


Abstract
Slime mold, plant and insect dihydropyrimidine amidohydrolases (DHPases, EC 3.5.2.2), which catalyze the second step of pyrimidine and several anti-cancer drug degradations, were cloned and shown to functionally replace a defective DHPase enzyme in the yeast Saccharomyces kluyveri. The yeast and slime mold DHPases were over-expressed, shown to contain two zinc ions, characterized for their properties and compared to those of the calf liver enzyme. In general, the kinetic parameters varied widely among the enzymes, the mammalian DHPase having the highest catalytic efficiency. The ring opening was catalyzed most efficiently at pH 8.0 and competitively inhibited by the reaction product, N-carbamyl-β-alanine. At lower pH values DHPases catalyzed the reverse reaction, the closing of the ring. Apparently, eukaryote DHPases are enzymatically as well as phylogenetically related to the de novo biosynthetic dihydroorotase (DHOase) enzymes. Modeling studies showed that the position of the catalytically critical amino acid residues of bacterial DHOases and eukaryote DHPases overlap. Therefore, only a few modifications might have been necessary during evolution to convert the unspecialized enzyme into anabolic and catabolic ones.
INTRODUCTION
The catabolic degradation of pyrimidines, together with the salvage and the de novo synthetic pathway, determines the size of the pyrimidine pool in the cell. In mammals, uracil, thymine and anti-cancer pyrimidine analogs are degraded in a three-step catabolic pathway, involving the enzymes dihydropyrimidine dehydrogenase, dihydropyrimidine amidohydrolase (DHPase) and β-alanine synthase (1,2). One of the end-products, β-alanine, is an essential precursor for the synthesis of pantothenate and coenzyme A, but in mammals it is also thought to have a neurotransmitter function due to its chemical similarity to the neural inhibitor γ-aminobutyrate (3). Pyrimidine catabolic enzymes are the major cause for the inactivation of clinically applied pyrimidines, such as 5-fluorouracil, used in treatment of several tumors and viral diseases (2). The degradation reduces the efficiency of the administered drug and requires the application of extremely high doses (4), while the accumulating fluorinated products are neurotoxic (5). Pyrimidine catabolic enzymes may also play a role in the degradation of pyrimidine-based biocides, such as bromacil (5-bromo-3sec-butyl-6-methyluracil) or lenacil (6).
DHPase, also known as dihydropyrimidinase, catalyzes the second step of the pyrimidine degradation, the reversible hydrolysis of 5,6-dihydrouracil (DHU) or 5,6-dihydrothymine (DHT) to N-carbamoyl-β-alanine (NCBA) or N-carbamyl-β-aminoisobutyrate, respectively. Various DHPases can also open five-membered cyclic ureides, like hydantoins or succinimides (7). So far DHPase has been isolated from various mammalian sources, such as bovine (8), rat (9,10), calf liver (11) and pig liver (12). Gene sequences coding for putative DHPase from Caenorhabditis elegans (13), rat (14) and human liver (15) have been cloned from cDNA libraries. The bacterial counterpart of DHPase, the so-called hydantoinases (HYDases), have also been cloned (16–18) or purified (19,20) from different sources. However, bacterial HYDases may not be directly involved in degradation of pyrimidines, but rather only in the synthesis of d- and l-amino acids (21,22). Sequence alignments suggest a close relationship between DHPases and several proteins involved in neuronal development. Among those are different forms of the so-called human DHPase-related protein (14), the rat turned- on-after division 64 kDa protein (23) and the collapsin-response-mediator proteins (24).
Mammalian DHPases are tetrameric enzymes and contain tightly bound zinc ions which can be removed by chelators (8,12). The pH dependencies of Vmax and Vmax/Km for native bovine amidohydrolase using 5-bromo-5,6-dihydrouracil as substrate showed the requirement for a single group that must be protonated for activity (25). The pH dependence of kinetic parameters and solvent deuterium isotope effects has been used to probe the mechanism of DHPase from calf and pig liver (12). The resting DHPase primed for hydrolysis of DHU has a general base (pK ∼ 7.5–8) and a zinc-bound water (pK ∼ 9–10). DHU binds to the enzyme displacing the Zn-OH2 (pK 9.6). The substrate likely binds with the 4-oxo group directly coordinated to the active site metal, so that the metal acts as a Lewis acid polarizing the carbonyl for the subsequent hydrolysis. The general base activates a water molecule for nucleophilic attack at C-4 to generate the tetrahedral intermediate. The latter in turn undergoes ring opening assisted by general acid protonation of the ring nitrogen using the same enzyme residue to give NCBA (12).
The reductive catabolism of pyrimidines has so far been characterized only in two eukaryotic groups, mammals and fungi. However, the fungal DHPase from Saccharomyces kluyveri has not been fully biochemically characterized (26). In this report, we describe novel DHPases from insect, plant and slime mold. Furthermore, the S.kluyveri and Dictyostelium discoideum DHPases were characterized for their substrate specificity and kinetics and compared with those of the mammalian DHPase. Modeling studies showed that DHPases have the same active center as dihydroorotases (DHOases).
MATERIALS AND METHODS
Materials
DHU, DHT, NCBA, 8-hydroxyquinoline and Chelex-100 were purchased from Sigma. Glutaric acid monoamide (GAMA) was prepared according to the method of Marquez et al. (27). All other reagents were of the highest purity available from different commercial sources.
Strains and growth media
The Escherichia coli strain XL1-blue was used for plasmid amplification and the E.coli BL21 (from Stratagene) for heterologous protein expression. Bacteria were grown at 37°C in Luria–Bertani medium supplemented with 100 mg l–1 of ampicillin for selection. The yeast strain S.kluyveri Y777 (MATα pyd2-1 ura3), deficient in DHPase (26,28), was grown at 25°C in the standard rich (YPD) and minimal (SD) media. The N-minimal DHU medium (1% succinic acid, 0.6% NaOH, 2% glucose, 0.17% yeast nitrogen base without amino acids and ammonia from Difco, and 0.1% DHU) was used for selection of yeast transformants. When necessary, the SD and DHU media were supplemented with 0.2 mM uracil, giving the SD+ura and DHU+ura media (26,29).
DNA sequences
Database searches to find novel putative DHPases were performed using the BLAST network services at the National Center for Biotechnology Information and provided partial sequences of the putative Arabidopsis thaliana (At), Drosophila melanogaster (Dm) and D.discoideum (Dd) open reading frames (ORFs). An expressed sequence tag (EST) cDNA clone (P397, 46F2T7, GenBank accession no. T14084) containing a putative At DHPase sequence was obtained from The Arabidopsis Information Resource (TAIR). The corresponding partial ORF sequence had a high similarity to that of the P1 clone: MXC9 from chromosome V (accession no. AB007727). The full ORF was afterwards rescued from a commercial Arabidopsis cDNA library (Stratagene). An EST cDNA clone (P639, LP11064, accession no. AI296940) carrying a putative Dm DHPase was obtained from Research Genetics (Birmingham, AL). An EST cDNA clone (P380, SLA867, accession no. AU060286) from Dd was obtained from the University of Tsukuba. The ORFs were determined by sequencing of the EST clones and novel cDNA clones and afterwards given new accession numbers.
Enzyme phylogenetic analysis and modeling
Nucleotide sequence analysis and protein sequence comparisons were performed with the ClustalW 1.7 program (30). The phylogenetic analysis was presented with the TreeCon version (Yves van de Peer, University of Antwerp). Comparison of the sequences of E.coli DHOase and human DHPase was performed with the Clustal algorithm and enzyme modeling of human DHPase into the DHOase structure was done using the Modeller program and for energy minimization the Charm program was employed.
DNA manipulation
All DNA manipulations were carried out following the standard laboratory procedures. Plasmid DNA was purified from E.coli transformants with Quantum prep columns (Bio-Rad) and the sequence of all plasmids verified using a commercial sequencing source.
Yeast expression plasmids
Fragments coding for putative ORFs were obtained by PCR using Pfu DNA polymerase (Stratagene). The At ORF was amplified from the commercial cDNA library, the Dm ORF from P639 and Dd DHPase was amplified from the Dictyostelium λZAP cDNA library (31). The PCR fragments containing appropriate terminal restriction sites were sub-cloned into the pre-cut yeast shuttle vector P403, containing the S.kluyveri PYD3 promoter followed by the multiple cloning sites: HindIII, KpnI, SacI, BamHI, BstXI, EcoRI, BstXI, NotI, XhoI and SphI (26). The following plasmids were obtained: P478 containing the At DHPase in the HindIII/EcoRI sites, P635 containing the Dm DHPase in the HindIII/XhoI sites and P633 containing the Dd DHPase in the KpnI/XhoI sites. Transformation of S.kluyveri (Sk) was done by electroporation (26) and followed by selection on the DHU medium. Putative transformants appeared on the plates after a week and they were tested for the plasmid stability and growth properties.
Over-expression plasmids
For heterologous expression in E.coli the C-terminal (His)8-tag vector P343 was used (29,32). PCR amplification of the DHPase ORFs was done in a way to create, upon restriction digestion, a fragment having XbaI and EcoRI sticky ends. The ORF for Sk DHPase was obtained by removing a 63 bp intron by a long PCR primer. The PCR fragments were sub-cloned into the XbaI/EcoRI pre-cut P343 plasmid, giving the expression plasmids P531 (Sk DHPase) and P634 (Dd DHPase).
Protein purification
For recombinant protein expression, E.coli cells were grown to a density of A600 nm = 0.5–0.6. Protein expression was induced by 200 µg l–1 of anhydrotetracycline hydrochloride (ACROS Organics, NJ) for 24 h at 25°C. Collected cells were resuspended in buffer A (50 mM sodium phosphate pH 8.0, 300 mM NaCl, 10% glycerol, protease inhibitors) and disrupted by French Press (4 × 1000 p.s.i.). After centrifugation at 13 000 g for 30 min, the supernatant was filtered on a 0.45 µm cellulose acetate filter and applied to a 10 ml Ni2+-NTA column (Qiagen). The column was washed with 10 vol of buffer A with 25 mM imidazole, 10 vol of buffer B (50 mM sodium phosphate pH 6.0, 300 mM NaCl) with 25 mM imidazole. The recombinant DHPases were eluted with a 10× volume of a linear gradient of 50–500 mM imidazole in buffer B. Active fractions were pooled and imidazole removed by filtration through Amicon Centriprep YM-10 (Millipore) using Tris buffer (100 mM Tris–HCl, pH 8.0). Proteins were stored at –20°C at a concentration of ∼10 mg ml–1. SDS– PAGE was performed according to the procedure of Laemmli (33) and proteins were visualized by Coomassie Blue staining. Protein concentration was quantified by the method of Bradford (34) and bovine serum albumin served as a protein standard.
Enzyme assays
DHPase activity was determined by measuring the decrease or increase in absorbance at 225 nm caused by the hydrolysis or formation of a 5,6-dihydropyrimidine ring using a Zeiss double beam spectrophotometer thermostated at 30°C. The molar absorption coefficients at 225 nm for DHU, DHT and glutarimide are 1287, 1059 and 400 M–1 cm–1, respectively. For routine assays, reaction mixtures containing 0.1 M potassium phosphate, pH 8.0, and 1.5 mM DHU were used. One unit of DHPase catalyzes the hydrolysis of 1 µmol DHU min–1 at 30°C under the above conditions. For pH studies the following buffers were used over the pH range indicated: potassium phosphate, 5.5–8.0; Tris–HCl, 8–9; potassium pyrophosphate, 8–10.
Molecular mass determination
The subunit molecular mass of the different amidohydrolases was estimated by SDS–PAGE in a discontinuous buffer system (33). The mass of the native proteins was determined by native gel electrophoresis on 4–10% gradient gels (Bio-Rad) in Tris–glycine buffer, pH 8.5 in a Gibco BRL Mini-V 8 × 10 vertical gel electrophoresis system (Life Technologies Inc.). Gels were stained for protein with Coomassie Blue or silver stain (35). Urease (trimer 272 kDa, hexamer 545 kDa), chicken egg albumin (45 kDa), bovine serum albumin (monomer 66 kDa, dimer 132 kDa) and carbonic anhydrase (29 kDa) were used as native protein standards, whereas phosphorylase b (94 kDa), bovine serum albumin (67 kDa), ovalbumin (43 kDa) and carbonic anhydrase (29 kDa) were standards for SDS–PAGE. BenchMark Protein Ladder (Gibco BRL/Life Technologies) was also used as molecular weight standards on the SDS–PAGE gels. The native size was determined as recommended by the kit producer (Sigma) (see also Supplementary Material).
Metal determination
Each of the two heterologous DHPases was over-expressed two times and isolated on the Ni2+-NTA column to give two independent enzyme sources. An aliquot of 200 µl of each sample, containing ∼2–5 mg of protein ml–1, was dialyzed against metal-free 100 mM Tris–acetate, pH 7.5. This buffer was purified by passing it through a Chelex 100™ column following a modified procedure of Himmelhoch et al. (36). The sulfur and metal content of the enzyme samples and the buffer was then determined in duplicates on biomolecular thin films by energy dispersive X-ray fluorescence analysis using the set-up at the Physics Laboratory of the Royal Veterinary and Agricultural University, Copenhagen, Denmark (37). The sulfur atoms in the protein served as an internal standard and helped to quantify the presence of metal ions relative to sulfur.
Resolution of metal and reconstitution of apoamidohydrolase
The metal ions were removed from Dd DHPase by incubating the enzyme (625 µg) in 0.1 M potassium phosphate buffer, pH 8.0 with 8-hydroxyquinoline 5-sulfonate (0.8 mM) overnight at room temperature. The chelator was removed by dialyzing the reaction mixture against 3 × 500 ml phosphate buffer. The resulting apoenzyme was inactive. Enzymatic activity could be restored by addition of increasing amounts of zinc ions and followed by plotting enzymatic activity versus amounts of zinc ions added.
Kinetic data processing
Reciprocal initial velocities were plotted versus reciprocal substrate concentrations. Data were fitted using the Fortran programs developed by Cleland (38). Reciprocal initial velocities were plotted versus reciprocal substrate concentrations. Data for single reciprocal plots were fitted using equation 1.
v = VA/(Ka + A)1
Data for linear competitive and non-competitive inhibition were fitted using equations 2 and 3, respectively.
v = VA/[Ka(1 + I/Kis) + A]2
v = VA/[Ka(1 + I/Kis) + A(1 + I/Kii)]3
In equations 1–3, V is the maximum velocity; A and I are concentrations of reactant and inhibitor, respectively; Ka is the Michaelis constant for reactant; Kis and Kii are slope and intercept inhibition constants, respectively.
RESULTS
Novel eukaryote DHPases
So far the pyrimidine catabolic genes and enzymes have been studied only in two eukaryote groups, mammals and yeast. When the sequence of the Sk DHPase gene was used as a query to screen EST databases, several positive hits from different organisms were obtained. In order to cover a whole range of eukaryote kingdoms, three sequences originating from a plant, an insect and a unicellular eukaryote were selected for further analysis. The ORF sequences, coding for putative DHPases, were deduced by sequencing of the corresponding EST cDNA clones, kindly provided by different laboratories. Since the At N-terminal sequence was missing in the EST clone (P397) it was assembled from the genomic sequence and afterwards the ORF was sub-cloned from the At cDNA library. Similarly, the Dd EST insert (P380) was also truncated at the 5′ end. The upstream sequence was deduced upon amplification from the Dd cDNA library, using an internal primer and a primer mapping to the vector. Apparently, the Dm EST clone (P639) contained the full ORF. The putative At ORF (accession no. AF465755) contains 1596 bp and encodes a 531 amino acid protein with an estimated molecular weight of 57.9 kDa. The putative Dm ORF (accession no. AF 465756) contains 1785 bp and encodes a 594 amino acid protein with an estimated molecular weight of 65 kDa. The putative DdPYD2 gene contains a 1512 bp long ORF (accession no. AF465757) and encodes a protein of 504 amino acids of 56 kDa.
Previously, a number of different mutants, pyd–, in the pyrimidine catabolic pathway have been isolated in the S.kluyveri yeast (26). When the three putative ORFs were expressed under the control of the PYD3 promoter in a yeast mutant deficient in DHPase, pyd2–, they could complement the mutation. While the S.kluyveri mutant strain (Y777) cannot grow in a medium with uracil or dihydrouracil as the sole nitrogen source, the transformants carrying P478, P633 or P635 could grow (Fig. 1). Therefore, the At, Dm and Dd ORFs indeed code for functional DHPases, which can catalyze the dihydrouracil ring opening in vivo. While the yeast mutants complemented with the slime mold and fruit fly PYD2 genes grew almost like the wild-type yeast strain, the mutant strain carrying the plant PYD2 was substantially slower on the selective media (data not shown).
Figure 1.
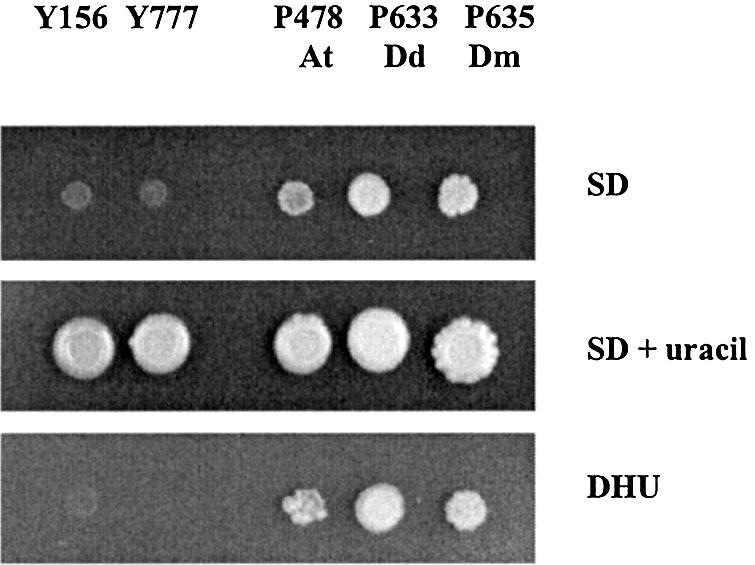
Spot assay. The S.kluyveri strain Y777, deficient in pyd2, was transformed with putative PYD2 genes from different eukaryotes, giving the following transformants: P478 (containing the A.thaliana PYD2 gene, At), P633 (containing the D.discoideum PYD2 gene, Dd) and P635 (containing the D.melanogaster PYD2 gene, Dm). The growth on the ‘rich’ medium, SD+uracil, and two selective media, SD and DHU is shown. Y156 is the parental strain of Y777, and both are uracil auxotrophs. Note that the plasmids also contain, apart from PYD2, the URA3 gene.
The sequences of the three novel DHPases were aligned with the yeast and mammalian sequences. A phylogenetic analysis including DHPases, as well as similar enzymes, shows that eukaryotic DHPases have the same origin (Fig. 2). However, they are also closely related to bacterial HYDases and animal dihydropyrimidinase-related proteins, and relatively close to DHOases.
Figure 2.

Phylogenetic analysis of DHPases and DHPase-like proteins. The DHPase-like proteins include dihydropyrimidinase-related proteins (DRP), hydantoinases (HYDA), allantoinases (ALLA), dihydroorotases (DHOase) and ureases (URE). The accession numbers of the amino acid sequences follow the protein names. The clustering method was used for inferring the phylogenetic tree topology. The numbers given are frequencies at which a given branch appeared in 100 bootstrap replications.
Recombinant Sk DHPase and Dd DHPase
Two ORFs, coding for Sk and Dd DHPases, were over-expressed in E.coli as His-tagged proteins. The proteins were purified on a Ni2+-NTA column and eluted with imidazole gradient buffer. The DHPase-active fractions were pooled, concentrated and imidazole removed by repeated addition of imidazole-free buffer followed by concentration. The purified enzymes were studied for their native and subunit size, metal content, substrate specificity and kinetics (Table 1).
Table 1. Properties of Sk and Dd DHPases.
| Sk | Dd | |
|---|---|---|
| Native molecular massa (kDa) | 255 | 220 |
| Subunit molecular mass (kDa) | 60 | 56 |
| Metal contentb per subunit | 2 Zn | 2 Zn |
| Substrate specificity | DHU, DHT | DHU, DHT |
| Glutarimide (very slowly) | ||
| NCBA | NCBA |
aThe native size was determined by native gel electrophoresis (see also Supplementary Material).
bMetal determination on Dd and Sk DHPases after removal of all metal ions and subsequent saturation with zinc ions.
The subunit size and the native size of both recombinant DHPases was estimated (Table 1; see also Supplementary Material). Dd DHPase was found to have a molecular mass of 220 kDa for the native enzyme and a subunit molecular mass of 56 kDa. The Sk enzyme (Sk DHPase) gave molecular masses of 255 and 60 kDa for the native and monomeric protein, respectively. These values are very similar to the size of native calf liver DHPase, 217 kDa, and its monomeric form, 54 kDa (11). Apparently, both DHPases are tetrameric, similar to their mammalian counterparts (Table 1). DHPases over-expressed in E.coli in Luria–Bertani medium were not initially saturated with zinc, they contained also iron and other metals, most likely because the growth medium became depleted of zinc. Metal determinations, however, showed that Dd and Sk DHPases fully saturated with zinc ions contained two metal ions per monomer (Table 1).
When all metal ions were removed from Dd DHPase, the enzymatic activity of the inactive apoenzyme could be fully recovered by zinc but not by any other metal ion. Titration of the inactive apoenzyme was performed by overnight incubation with increasing amounts of ZnCl2 at room temperature. Aliquots of each incubation were afterwards measured for DHPase enzymatic activity and plots of enzymatic activity versus Zn concentration revealed the titration end point. Two independent experiments showed that Dd DHPase contained 2.12 and 1.9 zinc ions per subunit, respectively.
Substrate specificity and kinetic parameters
In the forward reaction, the natural substrates for both Dd DHPase and Sk DHPase seem to be dihydrouracil and dihydrothymine (Table 2). The two DHPases exhibit their maximum enzymatic activity in the pH range 8.0–10.0. When the pH is decreased to 6.0, the enzymatic activity is reduced to ∼10% of the maximum value (data not shown). Substrate analogs, such as hydantoin, glutarimide and dihydroorotate (DHO), are not hydrolyzed by Sk DHPase. Hydantoin and DHO are not hydrolyzed by Dd DHPase, while glutarimide is hydrolyzed very slowly. These results indicate that the substrate binding site allows only a limited number of structural modifications for a potential substrate and that these two enzymes are highly specialized for degradation of pyrimidines.
Table 2. Kinetic parameters of DHPases from various organisms.
| Enzyme | Substrate | Km (µM) | Vmax (µM min–1 mg–1) | Vmax/Km (min–1 mg–1) |
|---|---|---|---|---|
| S.kluyveri | DHU | 710 ± 90 | 1340 ± 85 | 1.88 |
| DHT | 490 ± 80 | 700 ± 30 | 1.43 | |
| NCBAa | 10 000 ± 250 | 160 ± 20 | 0.016 | |
| D.discoideum | DHU | 400 ± 50 | 550 ± 20 | 1.38 |
| DHT | 37 ± 5 | 176 ± 6 | 4.8 | |
| NCBAa | 2400 ± 250 | 76 ± 2 | 0.032 | |
| Calf liver | DHU | 25 | 3670 | 146.8 |
| DHT | 85 | 8341 | 98.1 | |
| NCBAb | 4100 | 1500 | 0.36 |
From the point of view of structure and phylogenetics, as well as substrate specificity, the examined eukaryote DHPases represent a very uniform group, but the kinetic properties differ significantly from one enzyme to the other (Table 2). The Km values of Sk and Dd DHPase for DHU at pH 8.0 are about 28- and 16-fold, respectively, higher than that of the calf liver DHPase. In general, the catalytic efficiency of the two DHPases with DHU as substrate is about 78- or 106-fold less for the Sk and Dd DHPase, respectively, than that of the mammalian counterpart. The Km values for thymine are about 6-fold higher for Sk DHPase and about a factor of 2 smaller for Dd DHPase when compared with the calf liver enzyme.
In the reverse reaction direction, the formation of DHU from NBCA was determined. Maximal enzymatic activity in the direction of DHU formation was observed at pH 6.0 and 6.6 for Dd and Sk DHPase, respectively, whereas the calf liver DHPase is maximally active at pH 5.5. Negligible enzymatic activity was found for the calf liver enzyme above pH 6.5 (11), whereas the other two enzymes exhibit >50% of their activity at pH 7.3. The Km values for NCBA are lowest for the Dd and about 2.5-fold higher for Sk than that for calf liver DHPase. The catalytic efficiency of calf liver DHPase is 10 and 20 times lower for Dd and Sk DHPase, respectively.
For the direction of DHP hydrolysis, only a few, not very effective inhibitors have been reported so far (12). For all DHPases tested so far, the product NCBA was always found to inhibit the forward reaction competitively. The Ki values of Sk and Dd DHPase for NCBA are 23 ± 2 and 4.6 ± 0.6 mM, respectively. DHPases are also inhibited by GAMA; the Sk enzyme shows competitive inhibition with a Ki value of 15 ± 1 mM, whereas the inhibition of Dd DHPase is non-competitive with a Ki of 4.1 ± 0.4 mM. For the two DHPases discussed here, both inhibitors are less effective than for calf liver DHPase with Ki values of 0.21 mM for GAMA and 0.68 mM for NCBA (12). Both DHPases accept neither hydantoin nor DHO as substrates and these two compounds are not inhibitors.
DISCUSSION
Uracil and thymine were shown in mammals, yeast and some bacteria to be degraded in a three step catabolic pathway (2). In humans, this pathway is of crucial importance for degradation of several anti-cancer drugs and a detailed understanding of this pathway is therefore highly relevant from a clinical point of view. The second step is catalyzed by DHPase and this enzyme and the corresponding gene have so far been characterized only in a limited number of organisms. In this study, we isolated sequences coding for putative DHPases from different eukaryotes. The corresponding ORFs from a unicellular eukaryote (D.discoideum), a plant (A.thaliana) and an insect (D.melanogaster) were shown by complementation of a yeast pyd2 deficiency to encode functional DHPases (Fig. 1). These results demonstrate that DHPases, and thereby the reductive catabolism of pyrimidines, are likely to be present in all major eukaryotic kingdoms.
The sequences of the newly isolated DHPases were aligned with a number of DHPase-like enzymes, such as dihydropyrimidinase-related proteins, HYDases, DHOases, collapsin-response-mediator proteins, ureases and allantoinases, and analyzed for their phylogenetic relationship (Fig. 2). Dd DHPase and Dm DHPase group together with other animal DHPases, and these are closely related to dihydropyrimidinase-related proteins. We propose that both sub-groups originated from a common progenitor upon gene duplication taking place in an early animal ancestor. The dihydropyrimidinase-related protein lineage became involved in the early development and propagation of axons (15,22–24,39). A majority of bacterial HYDases belong to the same group as eukaryote DHPases, including plant and fungal DHPases, and animal dihydropyrimidinase-related proteins. Surprisingly, Sk DHPase is the least related member of this DHPases/ dihydropyrimidinase-related proteins/HYDases group (Fig. 2). Apparently, the ancient progenitor of this group, presumably already existing in the common ancestor of all prokaryotes and eukaryotes, was likely to be a catabolic enzyme. The groups, which are the closest phylogenetic relatives of the DHPases/dihydropyrimidinase-related proteins/HYDases group, consist of allantoinases and small (type II) DHOases (26), which catalyze the third reaction of the de novo pyrimidine biosynthetic pathway. The latter catalyzes the reverse of the reaction catalyzed by DHPases; it closes the dihydroorotate ring.
From a biochemical point of view DHPases belong to the amidohydrolase superfamily (40) containing proteins that catalyze various hydrolytic reactions at carbon and phosphorus centers. The superfamily can be divided into three subsets of three-dimensional structures of amidohydrolases differing in the presence of metal ions in the active site. Family I includes enzymes with binuclear metal centers, such as DHOase (41), phosphotriesterase (42) and urease (43). Family II contains proteins with a mononuclear metal center such as adenosine deaminase (44) whereas the third family includes proteins that carry out hydrolysis without a metal ion as shown for N-carbamyl-d-amino acid amidohydrolase from Agrobacterium sp. (45). In the latter, a triad of Glu-Cys-Lys executes hydrolysis. Family I and II enzymes have a cluster of four histidines and an aspartate in the metal binding site, but only family I proteins have an additional requirement for a carbamylated lysine to bridge the two metal ions. All proteins belonging to family I of the amidohydrolase superfamily, DHOase, urease and phosphotriesterase, each contain a lysine residue at this specific location.
At present no structural data are available for DHPase from any eukaryotic organism. Sequence alignment for eukaryote DHPases and E.coli DHOase, however, reveals the conservation of five, out of six, amino acid residues in the active site. Three histidine residues (H67, H69, H248), one aspartate (D326) and the crucial lysine (K165) (Fig. 3) are preserved in all DHPases with the exception of Sk DHPase. In addition, several other amino acid residues are conserved among the analyzed DHPases and E.coli DHOase (Fig. 3). DHOase was originally suggested to be a member of family II of the amidohydrolase superfamily because only a single divalent metal ion was found. Modeling of the DHPase sequence onto the DHOase structure shows clearly that the three histidines and the aspartate of both proteins are exactly in the same position (Fig. 4). The ε-amino group of K165 of human DHPase is within 4.5 Å of one of the metal ions. When the additional carboxyl group, however, is attached to the Nε of K165 it seems very likely that the two metal ions could be bridged by the carbamylated lysine (Fig. 4). Therefore, we propose that DHPases belong to family I of the superfamily and contain two metal ions in their active site, especially in the light of the fact that a second zinc ion in DHOase has been found only recently via the three-dimensional structure (41). The presented metal determinations of Sk and Dd DHPases also revealed two Zn ions per monomer (Table 1). The presented modeling data could be useful for design of inhibitors and activators, which can modify degradation of anti-cancer drugs.
Figure 3.

Aligment of the six eukaryote DHPases. The six eukaryote DHPases are human (JC5315), rat (Q63150), fruit fly (AF465756), slime mold (AF465757), thale cress (AF465755), Sk (AAF69237) and in addition E.coli DHOase (P05020). The preserved amino acid residues are shadowed and the catalytically critical DHOase residues are marked with asterisks. Note that in the Sk the ‘inserted’ sequences (26) were removed prior to alignment.
Figure 4.

Modeling of the human DHPase structure onto the known E.coli DHOase structure. The figure shows a close-up of the active site with amino acids within a 7.5 Å distance from the zinc ions. Amino acid side chains coordinating to the zinc ions (H69, K165, H248 and D326) are shown in the normal atomic colors. H67 is behind one of the zinc atoms and is therefore not visible in this figure. With the exception of the non-carboxylated K165 all other residues are in perfect overlap with the template structure of DHOase. The side chains of all other amino acids of the DHPase active center are in cyan, whereas the residues of template DHOase are in orange-red.
While DPHases show highly conserved sequences and most probably also structures, they differ in substrate specificity and kinetic parameters. The substrate specificity for Sk and Dd DHPases (Table 1) are more similar to each other than to the substrate specificity of calf liver DHPase, and more restrictive than that for rat liver imidase (46) and bacterial HYDases (22). The imidase hydrolyzes phthalimide, glutarimide, succinimide, adipimide, hydantoin and dihydropyrimidines. The pH optimum of the imidase for the different substrates ranges from pH 7.5 for the phthalimide to pH 9.5 for adipimide and pH > 10 for DHU (46), whereas the Sk and Dd DHPases hydrolyze preferentially DHU and DHT with a pH optimum around 9 (Table 2). It seems that the rat liver imidase functions more like a detoxifying enzyme due to its broad substrate specificity for cyclic and acyclic imides. Similarly, bacterial HYDases may also have a number of functions in the cell, while eukaryote DHPases are likely to be involved only in the catabolism of pyrimidines.
On the basis of the pH dependence of kinetic parameters and kinetic solvent deuterium isotope effects a reaction mechanism has been proposed for DHPases from livers of calf and pig (12). The mechanism was written assuming a single active site Zn2+ but now it has to be rewritten on the basis of the novel data of a binuclear Zn center (Fig. 5). This mechanism is a modified version of that proposed for DHOase (41). A general base, most likely D326 in human DHPase, which is homologous to D251 in DHOase, is required to activate a water molecule for nucleophilic attack on C-4 of the DHU ring (Fig. 5). While DHPases can catalyze a reaction similar to the third step of the de novo pyrimidine biosynthesis reaction, they cannot accept DHO, which is the usual substrate for DHOases. Apparently, the carboxyl group of DHO cannot be accommodated in the DHPase active site. However, it may be that only a limited number of amino acids have to be changed to increase the substrate specificity of DHPases and convert them into DHOases, and vice versa. A similar scenario could also have been followed in nature during the evolutionary history of small (type II) DHOases, DHPases and HYDases. Upon duplication of the progenitor enzyme, which could possibly catalyze both reactions, only a limited number of mutations was necessary to create the specialized, anabolic and catabolic, enzymes.
Figure 5.

Reaction mechanism of DHPase. The V/K for DHU decreases at both low and high pH giving pK values of about 7.5–8.0 and 9–10. The enzymic general base (most likely D326) with a pK of 7.5–8.0 is required to activate the water for nucleophilic attack on the C-4 of DHU which is directly coordinated to the active site zinc. The second group with a pK of 9–10 likely reflects Zn-water ionization of the free enzyme.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Wolfgang Knecht for his comments and Divine for moral support. We are indebted to Dr Thomas Müller (Physiologische Chemie II, Biozentrum, Universität Würzburg, Würzburg, Germany) for modeling the human DHPase onto the known structure of E.coli DHOase and providing Figure 4. This work was supported by a grant from the Danish Research Council to J.P. and a stipend from the Technical University of Denmark to K.D.S.
DDBJ/EMBL/GenBank accession nos+ AF465755–AF465757
REFERENCES
- 1.Wallach D.P. and Grisolia,S. (1957) The purification and properties of hydropyrimidine hydrase. J. Biol. Chem., 226, 277–288. [PubMed] [Google Scholar]
- 2.Wasternack C. (1980) Degradation of pyrimidines and pyrimidine analogs—pathways and mutual influences. Pharmacol. Ther., 8, 629–651. [DOI] [PubMed] [Google Scholar]
- 3.Sandberg M. and Jacobson,I. (1981) Beta-alanine, a possible neurotransmitter in the visual system? J. Neurochem., 37, 1353–1356. [DOI] [PubMed] [Google Scholar]
- 4.Milano G. and Etienne,M.-C. (1994) Potential importance of dihydropyrimidine dehydrogenase (DPD) in cancer chemotherapy. Pharmacogenetics, 4, 301–306. [DOI] [PubMed] [Google Scholar]
- 5.Okeda R., Shibutani,M., Matsuo,T., Kuroiwa,T., Shimokawa,R. and Tajima,T. (1990) Experimental neurotoxicity of 5-fluorouracil and its derivatives is due to poisoning by the monofluorinated organic metabolites, monofluoroacetic acid and α-fluoro-β-alanine. Acta Neuropathol. (Berl.), 81, 66–73. [DOI] [PubMed] [Google Scholar]
- 6.Chaudry G.R. and Cortez,L. (1988) Degradation of bromacil by Pseudomonas sp. Appl. Environ. Microbiol., 54, 2203–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dudley K.H., Butler,T.C. and Bius,D.L. (1974) The role of dihydropyrimidinase in the metabolism of some hydantoin and succinimide drugs. Drug Metab. Dispos., 2, 103–112. [PubMed] [Google Scholar]
- 8.Brooks K.P., Jones,E.A., Kim,B.D. and Sander,E.G. (1983) Bovine liver dihydropyrimidine amidohydrolase: purification, properties, and characterization as a zinc metalloenzyme. Arch. Biochem. Biophys., 226, 469–483. [DOI] [PubMed] [Google Scholar]
- 9.Traut T. and Loechel,S. (1984) Pyrimidine catabolism: individual characterization of the three sequential enzymes with a new assay. Biochemistry, 23, 2522–2539. [DOI] [PubMed] [Google Scholar]
- 10.Kikugawa M., Kaneko,M., Fujimoto-Sakata,S., Maeda,M., Kawasaki,K., Takagi,T. and Tamaki,N. (1994) Purification, characterization and inhibiton of dihydropyrimidinase from rat liver. Eur. J. Biochem., 219, 393–399. [DOI] [PubMed] [Google Scholar]
- 11.Kautz J. and Schnackerz,K.D. (1989) Purification and properties of 5,6-dihydropyrimidine amidohydrolase from calf liver. Eur. J. Biochem., 181, 431–435. [DOI] [PubMed] [Google Scholar]
- 12.Jahnke K., Podschun,B., Schnackerz,K.D., Kautz,J. and Cook,P.F. (1993) Acid–base catalytic mechanism of dihydropyrimidinase from pH studies. Biochemistry, 32, 5160–5166. [DOI] [PubMed] [Google Scholar]
- 13.Li W., Herman,R.K. and Shaw,J.E. (1992) Analysis of Caenorhabditis elegans axonal guidance and out-growth gene unc-33. Genetics, 132, 675–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuda K., Sakata,S., Kaneko,M., Hamajima,N., Nonaka,M., Sasaki,M. and Tamaki,N. (1996) Molecular cloning and sequencing of a cDNA encoding dihydropyrimidinase from the rat liver. Biochim. Biophys. Acta, 1307, 140–144. [DOI] [PubMed] [Google Scholar]
- 15.Hamajima N., Matsuda,K., Sakata,S., Tamaki,N., Sasaki,M. and Nonaka,M. (1996) A novel gene family defined by human dihydropyrimidinase and three related proteins with differential tissue distribution. Gene, 180, 157–163. [DOI] [PubMed] [Google Scholar]
- 16.Watabe K., Ishikawa,T., Mukohara,Y. and Nakamura,H. (1992) Cloning and sequencing of the genes involved in the conversion of 5-substituted hydantoins to the corresponding l-amino acids from the native plasmid Pseudomonas sp. strain NS671. J. Bacteriol., 174, 962–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LaPointe G., Viau,S., Leblanc,D., Robert,N. and Morin,A. (1994) Cloning, sequencing and expression in Escherichia coli of the hydantoinase gene from Pseudomonas putida and distribution of homologous genes in other microorganisms. Appl. Environ. Microbiol., 60, 888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim G.-J., Lee,S.-G., Park,J.H. and Kim,H.S. (1997) Direct detection of hydantoinase activity on solid agar plates and electrophoretic acrylamide gels. Biotechnol. Tech., 11, 511–513. [Google Scholar]
- 19.Runser S. and Meyer,P.C. (1993) Purification and biochemical characterization of the hydantoin hydrolyzing enzyme from Agrobacterium species, a hydantoinase with no 5,6-dihydropyrimidine amidohydrolase activity. Eur. J. Biochem., 213, 1315–1324. [DOI] [PubMed] [Google Scholar]
- 20.Durham D.R. and Weber,J.E. (1995) Properties of d-hydantoinase from Agrobacterium tumefaciens and its use for the preparation of N-carbamyl d-amino acids. Biochem. Biophys. Res. Commun., 216, 1095–1100. [DOI] [PubMed] [Google Scholar]
- 21.Kim G.J. and Kim,H.S. (1998) Identification of the structural similarity in the fuctionally related amidohydrolases acting on the cyclic amide ring. Biochem. J., 330, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Syldatk C., May,O., Altenbuchner,J., Mattes,R. and Siemann,M. (1999) Microbial hydantoinases—industrial enzymes from the origin of life? Appl. Microbiol. Biochtechnol., 51, 293–309. [DOI] [PubMed] [Google Scholar]
- 23.Minturn J.E., Fryer,H.J., Geschwind,D.H. and Hockfield,S. (1995) TOAD-64, a gene expressed early in neuronal differentiation in the rat, is related to unc-33, a C. elegans gene involved in axon outgrowth. J. Neurosci., 15, 6757–6766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goshima Y., Nakamura,F., Strittmatter,P. and Strittmatter,S.M. (1995) Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature, 376, 509–514. [DOI] [PubMed] [Google Scholar]
- 25.Lee M.H., Pettigrew,D.W., Sander,E.G. and Nowak,T. (1987) Bovine liver dihydropyrimidine amidohydrolase: pH dependence of the steady-state kinetic and proton relaxation rate properties of the Mn(II)-containing enzyme. Arch. Biochem. Biophys., 259, 597–604. [DOI] [PubMed] [Google Scholar]
- 26.Gojkovic Z., Jahnke,K., Schnackerz,K.D. and Piskur,J. (2000) PYD2 encodes 5,6-dihydropyrimidine amidohydrolase, which participates in a novel fungal catabolic pathway. J. Mol. Biol., 295, 1073–1087. [DOI] [PubMed] [Google Scholar]
- 27.Marquez V.E., Kelley,J.A. and Driscoll,J.S. (1980) 1,3-Diazepinones. 2. The correct structure of squamolone as 1-carbamoyl-2-pyrrolidine synthesis of authentic perhydro-1,3-diazepine-2,4-dione. J. Org. Chem., 45, 5308–5312. [Google Scholar]
- 28.Gojkovic Z., Paracchini,S. and Piskur,J. (1998) A new model organism for studying catabolism of pyrimidines and purines. Adv. Exp. Med. Biol., 431, 475–479. [DOI] [PubMed] [Google Scholar]
- 29.Gojkovic Z., Sandrini,M.P.B. and Piskur,J. (2001) Eukaryote β-alanine synthases are functionally related, but have a high degree of structural diversity. Genetics, 158, 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnitzler G.R., Fischer,W.H. and Firtel,R.A. (1994) Cloning and characterization of the G.box binding factor, an essential component of the developmental switch between early and late development in Dictyostelium. Genes Dev., 8, 502–514. [DOI] [PubMed] [Google Scholar]
- 32.Bader B., Knecht,W., Fries,M. and Löffler,M. (1998) Expression, purification and characterization of histidine-tagged rat and human flavoenzyme dihydroorotate dehydrogenase. Protein Expr. Purif., 13, 414–422. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli U. (1970) Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 34.Bradford M.M. (1967) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 35.Blum H., Beyer,H. and Gross,H.J. (1987) Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis, 8, 93–99. [Google Scholar]
- 36.Himmelhoch S.R., Sober,H.A., Vallee,B.L., Peterson,E.A. and Fuwa,K. (1966) Spectrographic and chromotographic resolution of metalloproteins in human serum. Biochemistry, 5, 2523–2530. [DOI] [PubMed] [Google Scholar]
- 37.Laursen J., Stikans,M., Karlsen,K. and Pind,N. (1999) A versatile and easy to handle EDXRF instrumentation. In Proceedings of the European Conference on Energy Dispersive X-Ray Spectrometry. Editrice Compositori, Bologna, Italy, pp. 139–144.
- 38.Cleland W.W. (1979) Statistical analysis of enzyme kinetic data. Methods Enzymol., 63, 103–108. [DOI] [PubMed] [Google Scholar]
- 39.Wang L.H. and Strittmatter,S.M. (1997) Brain CRMP forms heterotetramers similar to liver dihydropyrimidinase. J. Neurochem., 69, 2261–2269. [DOI] [PubMed] [Google Scholar]
- 40.Holm L and Sander,C. (1997) An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins Struct. Funct. Genet., 28, 72–82. [PubMed] [Google Scholar]
- 41.Thoden J.B., Phillips,G.N., Neal,T.M., Raushel,F.M. and Holden,H.M. (2001) Molecular structure of dihydroorotase: a paradigm for catalysis through the use of a binuclear metal center. Biochemistry, 40, 6989–6997. [DOI] [PubMed] [Google Scholar]
- 42.Benning M.M., Kuo,J.M., Raushel,F.M. and Holden,H.M. (1995) Three-dimensional structure of the binuclear metal center of phosphotriesterase. Biochemistry, 34, 7973–7978. [DOI] [PubMed] [Google Scholar]
- 43.Jabri E., Carr,M.B., Hausinger,R.P. and Karplus,P.A. (1995) The crystal structure of urease from Klebsiella aerogenes. Science, 268, 998–1004. [PubMed] [Google Scholar]
- 44.Wilson D.K., Rudolph,F.B. and Quiocho,F.A. (1991) Atomic structure of adenosine deaminase complexed with a transition-state analog: understanding catalysis and immunodeficiency mutations. Science, 252, 1278–1284. [DOI] [PubMed] [Google Scholar]
- 45.Nakai T., Hasegawa,T., Yamashita,E., Yamamoto,M., Kumasaka,T., Ueki,T., Nanba,H., Ikenaka,Y., Takahashi,S., Sato,M. and Tsukihara,T. (2000) Crystal structure of N-carbamyl-d-amino acid amidohydrolase with a novel catalytic framework common to amidohydrolases. Structure, 8, 729–739. [DOI] [PubMed] [Google Scholar]
- 46.Yang Y.S., Ramaswany,S. and Jacoby,W.B. (1993) Rat liver imidase. J. Biol. Chem., 268, 10870–10875. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.