capped primer binding complex is the same in both cases, but is present in different amounts.
Table 1. Dissociation of capped RNA from influenza polymerase–RNA complexes.
| kfast (min–1) | kslow (min–1) | Afast | Aslow | |
|---|---|---|---|---|
| 5′v3′v | 0.096 ± 0.066a | 0.0061 ± 0.0039a | 0.21 ± 0.08b | 0.49 ± 0.14b |
| 5′v | 0.126 ± 0.084 | 0.0078 ± 0.0041 | 0.20 ± 0.05 | 0.45 ± 0.11 |
aRNA dissociation rates for the fast and slow phases of the biphasic process were determined from two independent experiments, performed in duplicate, and from the analysis of released RNA as well as from the analysis of RNA remaining bound to the polymerase complex.
bAfast and Aslow are the amplitudes of the biphasic dissociation curve. They represent the relative fractions of RNA released during the fast and the slow phase, respectively.
Effect of assembly order on cap-primed transcription
The transcriptional activity of the 5′v/Pol and 5′v3′v/Pol complexes was determined by analysing the incorporation of [α-32P]GTP into the full-length product when the reactions were supplemented with nucleotides and globin mRNA as a source of capped RNA primer. The levels of activity were then compared to those obtained for reactions primed with the dinucleotide ApG, which functions as an efficient uncapped primer for the influenza virus polymerase. Globin mRNA is cleaved by the polymerase to produce a 12 nt long primer. Consequently, cap-primed transcription of the v34 3′v template RNA results in the production of a 46 nt product (34 + 12 nt). ApG is complementary to the first two bases of the 3′v template RNA and primes transcription initiation at position 3 to produce a 34 nt product from v34 RNA. 5′v/Pol showed only low levels of globin mRNA-primed synthesis when 10 pmol of the v34 template was added in combination with the primer RNA at the start of the transcription reaction (Fig. 4A, lane 3). Preincubation of the 5′v/Pol complex with v34 for 10 min prior to adding the primer and initiation of transcription resulted in a small increase in the level of transcription (Fig. 4A, lane 2). Both of these assembly orders resulted in high levels of ApG-primed transcription (Fig. 4A, lanes 5 and 6), showing that the sequentially assembled complex was transcriptionally competent and able to efficiently bind the 3′ vRNA template. In contrast, the 5′v3′v/Pol complex showed markedly increased levels of cap-primed transcription (Fig. 4A, lane 1) but reduced levels of ApG-primed transcription (Fig. 4A, lane 4). The increased electrophoretic mobility of the ApG-primed product is an artefact due to the lower level of v34 template RNA present in this reaction (1 pmol compared to 10 pmol, assuming a 1:1 ratio of 5′v:3′v in the 5′v3′v reactions). Addition of a further 9 pmol of v34 prior to electrophoresis eliminates this gel migration artefact (data not shown). Why the product of ApG-primed transcription was a doublet is not known (Fig. 4A, lane 4; it is also present but not apparent in lanes 5 and 6 because of the greater signal intensity). It is probably due to heterogeneity at the 5′-end of the T7 transcribed v34 template (29), as we have found its prevalence varies from one template preparation to another. For this reason we think it unlikely to be due to terminal transferase activity that is apparently common to non-proofreading polymerases (30). These results demonstrated that the polymerase initiated cap-primed transcription more efficiently on a template that was already annealed to the 5′-end than when the template was supplied after the polymerase had bound to the 5′-end, a result consistent with the enhanced primer binding activity observed under those circumstances. Polymerase that was bound to pre-annealed RNA generated significantly lower amounts of ApG-primed transcripts (Fig. 4B, lane 4). This was ascribed to the amount of v34 RNA limiting the ApG-primed reaction on pre-annealed template (1 pmol v34) as compared to sequentially added 3′ vRNA (10 pmol v34). To confirm this interpretation, a mixed template reaction was tested where the polymerase was bound to the pre-annealed 5′v16B/v34 RNA-coated beads, but the reaction was then supplemented with 9 pmol of a second template (v41) that had the identical sequence at the 3′-end but was extended by 7 nt at the 5′-end to allow the products from the two templates to be differentiated. In the absence of v41, the only products were the 34 nt ApG-primed doublet (Fig. 4B, lane 1) and the 46 nt cap-primed product (Fig. 4B, lane 2). When both v34 and v41 were present, a highly abundant 41 nt ApG-primed product was observed, whereas the level of the 34 nt product from v34 was not significantly affected (Fig. 4B, lane 3). This showed that the second template (v41) was able to associate efficiently with the polymerase attached to the beads. However, the v41 template was utilised very inefficiently for cap-primed synthesis as evidenced by the relatively small amount of the 53 nt product resulting from globin-primed synthesis on this template (Fig. 4B, lane 4, upper band). The level of the 46 nt capped product from the pre-annealed 3′v template in the mixed reaction was essentially unaffected by the inclusion of v41. These results confirm that the template that was pre-annealed to the 5′-end prior to binding the polymerase was preferentially used for cap-primed synthesis and that the reduced level of ApG-primed synthesis on the pre-annealed template was due to limited template availability rather than an intrinsic difference in the ApG priming activity of the 5′v/Pol complex.
Figure 4.
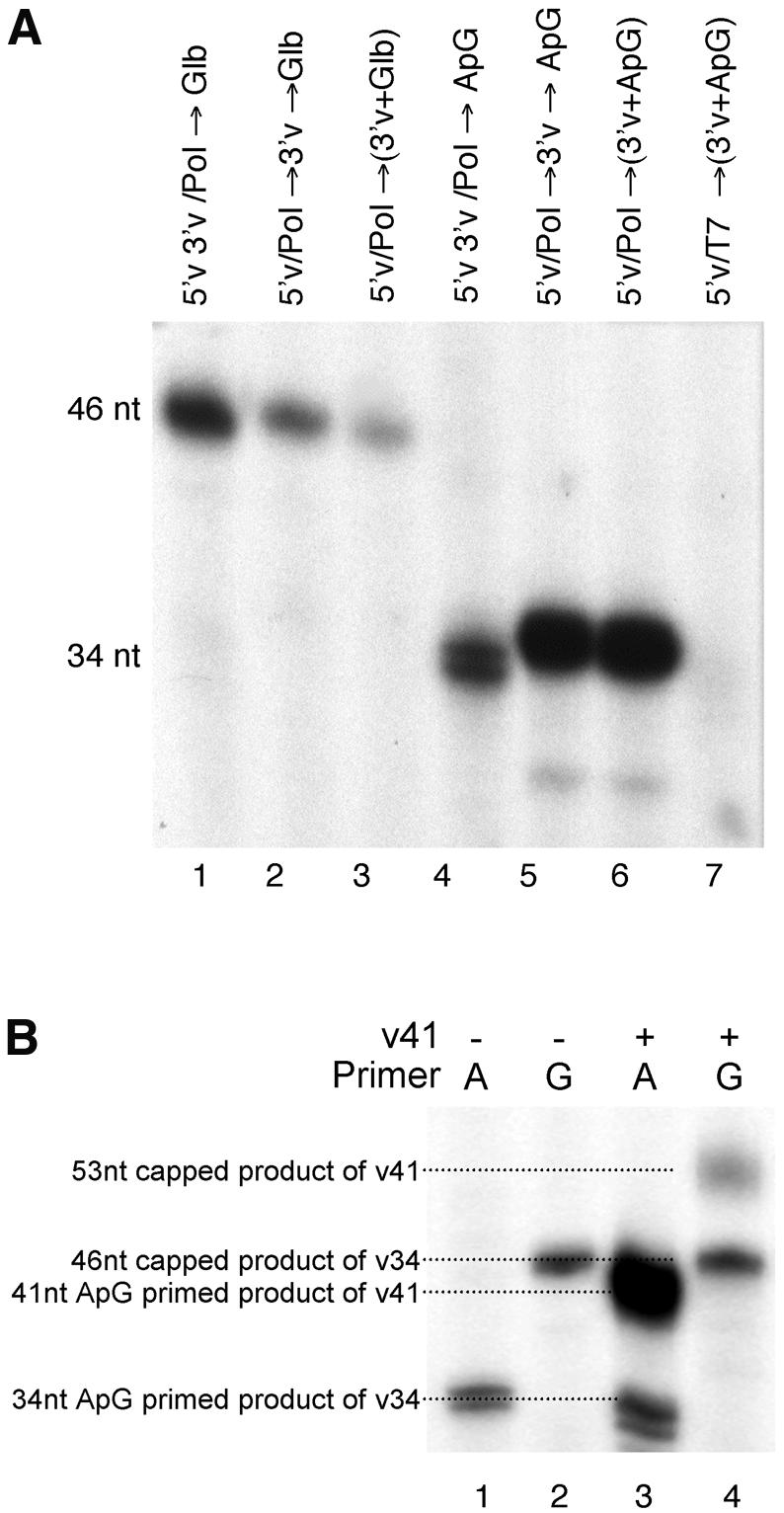
Cap-primed and ApG-primed transcription by the 5′v3′v/Pol and 5′v/Pol complexes. (A) 5′v3′v/Pol and 5′v/Pol (supplemented with v34 template after the polymerase binding reaction) were incubated with [α-32P]GTP, ATP, CTP and UTP in the presence of globin mRNA or ApG as a source of primer for 30 min at 30°C. The resultant reaction products were then resolved by 7 M urea, 18% PAGE. Globin-primed transcription (lanes 1–3) gives rise to a 46 nt reaction product. ApG-primed transcription (lanes 4–7) gives rise to a 34 nt reaction product, except for when the influenza polymerase is replaced with the T7 RNA polymerase as a specificity control (lane 7). Lanes 1 and 4, 5′v3′v/Pol reactions; lanes 2 and 5, 5′v/Pol reactions incubated with 3′v for 10 min prior to the addition of globin mRNA primer and nucleotides; lanes 3 and 6, 5′v/Pol reactions where v34, globin mRNA and nucleotides were added simultaneously. (B) 5′v3′v/Pol complexes were assembled using 1 pmol 5′v16B pre-annealed with v34 RNA. Transcription reactions were primed using ApG (A, lanes 1 and 3) or globin mRNA (G, lanes 2 and 4). Lanes 1 and 2, standard transcription reactions as described above; lanes 3 and 4, reactions supplemented with 9 pmol v41 RNA added simultaneously with the primer and nucleotides.
DISCUSSION
The current model for the formation of the influenza virus transcription complex proposes that in the absence of 5′- and 3′-ends, the viral polymerase is transcriptionally inert but is in the form of a complex competent for binding to the 5′-end of vRNA. Upon binding to 5′ vRNA the polymerase becomes competent to bind capped mRNAs but does not cleave them efficiently. The polymerase must be bound to both the 5′- and 3′-ends of vRNA to become fully endonucleolytically active (16). It has subsequently been suggested that this sequential order of assembly is obligatory and that the polymerase is unable to bind to the annealed 5′- and 3′-ends (1). This model must be revised in light of the data presented in this manuscript.
The polymerase binds to annealed 5′ and 3′ vRNA
The results presented herein show that the polymerase is able to bind to an RNA ligand composed of a pre-annealed 5′- and 3′-end and can initiate transcription on such templates. Indeed, this interaction is likely to be an important regulatory feature of the transcription process, as the polymerase that bound to the 5′v3′v ligand showed enhanced levels of cap-primed transcription. There is no evidence in the literature to support the contention that the sequential order of assembly is obligatory and all available evidence is consistent with the present results suggesting that the polymerase can bind to the annealed vRNA termini (16,18). Binding to pre-annealed 5′v3′v RNA is temperature dependent (it does not occur efficiently at 0°C; L.Tiley, unpublished observations). There is also evidence that the polymerase binds poorly to the 5′-end of vRNA if it is annealed to its perfect complement (i.e. 3′ cRNA) and that the polymerase is unable to bind to an annealed panhandle if the polymerase has already bound to a separate 5′ vRNA (15). These observations all suggest that the stability of the duplex is an important parameter for polymerase binding. It is likely that this region is dynamic and the range of conformations that it can adopt will be influenced by the presence and sequence of the opposite strand. The initial conformation of the template may affect how the RNA interacts with the polymerase and the subsequent (probably mutual) conformational changes that occur after binding (e.g. conversion of the RNA from a panhandle to a corkscrew conformation and allosteric changes in polymerase conformation). The apparent existence of distinct 5′-end binding sites for vRNA and cRNA (31) raises the additional possibility that the initial RNA conformation could influence which of these sites becomes occupied and the consequences this could have on polymerase activity.
Pre-annealed templates stimulate capped primer binding
Binding of the polymerase to pre-annealed 5′- and 3′-ends stimulated capped primer binding to a much greater extent than when the polymerase bound them sequentially. Furthermore, endonuclease activity was not absolutely dependent on the presence of the 3′-end of vRNA and the cleavage efficiency of G20 RNA was the same for both complexes. Although these data conflict with the current textbook model (1), previous papers have reported low but significant 5′ vRNA-dependent endonuclease activity in the absence of 3′ vRNA (16,17), although this has tended to be overlooked. All three studies agree that there is a significantly higher yield of endonuclease product associated with 5′v3′v-bound polymerase. The results presented here show that the higher yield of endonuclease product from panhandle RNA-bound polymerase correlated with the increased capped RNA binding activity of this complex. The results suggest the creation of a high affinity primer binding site coincident with the activation of the viral endonuclease. It is important to differentiate between cap binding and primer binding. Previously, cap binding was measured using a crosslinking assay performed in the absence of Mg2+ under saturating conditions. Our assay measures the stable association of the capped primer with the polymerase, which is able to survive multiple washing steps. This interaction was dependent on the primer being the correct length, and so was indirectly dependent on the endonuclease (and therefore on Mg2+). Substrate length, capped RNA (G20) was not stably bound to polymerase unless it was cleaved to primer length by the endonuclease. G11 RNA binding by 5′v3′v/Pol (± Mg2+) was stimulated to the same extent as that of G20 RNA when Mg2+ was present (Fig. 2A, lanes 1 and 8). Virion-associated polymerase shows a similar primer length requirement (32). The weaker association that manifested itself under conditions where the primer was not cleaved disproportionately affected the interaction of capped RNA with the 5′v3′v/Pol and thereby reduced the differential between the two types of polymerase complex (Fig. 2A, lane 2). Therefore, it is likely that the previous measurements of cap binding (16) were measuring the low affinity, readily dissociable binding that we observed here for both forms of polymerase. The activation of high affinity primer binding by the 5′- and 3′-ends would not have been detected previously and the effect would have been amalgamated into the activation of the endonuclease. The relatively low affinity for uncleaved capped RNAs may be a mechanism to limit the effects of poor endonuclease substrate RNAs that would be encountered in the infected cell. Tight binding would be restricted to those RNAs capable of efficient conversion into primers.
Pre-annealed templates are used preferentially for cap-primed transcription
The relative levels of cap-primed transcription (Fig. 4A) correlated with the levels of capped primer binding (Fig. 1A), whereas ApG-primed transcription was more abundant in the sequentially assembled reactions. Reactions containing two distinguishable templates, one of which is present in limiting quantities pre-annealed to the 5′-end and the other in excess, free in solution provided the explanation for this reciprocal effect (Fig. 4B). The level of ApG-primed synthesis was dependent upon the total amount of template RNA present in the reaction whereas the level of cap-primed transcription was primarily governed by the amount of RNA pre-annealed to the 5′-end before binding of the polymerase. The reduced level of ApG-primed synthesis observed for the 5′v3′v/Pol reactions was due to the limiting amounts of template RNA. Adding more template in solution resulted in greater levels of synthesis. The most likely reason for this is that the polymerase is then able to perform successive rounds of transcription on new template RNA. Alternatively, it could be due to saturation of a population of polymerase bound to the 5′ ligand on the beads that lacked the v34 template. We consider this to be unlikely as the v34 was present in excess in the annealing reaction and its interaction with the 5′-end is stable throughout many successive washing steps. Regardless, the significant result from this experiment is that the pre-annealed template was used preferentially for cap-primed synthesis even when an excess of secondary template was available in solution.
The pre-annealed template activates the polymerase more effectively
The amounts of polymerase bound by the two forms of RNA ligand were identical. Experiments using either the 5′v or 5′v3′v ligands to deplete the extract of active polymerase prior to performing the transcription assays have shown that both ligands deplete the extracts of cap-priming activity to the same extent (data not shown). Thus it is unlikely that structurally distinct forms of polymerase bind to the two ligands. The primer association and dissociation kinetics of stably bound primer were indistinguishable for polymerase complexes purified on 5′v3′v or 5′v ligands. Together, these results are most consistent with a single influenza polymerase complex, which can exist in two interchangeable conformational isoforms. Both forms are transcriptionally competent and bind capped RNA, but only one form exposes the high affinity primer binding site detected in our binding assays. This is also the endonucleolytically active form. The 5′v3′v and 5′v ligands differ in how efficiently they induce the adoption of the high affinity primer binding conformation. At equilibrium, the proportion of polymerase in the high affinity primer binding mode is higher for 5′v3′v/Pol.
Possible biological consequences of enhanced cap priming from pre-annealed templates
The observation that cap-primed transcription occurs more efficiently on pre-annealed templates can be viewed from two perspectives. From one perspective, sequential binding to the 5′-end followed by the 3′-end leads to low levels of cap priming. Thus the sequence of events likely to occur most often if the polymerase assembles on the 5′-end of one template and the 3′-end of another (i.e. on two linear templates) would result in inefficient cap priming. This is consistent with the hypothesis that linear templates are used for replication not transcription (15,25) and would be a mechanism to reduce the formation of inappropriately capped, full-length transcripts. From the perspective of mRNA synthesis, it suggests that cap priming will occur most efficiently on templates that are in a panhandle conformation at the time of polymerase binding as these are likely to be the predominant species that have annealed 5′- and 3′-ends. The current model for polyadenylation (see Introduction) requires that the template be in a panhandle conformation. Thus RNA structure-dependent activation of primer binding may be a way of coupling the processes of initiation and termination, ensuring the efficient formation of capped polyadenylated RNAs. Incoming virion RNPs are committed to a primary round of mRNA synthesis, which suggests that nascent virion RNPs assemble from a pre-annealed panhandle precursor. Against this is the argument that for genome length RNAs, binding of the polymerase is necessary for the formation of the panhandle in the first place (24). Perhaps sequentially assembled complexes gradually mature into complexes capable of cap priming (an indication of this was seen in Fig. 1A, lane 4, where preincubating with v34 RNA restored a small amount of the primer binding activity). The assembly of RNPs in vivo is very poorly understood and the role of the polymerase in this process is clearly an area requiring further study.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by grants from the Wellcome Trust (059151 to P.D. and L.T.), MRC (G9901213 to P.D.) and Royal Society (to P.D.). P.D. is a Royal Society University Research Fellow.
REFERENCES
- 1.Lamb R.A. and Krug,R.M. (2001) Orthomyxoviridae: the viruses and their replication. In Howley,P.M. (ed.), Fields Virology. Lippincott Williams and Wilkins, Philadelphia, PA, Vol. 1, pp. 1487–1531.
- 2.Biswas S.K. and Nayak,D.P. (1994) Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J. Virol., 68, 1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braam J., Ulmanen,I. and Krug,R.M. (1983) Molecular model of a eukaryotic transcription complex: functions and movements of influenza P proteins during capped RNA-primed transcription. Cell, 34, 609–618. [DOI] [PubMed] [Google Scholar]
- 4.Li M.L., Rao,P. and Krug,R.M. (2001) The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J., 20, 2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaas D., Patzelt,E. and Kuechler,E. (1982) Identification of the cap binding protein of influenza virus. Nucleic Acids Res., 10, 4803–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ulmanen I., Broni,B.A. and Krug,R.M. (1981) Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl Acad. Sci. USA, 78, 7355–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang T.S., Palese,P. and Krystal,M. (1990) Determination of influenza virus proteins required for genome replication. J. Virol., 64, 5669–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee M.T., Bishop,K., Medcalf,L., Elton,D., Digard,P. and Tiley,L. (2002) Definition of the minimal viral components required for the initiation of unprimed RNA synthesis by influenza virus RNA polymerase. Nucleic Acids Res., 30, 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perales B. and Ortin,J. (1997) The influenza A virus PB2 polymerase subunit is required for the replication of viral RNA. J. Virol., 71, 1381–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fodor E., Crow,M., Mingay,L.J., Deng,T., Sharps,J., Fechter,P. and Brownlee,G.G. (2002) A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol., 76, 8989–9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahy B.W.J., Barrett,T., Nichol,S.T., Penn,C.R. and Wolstenholme,A.J. (1981) Analysis of the functions of influenza virus genome segments by use of temperature-sensitive mutants of Fowl Plague virus. In Compans,R.W. (ed.), The Replication of Negative Strand Viruses. Elsevier North Holland, New York, NY, pp. 379–394.
- 12.Honda A., Mizumoto,K. and Ishihama,A. (2002) Minimum molecular architectures for transcription and replication of the influenza virus. Proc. Natl Acad. Sci. USA, 99, 13166–13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brownlee G.G. and Sharps,J.L. (2002) The RNA polymerase of influenza a virus is stabilized by interaction with its viral RNA promoter. J. Virol., 76, 7103–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fodor E., Pritlove,D.C. and Brownlee,G.G. (1994) The influenza virus panhandle is involved in the initiation of transcription. J. Virol., 68, 4092–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tiley L.S., Hagen,M., Matthews,J.T. and Krystal,M. (1994) Sequence-specific binding of the influenza virus RNA polymerase to sequences located at the 5′ ends of the viral RNAs. J. Virol., 68, 5108–5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cianci C., Tiley,L. and Krystal,M. (1995) Differential activation of the influenza virus polymerase via template RNA binding. J. Virol., 69, 3995–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M.L., Ramirez,B.C. and Krug,R.M. (1998) RNA-dependent activation of primer RNA production by influenza virus polymerase: different regions of the same protein subunit constitute the two required RNA-binding sites. EMBO J., 17, 5844–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagen M., Chung,T.D., Butcher,J.A. and Krystal,M. (1994) Recombinant influenza virus polymerase: requirement of both 5′ and 3′ viral ends for endonuclease activity. J. Virol., 68, 1509–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu M.-T., Parvin,J.D., Gupta,S., Krystal,M. and Palese,P. (1987) Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc. Natl Acad. Sci. USA, 84, 8140–8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bae S.H., Cheong,H.K., Lee,J.H., Cheong,C., Kainosho,M. and Choi,B.S. (2001) Structural features of an influenza virus promoter and their implications for viral RNA synthesis. Proc. Natl Acad. Sci. USA, 98, 10602–10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flick R. and Hobom,G. (1999) Interaction of influenza virus polymerase with viral RNA in the ‘corkscrew’ conformation. J. Gen. Virol., 80, 2565–2572. [DOI] [PubMed] [Google Scholar]
- 22.Pritlove D.C., Poon,L.L., Devenish,L.J., Leahy,M.B. and Brownlee,G.G. (1999) A hairpin loop at the 5′ end of influenza A virus virion RNA is required for synthesis of poly(A)+ mRNA in vitro. J. Virol., 73, 2109–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leahy M.B., Pritlove,D.C., Poon,L.L. and Brownlee,G.G. (2001) Mutagenic analysis of the 5′ arm of the influenza A virus virion RNA promoter defines the sequence requirements for endonuclease activity. J. Virol., 75, 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klumpp K., Ruigrok,R.W. and Baudin,F. (1997) Roles of the influenza virus polymerase and nucleoprotein in forming a functional RNP structure. EMBO J., 16, 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poon L.L., Pritlove,D.C., Sharps,J. and Brownlee,G.G. (1998) The RNA polymerase of influenza virus, bound to the 5′ end of virion RNA, acts in cis to polyadenylate mRNA. J. Virol., 72, 8214–8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo Y., Mao,X., Deng,L., Cong,P. and Shuman,S. (1995) The D1 and D12 subunits are both essential for the transcription termination factor activity of vaccinia virus capping enzyme. J. Virol., 69, 3852–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klumpp K., Hooker,L. and Handa,B. (2001) Influenza virus endoribonuclease. Methods Enzymol., 342, 451–466. [DOI] [PubMed] [Google Scholar]
- 28.Doan L., Handa,B., Roberts,N.A. and Klumpp,K. (1999) Metal ion catalysis of RNA cleavage by the influenza virus endonuclease. Biochemistry, 38, 5612–5619. [DOI] [PubMed] [Google Scholar]
- 29.Pleiss J.A., Derrick,M.L. and Uhlenbeck,O.C. (1998) T7 RNA polymerase produces 5′ end heterogeneity during in vitro transcription from certain templates. RNA, 4, 1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fodor E., Pritlove,D.C. and Brownlee,G.G. (1995) Characterization of the RNA-fork model of virion RNA in the initiation of transcription in influenza A virus. J. Virol., 69, 4012–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez S. and Ortin,J. (1999) Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. EMBO J., 18, 3767–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung T.D., Cianci,C., Hagen,M., Terry,B., Matthews,J.T., Krystal,M. and Colonno,R.J. (1994) Biochemical studies on capped RNA primers identify a class of oligonucleotide inhibitors of the influenza virus RNA polymerase. Proc. Natl Acad. Sci. USA, 91, 2372–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]