


Abstract
The expression of mitochondrial HMG-CoA synthase in the colon has been correlated with the levels of butyrate present in this tissue. We report here that the effect of butyrate on mitochondrial HMG-CoA synthase gene expression is exerted in vivo at the transcriptional level, and that trichostatin A (TSA), a specific histone deacetylase inhibitor, also induces transcriptional activity and mRNA expression of the gene in human cell lines derived from colon carcinoma. Using chromatin immunoprecipitation assays, we show that histone deacetylase 1 (HDAC1) is associated with the endogenous mitochondrial HMG-CoA synthase promoter and that TSA induction correlates with hyperacetylation of H4 histone associated with the 5′ flanking region of the gene. Overexpression of HDAC1 activity leads consistently to mitochondrial HMG-CoA synthase promoter hypoacetylation and reduces its transcriptional activity. The effect of butyrate and TSA maps to a single Sp1 site present in the proximal promoter of the gene, which is able to bind Sp1 and Sp3 proteins. Interestingly, the binding affinity of Sp1 and Sp3 proteins to the Sp1 site correlates with the TSA responsiveness of the promoter. Using a one-hybrid system (GAL4-Sp1 and GAL4-Sp3), we show that both proteins can mediate responsiveness to TSA in CaCo-2 cells employing distinct mechanisms.
INTRODUCTION
Mitochondrial 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) synthase catalyzes the condensation of acetoacetyl-CoA and acetyl-CoA to produce HMG-CoA plus free CoA. The HMG-CoA is then converted, through the actions of HMG-CoA lyase and D-3-hydroxybutyrate dehydrogenase, into the ketone bodies acetoacetate and β-hydroxybutirate, which are used as a source of oxidative fuels in several non-hepatic tissues (reviewed in 1). The mitochondrial carnitine palmitoyltransferase system has been considered important in regulating the synthesis of ketone bodies by means of substrate supply, the acetyl-CoA (2,3). However, over the last 20 years compelling evidence has been provided to show that mitochondrial HMG-CoA synthase is the key enzyme in ketogenesis in various physiological situations (e.g. starvation, high fat diet, fetal-suckling transition, sustained exercise). Thus, in tissues such as liver and intestine, ketogenesis and mitochondrial HMG-CoA synthase gene expression are regulated in a coordinated fashion (1).
However, ketogenic tissues do not present an identical pattern of ketogenesis or mitochondrial HMG-CoA synthase gene expression. For instance, in the liver and colon, the gene is expressed throughout an animal’s life, while in the small intestine the gene is only expressed during the neonatal period, disappearing at weaning (4,5). Therefore, despite certain common hormonal and nutritional factors (6), the expression pattern of the mitochondrial HMG-CoA synthase gene depends on a number of unknown tissue-specific mechanisms.
In the colon, the expression of the mitochondrial HMG-CoA synthase gene has been correlated with the butyrate levels in this tissue, where it is produced by the bacterial fermentation of dietary fiber (7). Thus, the experimental germ-free status—the absence of intestinal microflora and, consequently, the absence of short fatty acids such as n-butyrate—correlates with a 75% reduction in the colon-expressed levels of the mitochondrial HMG-CoA synthase gene (7). As expected, the capacity to generate ketone bodies from n-butyrate is lower in colonocytes of germ-free animals.
n-Butyrate plays an important role in the homeostasis of the colonic mucosa by inducing pathways of cell maturation, differentiation and apoptosis (8–10). Butyrate is able to modulate the gene expression of a small number of genes through the inhibition of histone deacetylase (HDAC) enzymes (11). Histone acetylation and deacetylation involve the reversible transfer of the acetyl moiety of acetyl-CoA to the ε-amino group of lysine residues in histone proteins. This reaction is catalyzed by histone acetyltransferases (HATs) and can be reversed by HDACs. Thus, these two activities determine the acetylation levels of a given promoter. Butyrate induces histone hyperacetylation by reducing the rate of histone deacetylation, through the inhibition of HDAC. A correlation has been established between hyperacetylated histones and transcriptionally active chromatin, since acetylation makes chromatin less condensed and more accessible to trans-acting factors. In fact, some co-activators have intrinsic HAT activities while HDAC activities are present in co-repressor complexes, as independent entities recruited on gene promoters (12,13).
The rat mitochondrial HMG-CoA synthase promoter contains sequences that mediate its multihormonal regulation and tissue specificity (6). Two of these sequences, a Sp1 binding site and a nuclear receptor responsive element, are located in the proximal promoter region of the gene (14 and unpublished data). Both elements are able to recruit HDAC through different trans-acting factors (15–18). For this reason, we wished to investigate the role of butyrate in mitochondrial HMG-CoA expression in rat colon and human cell lines derived from colon.
In this paper we demonstrate that acetylation level of histones associated with mitochondrial HMG-CoA synthase promoter correlates with the transcriptional activity of the gene and we show that HDAC1 activity is involved in regulating the acetylation levels of the promoter. We also show that HDAC inhibitors, such as butyrate and trichostatin A (TSA), can strongly activate the transcription of mitochondrial HMG-CoA synthase in a CaCo-2 cell line through a Sp1 site located at –57 to –48 relative to the transcription start site. Interestingly, we show that the affinity for the Sp1 site of Sp1 and Sp3 proteins is critical for the TSA responsiveness.
Thus, our results suggest that in the colon, butyrate stimulates the transcription of the mitochondrial HMG-CoA synthase gene by inhibiting the HDAC activities recruited, mainly, through the Sp1 site present in the proximal promoter of the gene. Since the mitochondrial HMG-CoA synthase gene is expressed in tissues with a high capacity for fatty acid β-oxidation, we propose that butyrate is involved in a positive feed-forward regulation on its own β-oxidation rate and accordingly on the levels of this metabolite in the colon.
MATERIALS AND METHODS
Cell culture
The human colon cancer cell lines CaCo-2 and HT-29, obtained from the American Type Culture Collection (Rockville, MD), were maintained in Dulbecco’s modified Eagle’s medium supplemented with antibiotics and 10% fetal bovine serum at 37°C and 5% CO2.
Reagents
Sodium butyrate and TSA were obtained from Sigma (St Louis, MO) and Calbiochem (La Joya, CA), respectively.
Production and characterization of the SMCAT1 transgenic line
The SMCAT1 DNA fragment, containing from –1148 to +28 of mitochondrial HMG-CoA synthase promoter fused to CAT, was isolated as SalI–NdeI restriction fragment from pSMCAT1 plasmid described previously (6) and purified by electroelution into dialysis bags (19). The DNA was microinjected into fertilized ovocytes of hybrid mice (C57BL/6xSJLF2). Founders and positive offspring were bred with nontransgenic mice (C57BL/6xSJLF2) to generate hemizygous offspring. Transgenic mice were identified by genomic Southern blot. Isolation of tail DNA and genomic Southern blot were performed by standard protocols. The SalI–NdeI fragment used for microinjection, labeled with [α-32P]dCTP, was used as a probe. Animal care and euthanasia protocols were all approved by the ethical committee for animal research at the University of Barcelona.
Tissue extraction for CAT assays
Protein extracts from three different lines of transgenic animals fed over 5 days with a normal diet or a diet supplemented with butyrate (10%, w/w) were prepared by homogenization of the tissue in 0.25 M Tris–HCl buffer pH 7.5. The suspension was cleared by centrifugation, and the total amount of protein content was determined by the Bradford method (20). All samples assayed for CAT activity were first incubated at 65°C for 5 min. CAT assays were performed for either 60 min or 8–10 h, in order to increase the sensitivity of the method (19). Radioactivity of samples was measured on an LKB-1217 liquid scintillation counter.
Plasmids
The rat mitochondrial HMG-CoA synthase promoter- luciferase fusion plasmids –116pGl3, –116pGl3(M-PPRE), –116pGl3(M-Sp1) and –62pGl3 were constructed by HindIII subcloning of the –116 to +28 fragments of pSMpCAT6A, pSMpCAT6AM and pSMpCAT116(M-Sp1), and the –62 to +28 fragment of pSMpCAT6D into the pGL3-BASIC firefly luciferase reporter gene (Promega). The plasmids pSMpCAT6A, pSMpCAT6AM and pSMpCAT6D were obtained as previously described (14,21). Mutant pSMPCAT116(M-Sp1) was generated from pSMPCAT6A in which the Sp1 site was mutated by site-directed mutagenesis using an overlap extension PCR (22). Briefly, 100 ng of pSMPCAT6A plasmid was used as a target in two independent PCRs. The first PCR used a forward primer, 5′-AGC TGACTTGTTCTGAGACCTTTG-3′, which corresponds to co-ordinates –116 to –96, and a reverse primer 5′-CTC TGCAAGCtctaGaTCCACTTAGCGAGG-3′, which corresponds to co-ordinates –39 to –69 (nucleotides designated in underlined lower case correspond to those that were changed from the wild-type sequence). The second PCR used a forward primer, 5′-GCTAAGTGGAtctaGaGCTTGCAGAGGCTGG-3′), which corresponds to co-ordinates –64 to –34, and a reverse primer, CAToligo30 (which annealed with the vector downstream of the polylinker). In the subsequent overlap extension reaction, 200 ng of each PCR product was used as a target and the primer corresponding to coordinates –116 to –96 and CAToligo30 were used as primers. The PCR product was cloned in the p-GEM-T vector and subsequently subcloned into pCAT-Basic, thus generating pSMPCAT116(M-Sp1).
The construct –62pGl3(Sp1-la) was generated as follows: two oligonucleotides, 5′-TAAGTGGgGGCaGGGCTTGCA GAGGCTG-3′ and 5′-CAGCCTCTGCAAGCCCtGCCgCC AC TTAAGCT-3′, were annealed together. The nucleotides corresponding to the mutated Sp1 site are shown in underlined lower case letters. A second pair of oligonucleotides, 5′-GGG TCTTATAAAGCCCAACCCACAGTCGCTGGGCATCTC TCCCAGGGGCTGTGGACTGCTGG-3′ and 5′-TCGACC AGCAGTCCACAGCCCCTGGGAGAGATGCCCAGCGA CTGTGGGTTGGGCTTTATAAGACCC-3′, were also annealed together. The two resulting double-stranded DNA fragments were ligated together (blunt-end ligation). This ligation generated a double-stranded DNA fragment with 5′ and 3′ ends compatible with SacI and XhoI, respectively, which were used to introduce it into SacI/XhoI-cut pGL3Basic.
The pFA-CMV-LBDγ2 plasmid expresses the GAL4 binding domain fused to the ligand binding domain (LBD) of the human nuclear receptor PPARγ2 (amino acids 203 to end) (23). PPARγ2 was amplified by PCR using the Human Fat Cell Quick-Clone™ cDNA (Clontech). Oligonucleotides for PCR amplification were designed in agreement with the human described sequence PPARγ2 (U79012) and were 5′-CTG CAGCCCGGGGCTGTTATGGGTGAAAC-3′ (positions –6 to +11) and 5′-CTGCAGGCGGCCGCCTAGTACAAGT CCTTGTAG-3′ (positions +1517 to +1498). PPARγ2 PCR product was digested with SmaI and NotI, cloned into pBSSK+ SmaI/NotI sites and sequenced. The region of PPARγ2 cloned into pBSSK+ was then amplified by PCR using the primers 5′-GCAGCTGGATCCATGTCTCATA ATGCCATCAG-3′ (positions +676 to +695) and 5′-GCA GCTTCTAGACTAGTACAAGTCCTTG-3′ (positions +1517 to +1502). The PCR product was digested with BamHI–XbaI and cloned into the pFA-CMV expression plasmid.
All construction sequences were confirmed by the dideoxynucleotide-chain-termination method with the use of an automated fluorescence-based system (Perkin-Elmer).
The pCINeoHDAC1 expression plasmid was a kind gift from Dr Christian Seiser (University of Vienna) (24). Dr Yoshihiro Sowa (Kyoto Prefectural University of Medicine) kindly provided the pM-Sp1, pM-Sp3, pM-DNSp1 and pM-DNSp3 plasmids (17). The pFR-Luc plasmid (Stratagene) is a reporter plasmid that contains five GAL4 binding sites upstream TATA box.
Northern blot analysis
Total RNA was extracted with Trizol (Life Technologies) and northern blot analyses were performed with 15 µg of total RNA per lane. The 2.2 kb human mitochondrial HMG-CoA synthase cDNA used as a probe was obtained by digesting pBS-MSH (25) with EcoRI. Northern blot analysis was performed following standard methods (19).
Transient transfection
CaCo-2 cells were seeded at a density of 2 × 104 cells/cm2 12 h before transfection. Cells were transfected by using FuGENE-6 reagent according to the manufacturer’s instructions (Roche, IN). Twenty-four hours after transfection, half of the cells were treated with 0.3 µM TSA, 5 mM butyrate or vehicle when indicated. Twenty-four hours later cell lysates were collected for luciferase assay. pRL-CMV plasmid was included in the transfection procedure to correct for variation in transfection efficiency. In all the transfection experiments in which HDAC1 was overexpressed, the luciferase activity was corrected by the amount of protein assayed (19), because of the regulation of the pRL-CMV control plasmid expression by the expression of HDAC1. In co-transfection experiments, the amount of transfected DNA was kept constant by including salmon sperm DNA.
Luciferase and CAT assays
For CAT assays, extracts of harvested cells were prepared by liquid nitrogen freeze/thaw disruption (three times) after resuspension in 50 µl of 0.25 M Tris–HCl, pH 7.5. All samples assayed for CAT activity were first incubated at 65°C for 5 min. CAT assays were performed for 60 min (20). Radioactivity of samples was measured on an LKB-1217 liquid scintillation counter. For luciferase assays, cells were harvested using the passive lysis method (Promega) and luciferase assays were performed following the instructions of the Dual Luciferase Reporter Assay System (Promega). Luciferase activity was measured using a TD-20/20 Luminometer (Turner Designs).
Chromatin immunoprecipitation (ChIP) assay
In order to cross-link proteins to DNA, 1.5 × 106 of CaCo-2 and HT-29 cells were treated with formaldehyde to a final concentration of 1% and incubated at 37°C for 10 min. Cells were washed with PBS and 1 mM PMSF, scraped and collected by centrifugation. Cells were then resuspended in lysis buffer (85 mM KCl, 0.5% NP-40, 1 mM PMSF, 1 µg/ml leupeptin and aprotinin at pH 8) and homogenized. Nuclei were isolated by centrifugation at 250 g for 10 min, then resuspended in sonication buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl at pH 8.1) and sonicated for four cycles of 10 s each. The extracts were then centrifuged at 13 000 r.p.m. for 10 min at 4°C. The cross-linked preparations were diluted 10-fold in ChIP dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris–HCl plus the above indicated proteases inhibitors, at pH 8.1). The chromatin solution was pre-cleared with 80 µl of a 50% slurry prot A-Agarose (Upstate Biotechnology) for 1 h at 4°C. An aliquot (20 µl) was removed as a control (input). Immunoprecipitation was performed overnight with antibodies against acetylated histone H4, HDAC1 (Upstate Biotechnology), Sp1 and Sp3 (Santa Cruz Biotechnology) or preimmune serum (IgG). Following immunoprecipitation, 60 µl of 50% slurry prot A-Agarose was added, and incubation continued for 1 h. Agarose beads were then collected and washed sequentially for 5 min each time in low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, 150 mM NaCl at pH 8.1), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, 500 mM NaCl at pH 8.1) and LiCl buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris–HCl at pH 8.1). Beads were then washed twice with TE buffer and extracted twice with 250 µl of 1% SDS–0.1 M NaHCO3. Eluates were pooled and heated at 65°C for 4 h to reverse the cross-linking. The chromatin associated proteins were digested with proteinase K and samples were extracted with phenol/CHCl3 followed by precipitation with ethanol. Pellets were resuspended in water and subjected to PCR. Primers for amplification of the human mitochondrial HMG-CoA synthase promoter were: (i) 5′-GGGTTACCTTGG GATGTGATAC-3′ (positions –305 to –284) and (ii) 5′-GAAAGAGTGCCCAGTGGTGCC (positions +8 to –14).
The ChIP assay of transfected CaCo-2 cells was performed using antibodies against Flag peptide (M2 monoclonal antibody, Sigma) and acetylated H4 histone antibody (Upstate Biotechnology). The primers used for amplification of transfected mitochondrial HMG-CoA synthase promoter in PCRs were: (i) 5′-GACTTGTTCTGAGACCTTTGGC-3′ (positions –115 to –94) and (ii) 5′-CTTTATGTTTTT GGCGTCTTCC-3′ (corresponding to a sequence in the pGl3basic vector).
Protein isolation and immunoblot analysis
CaCo-2 cells transfected with the expression plasmid pCDNA3FlagHADC1 or salmon sperm DNA were harvested 48 h after transfection, and whole cell extracts were obtained by the following procedure: cells were lysed in RIPA buffer (50 mM Tris–HCl pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM NaF and Na3VO4) plus protease inhibitors in a rotating platform for 15 min at 4°C. The lysates were then centrifuged at 14 000 g for 10 min and the supernatants collected, protein quantified and stored at –80°C. SDS–PAGE (10%) was carried out (60 µg of protein/lane). Western blot analysis was performed with monoclonal M2 anti-flag antibody (Sigma) as recommended by the manufacturers. Proteins were detected using the ECL chemiluminescence system (Amersham Pharmacia Biotech).
Electrophoretic mobility shift assay
CaCo-2 cells were harvested in 1.5 ml of ice-cold PBS and nuclear extracts obtained as described (26). Briefly, after centrifugation the supernatant was discarded and cells were resuspended in 400 µl of cold hypotonic buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 2 mM Na3VO4, 2 mM NaF, 0.5 mM dithiothreitol, 0.5 mM PMSF, 5 µM leupeptin and 200 U/ml aprotinin, at pH 7.9). After 10 min on ice, cells were vortexed for 10 s and centrifuged. The pellet was resuspended in 50 µl of cold high salt buffer (20 mM HEPES, 25% glycerol, 1.5 mM MgCl2, 420 mM KCl, 2 mM EDTA, 0.5 mM dithiothreitol, 0.5 mM PMSF, 5 µM leupeptin and 200 U/ml aprotinin, at pH 7.9). The lysate was centrifuged for 2 min and the supernatant containing the nuclear extracts was aliquoted and stored at –70°C.
A doubled-stranded 5′-overhang oligonucleotide, comprising from –57 to –48 of rat mitochondrial HMG-CoA synthase gene (5′-agcttGTGGAGGCGGGGCg-3′; referred to as Sp1 wild-type probe; in bold and capital letters the consensus Sp1 binding site) was used as a probe. Nuclear proteins were preincubated on ice for 10 min in 10 mM Tris–HCl, pH 8.0, 40 mM KCl, 0.05% (v/v) NP-40, 6% glycerol, 1 mM dithiothreitol, and 1 µg of poly(dI–dC). Then, 4 ng of Sp1 wild-type probe, 32P-labeled by fill-in with Klenow polymerase, was added, and the incubation was continued for 20 min at room temperature. The reaction mixture was further incubated for 20 min in the presence or absence of anti-Sp1 or anti-Sp3 antibody (Santa Cruz Biotechnology). Samples were electrophoresed at 4°C on non-denaturing 4.5% (w/v) polyacrylamide gel in 0.5% TBE buffer (45 mM Tris/45 mM boric acid/1 mM EDTA pH 8).
The affinity of Sp1 and Sp3 proteins to the Sp1 wild-type (see above) and Sp1 mutated probe (5′-agcttGTG GgGGCaGGGCg-3′; in bold and capital letters the nucleotides corresponding to the consensus Sp1 binding site; in bold underlined lowercase those nucleotides that have been changed from the wild-type sequence) was evaluated using Scatchard assays. Ten micrograms of CaCo-2 nuclear proteins were incubated, as described above, with increasing amounts of either labeled wild-type or point-mutated Sp1 probe (Sp1-la).
RESULTS
Sodium butyrate induces mitochondrial HMG-CoA synthase gene transcription
To determine whether butyrate plays a direct role in mitochondrial HMG-CoA synthase gene transcription, we used transgenic animals in which the reporter CAT gene was controlled by the rat mitochondrial HMG-CoA synthase promoter. Figure 1 shows that butyrate induces the transcriptional activity of mitochondrial HMG-CoA synthase promoter in the liver (Fig. 1A) and the colon (Fig. 1B) independently of the CAT activity of each independent transgenic line.
Figure 1.

Butyrate stimulates reporter CAT activity gene expression driven by rat mitochondrial HMG-CoA synthase promoter in transgenic mice. CAT assays of mouse liver (A) and colon (B) from transgenic animals of three different lines fed with a normal diet (–) or a diet supplemented with butyrate (+). CAT assays were performed in triplicate as described in Materials and Methods.
TSA induces HMG-CoA synthase mRNA expression in human colon carcinoma cell lines through hyperacetylation of histone H4 associated with the mitochondrial HMG-CoA synthase promoter
To study if the mitochondrial HMG-CoA gene transcription induction by butyrate was related with a change in the acetylation levels of histones associated with its promoter we use TSA, a specific inhibitor of histone deacetylase enzymes, and therefore a hyperacetylating agent. Figure 2A shows that TSA induces mRNA expression of mitochondrial HMG-CoA synthase in HT-29 and CaCo-2 cell lines. We next performed ChIP assays using chromatin isolated from HT-29 and CaCo-2 cells both treated and untreated with TSA. Figure 2B shows that the mitochondrial HMG-CoA synthase promoter sequence was enriched in the immunocomplex containing acetylated H4 from TSA-treated cells compared with that from control cells. These results suggest that mitochondrial HMG-CoA synthase promoter is able to recruit co-repressor complexes harboring HDAC activities. To confirm this observation, we performed a new ChIP assay in which chromatin isolated from CaCo-2 cells was incubated with antibodies against human HDAC1. Figure 2C shows that this antibody is specifically able to immunoprecipitate human mitochondrial HMG-CoA synthase promoter, indicating the presence of this protein in the complexes present on the endogenous promoter of the gene.
Figure 2.
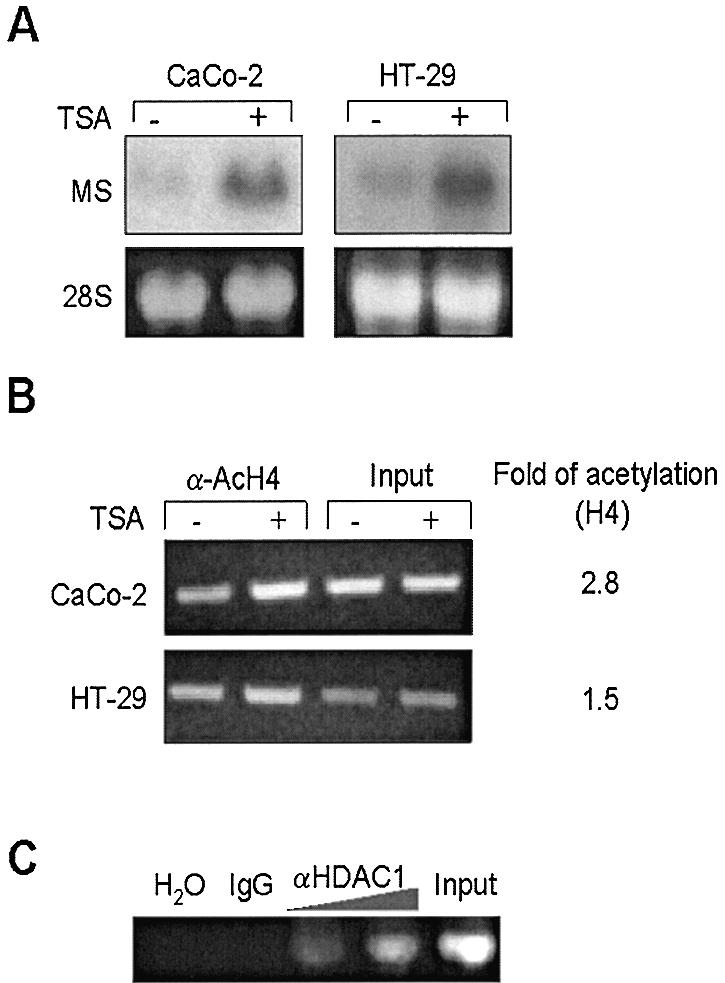
Mitochondrial HMG-CoA synthase, a target gene for HDAC inhibitors: TSA induces mitochondrial HMG-CoA synthase mRNA expression through mitochondrial HMG-CoA synthase promoter acetylation in the chromatin complex. (A) HT-29 and Caco-2 cells were treated with 0.3 µM TSA (+) or DMSO (–) for 24 h. The mitochondrial HMG-CoA synthase mRNA levels were analyzed by northern blot. Levels of 18S rRNA are also shown to correct for gel loading variation. (B) ChIPs were performed using chromatin isolated from HT-29 and CaCo-2 cells either treated with TSA (+) for 24 h or untreated (–). The chromatin preparations were immunoprecipitated with antibodies against acetylated histone H4 (α-AcH4) and analyzed by PCR to detect the HMG-CoA synthase promoter associated to acetylated histones. Aliquots of chromatin were also analyzed before immunoprecipitation (Input). Relative histone acetylation levels (Fold of acetylation) were determined by quantitation of PCR product using BIOPROFIL® image analysis software, BIO-1D (Vilber Lourmat) and by correction with the input data and represented as the ratio of relative acetylation in the presence versus the absence of TSA treatment. (C) ChIP assay of HDAC1 bound to endogenous mitochondrial HMG-CoA synthase promoter. CaCo-2 chromatin, isolated as described above, was immunoprecipitated with increasing amounts of anti-HDAC1 or preimmune serum (IgG). PCRs shown in (B) and (C) were performed with 5 µl of DNA and 30 cycles of amplification were used.
Overexpression of histone deacetylase 1 (HDAC1) leads to HMG-CoA synthase promoter hypoacetylation and reduces its transcriptional activity
The role of histone hypoacetylation in mitochondrial HMG-CoA synthase expression was next examined by co-transfection of a reporter luciferase gene under the control of mitochondrial HMG-CoA synthase promoter and increasing amounts of a plasmid encoding the HDAC1 in CaCo-2 cells. Figure 3A shows that the overexpression of HDAC1 reduces the transcriptional activity of mitochondrial HMG-CoA synthase promoter. The effect of HDAC1 expression on mitochondrial HMG-CoA synthase promoter hypoacetylation is demonstrated by ChIP assay using antibodies against acetylated histone H4 (Fig. 3B, lanes 2 and 3). The recruitment of HDAC1 over the transfected mitochondrial HMG-CoA synthase promoter is demonstrated by using antibodies against the Flag-epitope-tagged HDAC1 (Fig. 3B, lanes 4 and 5). The expression of HDAC1 is shown by western blot of whole cell extracts (Fig. 3B, right).
Figure 3.
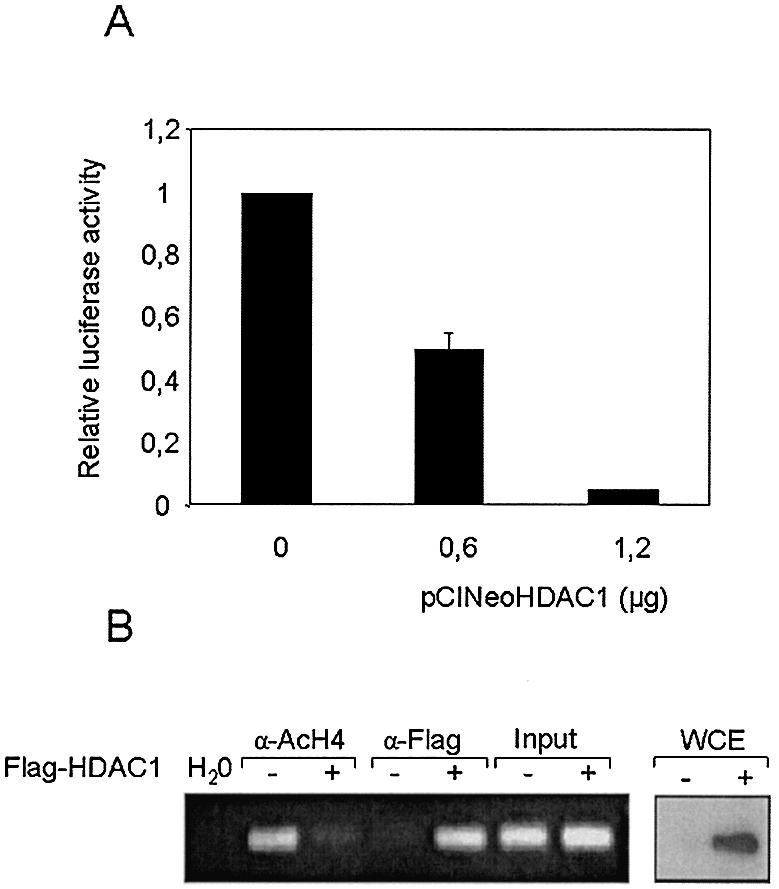
Recruitment of HDAC1 over transfected mitochondrial HMG-CoA synthase promoter: deacetylation of promoter associated histones and down-regulation of mitochondrial HMG-CoA synthase transcriptional activity. (A) Caco-2 cells were transiently co-transfected with 4 µg of the reporter plasmid –116pGl3 in combination with increasing amounts of an expression vector for the HDAC1 (pCINeoHDAC1). Luciferase activities are normalized by micrograms of protein assayed. Data are means and standard deviations from four independent experiments with two plates each. (B) ChIP assay to demonstrate histone deacetylation as a result of expression and recruitment of HDAC1 over mitochondrial HMG-CoA synthase proximal promoter. Caco-2 cells were transiently transfected with 10 µg of the reporter plasmid –116pGl3 (–) or in combination with 5 µg of an expression vector for the Flag-epitope-tagged HDAC1 (pCDNA3FlagHDAC1) (+). Thirty-six hours after transfection chromatin preparations were obtained and immunoprecipitated with antibodies against acetylated histone H4 (α-AcH4) and Flag epitope (M2). The immunoprecipitates were then analyzed by PCR using specific primers for mitochondrial HMG-CoA synthase promoter and luciferase gene. Aliquots of chromatin were also analyzed before immunoprecipitation (Input). Thirty cycles of PCR were used. Western blot analysis of Flag-HDAC1 protein in whole cell extracts from control (–) or pCDNA3FlagHDAC1 transfected (+) CaCo-2 cells is shown on the right.
TSA activates transcription from the rat HMG-CoA synthase promoter: analysis of the histone deacetylase inhibitor responsive elements in CaCo-2 cells
The proximal promoter of the mitochondrial HMG-CoA synthase gene contains a peroxisome proliferator responsive element (PPRE, located in position –104 to –92) and a Sp1 binding site (located in position –57 to –48) (14 and unpublished data). To analyze the role of these elements on promoter TSA mediated stimulation, we performed transient transfection experiments with several constructs containing the reporter luciferase gene under the control of mitochondrial HMG-CoA synthase promoter variants (Fig. 4A). Both elements appear to be critical for promoter activity, since mutation of either of the two elements, as well as deletion of the PPRE, dramatically diminished the promoter transcriptional activity in CaCo-2 cell line (Fig. 4B). These constructs are still active, and in agreement with ChIP assay results (Figs 2C and 3), TSA is able to induce the reporter luciferase activity driven by the mitochondrial HMG-CoA synthase promoter variants (Fig. 4C).
Figure 4.




TSA activates transcription of rat mitochondrial HMG-CoA synthase promoter: 5′ deletion and mutational analysis of the proximal HMG-CoA synthase promoter in CaCo-2 cells. (A) The promoter constructs used. (B) The promoter constructs were transiently transfected into CaCo-2 cells. In each transfection experiment half of the cells were treated with 0.3 µM TSA or 5 mM butyrate for 24 h. Luciferase activities of untreated cells, and TSA or butyrate treated cells are depicted relative to the respective Renilla activities. (C) Mean fold promoter activation for each construct by normalizing TSA treated relative luciferase activity to relative luciferase activity from untreated cells. (D) Mean fold promoter activation for each construct by normalizing butyrate treated relative luciferase activity to relative luciferase activity from untreated cells. Data are means and standard deviations from three independent experiments with two plates each.
Mutation of mitochondrial HMG-CoA synthase Sp1 site [–116pGl3(M-Sp1)] causes a dramatic loss in TSA responsiveness, while a construct containing only the Sp1 binding site (–62pGl3) or the Sp1 site and a mutated PPRE [–116pGl3(M-PPRE)] were highly responsive to TSA. Therefore, these results suggest that the critical element in the recruitment of HDAC activities to mitochondrial HMG-CoA synthase promoter is the Sp1 binding site. Surprisingly, the wild-type promoter containing the PPRE was only weakly sensitive (∼9-fold induction) to TSA induction. The same results were obtained when cells were treated with 5 mM butyrate (Fig. 4D). The low induction of –116 promoter produced by TSA treatment was observed even when 5- or 10-fold lower amounts of plasmid were used in the transfection assay (data not shown).
High affinity interaction of Sp1 and Sp3 with the Sp1 binding site is required for the trans-activation of mitochondrial HMG-CoA synthase promoter by TSA
Previous studies performed with HepG2 cell nuclear extracts showed that Sp1 and Sp3 proteins can interact specifically with the Sp1 binding site present in position –57 to –48 of mitochondrial HMG-CoA synthase promoter (data not shown). Figure 5A shows that this sequence is also able to form shift complexes in the presence of CaCo-2 cell nuclear extracts. The specificity of these complexes was assessed by competition with an excess of cold probe (lane 6). The addition of anti-Sp1 (lane 3), anti-Sp3 (lane 4) or both antibodies (lane 5) to the binding reaction results in the supershift of the upper and the two lower complexes. Therefore, Sp1 and Sp3 proteins present in the nuclear extract of the CaCo-2 cell line are able to bind to the Sp1 site of the mitochondrial HMG-CoA synthase. The shift pattern of the Sp1 binding site was similar in the presence of nuclear extracts from CaCo-2 cells regardless of whether they had been treated or not with butyrate or TSA (data not shown). To directly address whether Sp1 and Sp3 are associated with the endogenous mitochondrial HMG-CoA synthase promoter, we performed a new ChIP assay using antibodies against either Sp1 or Sp3 proteins. Figure 5B shows that in the CaCo-2 cell line both Sp1 and Sp3 are associated with the proximal promoter of the gene.
Figure 5.
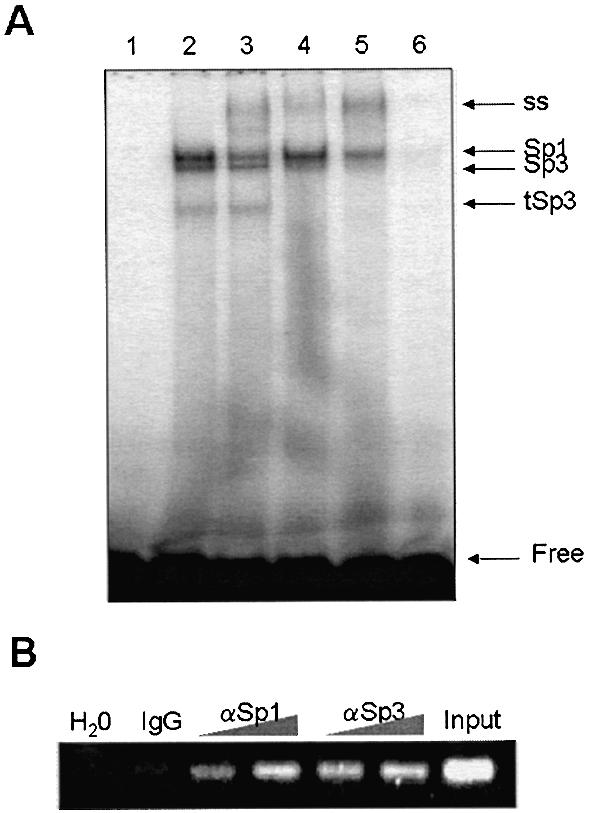
Sp1 and Sp3 proteins interact with the main HDAC inhibitor responsive element. (A) EMSA of the mitochondrial HMG-CoA synthase Sp1 site probe (GAGGCGGGGC) in the presence of CaCo-2 cell nuclear extracts (lanes 2–6) and anti-Sp1 (lanes 3 and 5) and anti-Sp3 (lanes 4 and 5) antibodies. Lane 6 contains a 200-fold molar excess of unlabeled probe. ss, supershifted; tSp3, truncated Sp3. (B) In vivo interaction of Sp1 and Sp3 with mitochondrial HMG-CoA synthase promoter is demonstrated by chromatin immunoprecipitation assay. Chromatin isolated from CaCo-2 cells was immunoprecipitated with increasing amounts of antibodies against Sp1, Sp3 or pre-immune serum (IgG) and analyzed by PCR. Thirty cycles of PCR were used. Input sample containing crude chromatin extract prior to immunoprecipitation and a negative control (water) are also shown.
To investigate further the role of such binding in the TSA-mediated induction on the promoter, we used previously characterized orthologous genes (25,26). Thus, we knew that at similar positions from the transcriptional start site, the rat (from –57 to –48), human (from –63 to –54), and pig (from –67 to –58) HMG-CoA synthase genes have a Sp1 binding site sequence; and that the pig gene was less responsive to TSA (data not shown). Since the human and rat Sp1 sequences are identical, but two nucleotides differ between pig and rat or human, we introduced two single point mutations in the rat Sp1 binding site to recreate the orthologous Sp1 site sequence, and then proceeded to study the binding of Sp1 and Sp3 proteins as well as the TSA responsiveness of the mutated Sp1 site. A Scatchard assay performed with CaCo-2 cell nuclear extracts with the wild-type or the mutated Sp1 site probe shows that the mutated Sp1 sequence has less affinity for Sp1 and Sp3 proteins than the wild-type probe (Fig. 6A). Interestingly, the mutated low affinity Sp1 sequence (Sp1-la) is less responsive to TSA than is the wild-type (Sp1-wt) (Fig. 6B). These data show that the affinity binding of Sp1 and Sp3 proteins to the Sp1 site correlate with the strength of activation produced by TSA.
Figure 6.


High affinity interaction of Sp1 and Sp3 is required for the TSA trans-activation. (A) Assessment of the dissociation constant (Kd) of wild-type Sp1 (GAGGCGGGGC) and Sp1 mutated (GGGGCAGGGC) probes by Scatchard analysis. EMSA was performed using nuclear extracts of CaCo-2 cells and increasing amounts of radiolabeled Sp1 wild-type [Sp1-wt (black squares)] or mutated [Sp1-la (black dots)] probes. Complexes [Sp1, Sp3 and the truncated form of Sp3 (tSp3)] and free probe were quantified by a Storm PhosphorImager® (Amersham Pharmacia Biotech). Scatchard analysis revealed that the Kd was higher for the mutated Sp1 site probe (761.28 pM for Sp1, 189.68 pM for Sp3 and 791.04 pM for tSp3) than for the wild-type probe (43.90 pM for Sp1, 27.61 pM for Sp3 and 18.20 pM for tSp3). (B) Analysis of the induction by TSA of HMG-CoA synthase promoter containing Sp1-wt or Sp1-la sequences. CaCo-2 cells were transiently transfected with 4 µg of the rat mitochondrial HMG-CoA synthase promoter-luciferase constructs containing either Sp1-wt sequence (–62pGl3) or Sp1-la sequence [–62pGl3(Sp1-la)] in combination with the control plasmid pRL-CMV. In each transfection experiment half of the cells were treated with 0.3 µM TSA for 24 h. Luciferase activities of untreated cells and TSA-treated cells are depicted relative to the respective Renilla activities.
Sp1 and Sp3 proteins mediate responsiveness to TSA in CaCo-2 cells
In order to clarify the possible role of Sp1 and Sp3 in the TSA induction observed on HMG-CoA synthase promoter, we used a one-hybrid system in which each of these proteins is fused to the DNA binding domain of the bacterial transcription factor GAL4. The ability of these recombinant proteins to trans-activate the (GAL4)5TATALuc-reporter gene was assayed in CaCo-2 cell line. Table 1 shows that both recombinant proteins are able to trans-activate the reporter luciferase gene. GAL4-Sp1 is a stronger trans-activator than GAL4-Sp3. However, transcription activity mediated by both recombinant proteins is similar after TSA treatment, indicating that the activation induced by TSA is stronger for the GAL4-Sp3 than it is for the GAL4-Sp1 system (see also Fig. 7). As a control for the specificity of the TSA effect over Sp1 or Sp3 recombinant proteins, we also transfected the fusion protein constituted by the LBD of the nuclear receptor PPARγ and the DNA binding domain of GAL4. This fusion protein (GAL4-PPARγ) is also able to activate transcription from the luciferase reporter plasmid in the presence of Brl49653, a specific PPARγ ligand, and this activity is not affected by the presence of TSA. Table 1 also shows the effect of HDAC1 co-transfection in both systems. Interestingly, while HDAC1 represses the GAL4-Sp1 system, no effect was observed in the GAL4-Sp3 system, thus indicating that Sp1 could play a role in the down-regulation of mitochondrial HMG-CoA synthase by HDAC1 (see Fig. 3). In addition, HDAC1 co-transfection is able to reduce the effect of TSA in both systems, but HDAC1 down-regulation is complete only in the GAL4-Sp3 system (Table 1) (see Discussion).
Table 1. Activation of the luciferase reporter gene by co-transfection with different GAL4-recombinant proteins.
| (GAL4)5TATALuc reporter fold induction | ||
|---|---|---|
| –TSA | +TSA | |
| Gal4-Sp1 | 17.45 ± 5.4 | 84.26 ± 9.83 |
| Gal4-Sp1 + HDAC1 | 4.70 ± 1.2 | 32.0 ± 6.8 |
| Gal4-Sp3 | 3.81 ± 0.63 | 56.82 ± 11.3 |
| Gal4-Sp3 + HDAC1 | 3.10 ± 0.8 | 4.15 ± 0.43 |
| Gal4-PPARγ | 63.9 ± 25.5 | 69.13 ± 24.3 |
CaCo-2 cells were transfected with 4 µg of pFRLuc reporter plasmid in combination with 1.3 µg of the effector plasmids (pM, pM-Sp1, pM-Sp3 GAL4-PPAR) and, when indicated, 0.2 µg of pCDNA3FlagHDAC1. In each transfection experiment half of the cells were treated with 0.3 µM TSA for 24 h. Luciferase activities were measured as relative light mU normalized per µg of protein assayed, and expressed as GAL4 reporter fold induction. Data are means and standard deviations from three independent experiments with two plates each.
Figure 7.

GAL4 dominant negative Sp1 and Sp3 proteins are not activated by TSA treatment. CaCo-2 cells were transiently transfected with 4 µg of pFRLuc reporter plasmid in combination with 1.3 µg of the effector plasmids pM, pM-Sp1, pM-Sp3, pM-DNSp1 and pM-DNSp3. In each transfection experiment half of the cells were incubated with 0.3 µM TSA for 24 h. Relative luciferase activities, normalized per microgram of protein, are represented as fold induction of the activity in the presence of TSA over that observed in the absence of the TSA. All luciferase assays were carried out in triplicate.
To further determine the TSA response region of Sp1 or Sp3, we used dominant negative forms of both proteins fused to the DNA binding domain of GAL4 (GAL4-DNSp1 and GAL4-DNSp3, respectively) (17). Since these proteins lack their respective trans-activation domains, both showed low trans-activation capacity, of the (GAL4)5TATALuc-reporter gene, when compared with their full-length precursors (9.6 ± 3.2% of GAL4-DNSp1 versus GAL4-Sp1; and 4.1 ± 1.2% of GAL4-DNSp3 versus GAL4-Sp3). Figure 7 shows that GAL4-DNSp1 or GAL4-DNSp3 fusion proteins could not be activated by TSA treatment. These results suggest that the trans-activation domain of Sp1 or Sp3 mediated the transcriptional activation though the Sp1 site by TSA.
DISCUSSION
The results presented here clearly demonstrate that mitochondrial HMG-CoA synthase gene expression is regulated by the acetylation/deacetylation of the histones associated with its promoter. We have shown that butyrate affects the transcription of the gene in vivo (Fig. 1) and in vitro (Fig. 4D). In addition, a highly specific inhibitor of HDAC activity, as is the case of TSA, is also a potent stimulator of the transcriptional activity of mitochondrial HMG-CoA synthase promoter (Fig. 4C), and this up-regulation correlates with an increase in the hyperacetylation status of histone H4 (Fig. 2B).
Following transient transfection experiments on CaCo-2 cells, we have also shown that HDAC1 is recruited through the proximal mitochondrial HMG-CoA synthase promoter (Fig. 3B), resulting in the hypoacetylation of the transfected promoter and consequently in a drastic reduction of its transcriptional activity (Fig. 3A). The results obtained from these experiments are in agreement with the ChIP assays performed to detect HDAC1 on endogenous HMG-CoA synthase promoter in CaCo-2 cells. (Fig. 2C). All these data demonstrate that the transcriptional activity of both the endogenous gene and a transfected reporter can be repressed by recruitment of HDAC1 to the promoter.
The selectivity of the HDAC inhibition effect on gene expression has been suggested by the small number of genes whose expression is altered in response to HDAC inhibitors. Approximately 2% of genes were changed upon TSA treatment in human lymphoid cell lines (11). Recently, mitochondrial HMG-CoA synthase has been identified within a small group of genes up-regulated by demethylation and HDAC inhibition in human colorectal cancer cells (27). This is a relevant finding since it has been previously suggested that impaired β-oxidation could be caused by the lack of induction of the HMG-CoA mitochondrial synthase (28). Therefore, up-regulation of mitochondrial HMG-CoA synthase by butyrate could play a significant role in determining the butyrate oxidation rate (see below).
Mutational analysis of the proximal mitochondrial HMG-CoA synthase promoter (Fig. 4) has revealed that the main deacetylase inhibitor responsive element is the Sp1 site located between –57 and –48 relative to the transcription start site. This Sp1 site is able to bind Sp1 and Sp3 proteins from nuclear extracts of HepG2 (data not shown) or CaCo-2 cell lines (Fig. 5A). However, the intensity and the pattern of the protein complexes found in the electrophoretic mobility shift assay (EMSA) experiments did not reveal any substantial differences between the complexes obtained when using nuclear extracts from control or butyrate/TSA-treated cells (data not shown). Complete mutation of this element in its natural context removes most of the effect of the TSA (Fig. 4C) or butyrate (Fig. 4D) treatment. The role of Sp1 and Sp3 proteins in the TSA response of the mitochondrial promoter was further confirmed using a two-single point mutation of the rat Sp1 binding site. This mutation (Sp1-la) allows the binding of Sp1 and Sp3 proteins, but with a lower affinity than the wild-type Sp1 site (Sp1-wt in Fig. 6A). This change in affinity (see legend of Fig. 6A for complexes Kd) clearly diminished the TSA response of the mitochondrial HMG-CoA proximal promoter (Fig. 6B). Therefore, the strength of the Sp1 and Sp3 protein binding to the Sp1 site correlates with the TSA effect on promoter transcription. These data suggest that only the Sp1 binding site with high affinity for the Sp1 family of proteins are candidates for recruiting HDAC-containing complexes.
Recent studies have involved GC-rich Sp1 binding sites as targets of the butyrate effect over a small list of butyrate regulated genes, including WAF1/Cip gene (17), thymidine kinase gene (24), the mouse ferritin H gene (29), Gαi2 gene (30), IGF binding protein-3 gene (31), cyclooxigenase-1 gene (32), human telomerase gene (33), and the more recently described human luteinizing hormone receptor (hLHR) gene (34). Nevertheless, a strict correlation between the chromatin acetylation level and transcriptional activity of the gene has not always been shown. The involvement of Sp1 and/or Sp3 proteins, in the role of HDAC activities over these GC-rich Sp1 binding site sequences, has also been described for promoters of some of the genes transcriptionally activated by butyrate. This is the case of the human telomerase gene (33) or the hLHR gene (34). Interestingly, a differential participation of Sp1 and Sp3 proteins has been described in the induction of WAF1/Cip gene by TSA, which seems to be dependent on the cellular context. Therefore, while only Sp3 is responsible in mediating the TSA induction of WAF1/Cip gene in MG63 cells (17), both proteins are involved in the regulation of the same gene by TSA in NIH3T3 cells (35). A similar situation has been described for the IGF binding protein-3 gene, since induction by butyrate implies different responsive elements depending on the cellular context (31,36).
Using one-hybrid assays in CaCo-2 cells (Table 1), we have shown that Sp1 and Sp3 proteins could be differentially involved in the regulation of mitochondrial HMG-CoA synthase promoter by HDAC1 activity because: (i) GAL4-Sp3 acts only as a weak trans-activator factor whereas GAL4-Sp1 protein is able to strongly activate the transcription of the reporter gene; (ii) overexpression of HDAC1 is able to block the trans-activation effect of GAL4-Sp1 protein without affecting the low activity of the GAL4-Sp3 system; (iii) in the presence of TSA, we observed an inhibitory effect of HDAC1 especially in the GAL4-Sp3 system.
The direct interaction of Sp1 and HDAC1 has been described, and a model in which Sp1 transcription regulation involves a competition between HDAC1 and transcription factors has been proposed (24). Therefore, Sp1 is able to cooperate, in addition with HDAC1, with proteins such as p300 (37,38) with HAT activities. In the case of Sp3, the interaction with HDAC1 and HDAC2 has been recently described (39,40). Moreover, HDAC activities could be involved in the regulation of Sp3 transcription activity since Sp3 activity is modulated by the acetylation/deacetylation process (41). Our data will be compatible with a model in which, in the absence of TSA, GAL4-Sp3 protein is present mainly in an inactive or weakly active form, which is converted into a strong activator of transcription in the presence of TSA. However, the HDAC1 down-regulation, observed in the GAL4-Sp3 system in the presence of TSA (Table 1), also suggests a protein–protein interaction, or protein displacement, in the HDAC1-containing complex that mediated down-regulation of the Sp3 activity.
Although the Sp1 binding element seems to be the critical cis element involved in the recruitment of HDAC activities over mitochondrial HMG-CoA synthase promoter, it would seem possible that the PPRE-bound proteins could somehow balance the acetylation status of the histones associated with mitochondrial HMG-CoA synthase promoter. In fact, the acetylation levels of histone H4 associated with mitochondrial HMG-CoA synthase promoter increases following treatment with PPARγ ligands (data not shown). These data could explain why the promoter construct that has the PPRE and Sp1 site (–116 in Fig. 4) was less responsive to TSA than the constructs that did not have the element [–116 (M-PPRE) and –62]. Nevertheless, the identity and relevance of the co-activators involved in the induction of mitochondrial HMG-CoA synthase promoter acetylation in response to PPAR ligands remains to be studied.
The study of mitochondrial HMG-CoA synthase gene regulation by butyrate in the colon, and the molecular mechanisms involved in this up-regulation, could be important in understanding the contrasting effects of butyrate in normal colonic epithelial cells (in vivo and ex vivo) and in transformed colonic epithelial cells in vitro. Butyrate has been described as tending to stimulate cell proliferation in normal colonic epithelial cells. However, butyrate reduces this proliferation and stimulates cell differentiation and apoptosis in colon cancer cell lines (8–10). One of the hypotheses that seeks to explain the ‘butyrate paradox’ is that normal colonocites are able to metabolize butyrate efficiently, which could result in a reduction in the intracellular concentration and, therefore, in a reduction in its growth inhibition capacity (42). This butyrate metabolizing capability could be lost in colon cancer cell lines (43). In this context, it has recently been described that a lack of induction of the mitochondrial HMG-CoA synthase, which leads to the accumulation of acetyl-CoA and/or β-oxidation intermediates which are strong inhibitors of acyl-CoA dehydrogenases, leads to an impaired β-oxidation (28).
As one of the metabolic destinies of the butyrate present in the colon is the production of ketone bodies (7), we are currently trying to determine whether the differential expression of the ketogenic key enzyme, mitochondrial HMG-CoA synthase, might be responsible in part for these differences in the metabolism of butyrate. What we do describe in this paper though is that butyrate up-regulates the expression of a gene which could modulate the levels of this short fatty acid inducing its metabolization.
Acknowledgments
ACKNOWLEDGEMENTS
We are indebted to Dr C. Seiser for supplying the expression vector for the mouse HDAC1 and Dr Y. Sowa for providing the GAL4-Sp1, GAL4-Sp3, GAL4-DNSp1 and GAL4-DNSp3 constructs. We thank the Language Service at the University of Barcelona for valuable assistance in the preparation of the English manuscript. Grants BM2001-2891 (to P.F.M.) and PB97-0958 (to D.H.), from the Dirección General de Investigación, supported this study.
REFERENCES
- 1.Hegardt F.G. (1999) Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem. J., 338, 569–582. [PMC free article] [PubMed] [Google Scholar]
- 2.Girard J., Ferré,P., Pegorier,J.P. and Duée,P.H. (1992) Adaptations of glucose and fatty acid metabolism during perinatal period and suckling-weaning transition. Physiol. Rev., 72, 507–562. [DOI] [PubMed] [Google Scholar]
- 3.McGarry J.D. and Brown,N.F. (1997) The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem., 244, 1–14. [DOI] [PubMed] [Google Scholar]
- 4.Thumelin S., Forestier,M., Girard,J. and Pegorier,J.P. (1993) Developmental changes in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene expression in rat liver, intestine and kidney. Biochem. J., 292, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serra D., Asins,G. and Hegardt,F.G. (1993) Ketogenic mitochondrial 3-hydroxy 3-methylglutaryl-CoA synthase gene expression in intestine and liver of suckling rats. Arch. Biochem. Biophys., 30, 445–448. [DOI] [PubMed] [Google Scholar]
- 6.Gil-Gomez G., Ayte,J. and Hegardt,F.G. (1993) The rat mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme-A-synthase gene contains elements that mediate its multihormonal regulation and tissue specificity. Eur. J. Biochem., 213, 773–779. [DOI] [PubMed] [Google Scholar]
- 7.Cherbuy C., Darcy-Vrillon,B., Morel,M.T., Pegorier,J.P. and Duée,P.H. (1995) Effect of germfree state on the capacities of isolated rat colonocytes to metabolize n-butyrate, glucose and glutamine. Gastroenterology, 109, 1890–1899. [DOI] [PubMed] [Google Scholar]
- 8.Hass R., Busche,R., Luciano,L., Reale,E. and Engelhardt,W.V. (1997) Lack of butyrate is associated with induction of Bax and subsequent apoptosis in the proximal colon of guinea pig. Gastroenterology, 112, 875–881. [DOI] [PubMed] [Google Scholar]
- 9.Hague A., Singh,B. and Paraskeva,C. (1997) Butyrate acts as a survival factor for colonic epithelial cells: further fuel for the in vivo versus in vitro debate. Gastroenterology, 112, 1036–1040. [DOI] [PubMed] [Google Scholar]
- 10.Heerdt B.G., Houston,M.A. and Augenlicht,L.H. (1994) Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res., 54, 3288–3294. [PubMed] [Google Scholar]
- 11.Van Lint C., Emiliani,S. and Verdin,E. (1996) The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Express., 5, 245–253. [PMC free article] [PubMed] [Google Scholar]
- 12.Kuo M.H. and Allis,C.D. (1998) Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays, 20, 615–626. [DOI] [PubMed] [Google Scholar]
- 13.Spencer V.S. and Davie,J.R. (1999) Role of covalent modifications of histones in regulating gene expression. Gene, 240, 1–12. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez J.C., Gil-Gomez,G., Hegardt,F.G. and Haro,D. (1994) Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J. Biol. Chem., 269, 18767–18772. [PubMed] [Google Scholar]
- 15.Gelman L., Zhou,G., Fajas,L., Raspe,E., Fruchart,J.C. and Auwerx,J. (1999) p300 interacts with the N- and C-terminal part of PPARgamma2 in a ligand-independent and -dependent manner, respectively. J. Biol. Chem., 274, 7681–7688. [DOI] [PubMed] [Google Scholar]
- 16.Xu H.E., Stanley,T.B., Montana,V.G., Lambert,M.H., Shearer,B.G., Cobb,J.E., McKee,D.D., Galardi,C.M., Plunket,K.D., Nolte,R.T., Parks,D.J., Moore,J.T., Kliewer,S.A., Willson,T.M. and Stimmel,J.B. (2002) Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature, 415, 813–817. [DOI] [PubMed] [Google Scholar]
- 17.Sowa Y., Orita T., Minamikawa-Hiranabe,S., Mizuno,T., Nomura,H. and Sakai,T. (1999) Sp3, but not Sp1, mediates the transcriptional activation of the p21/WAF1/Cip1 gene promoter by histone deacetylase inhibitor. Cancer Res., 59, 4266–4270. [PubMed] [Google Scholar]
- 18.Walker G.E., Wilson,E.M., Powel,L.D. and Oh,Y. (2001) Butyrate, a histone deacetylase inhibitor, activates the human IGF binding protein-3 promoter in breast cancer cells molecular mechanism involves an Sp1/Sp3 multiprotein complex. Endocrinology, 142, 3817–3827. [DOI] [PubMed] [Google Scholar]
- 19.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 20.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 21.Rodriguez J.C., Ortiz,J.A., Hegardt,F.G. and Haro,D. (1997) Chicken ovalbumin upstream-promoter transcription factor (COUP-TF) could act as a transcriptional activator or repressor of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene. Biochem. J., 326, 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction Gene, 77, 51–59. [DOI] [PubMed] [Google Scholar]
- 23.Elbrecht A., Chen,Y., Adams,A., Berger,J., Griffin,P., Klatt,T., Zhang,B., Menke,J., Zhou,G., Smith,R.G. and Moller,D.E. (1999) L-764406 is a partial agonist of human peroxisome proliferator-activated receptor gamma. The role of Cys313 in ligand binding. J. Biol. Chem., 274, 7913–7922. [DOI] [PubMed] [Google Scholar]
- 24.Doetzlhofer A., Rotheneder,H., Lagger,G., Koranda,M., Kurtev,V., Brosch,G., Wintersberger,E. and Seiser,C. (1998) Histone deacetylase 1 can repress transcription by binding to Sp1. Mol. Cell. Biol., 19, 5504–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mascaro C., Buesa,C., Ortiz,J.A., Haro,D. and Hegardt,F.G. (1995) Molecular cloning and tissue expression of human mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase. Arch. Biochem. Biophys., 317, 385–390. [DOI] [PubMed] [Google Scholar]
- 26.Ortiz J.A., Mallolas,J., Nicot,C., Bofarull,J., Rodríguez,J.C., Hegardt,G.F., Haro,D. and Marrero,P.F. (1999) Isolation of pig mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene promoter: characterization of a peroxisome proliferator-responsive element. Biochem. J., 337, 329–335. [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki H., Gabrielson,E., Chen,W., Anbazhagan,R., van Engeland,M., Weijenberg,M.P., Herman,J.G. and Baylin,S.B. (2002) A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nature Genet., 31, 141–149. [DOI] [PubMed] [Google Scholar]
- 28.Le May C., Pineau,T., Bigot,K., Kohl,C., Girard,J. and Pégorier,J.P. (2000) Reduced hepatic fatty acid oxidation in fasting PPARalpha null mice is due to impaired mitochondrial hydroxymethylglutaryl-CoA synthase gene expression. FEBS Lett., 475, 163–166. [DOI] [PubMed] [Google Scholar]
- 29.Tsuji Y., Moran,E., Torti,S.V. and Torti,F.M. (1999) Transcriptional regulation of the mouse ferritin H gene. Involvement of p300/CBP adaptor proteins in FER-1 enhancer activity. J. Biol. Chem., 274, 7501–7507. [DOI] [PubMed] [Google Scholar]
- 30.Yang J., Kawai,Y., Hanson,R.W. and Arinze,I.J. (2001) Sodium butyrate induces transcription from the G alpha(i2) gene promoter through multiple Sp1 sites in the promoter and by activating the MEK-ERK signal transduction pathway. J. Biol. Chem., 276, 25742–25752. [DOI] [PubMed] [Google Scholar]
- 31.Walker G.E., Wilson,E.M., Powell,D. and Oh,Y. (2001) Butyrate, a histone deacetylase inhibitor, activates the human IGF binding protein-3 promoter in breast cancer cells: molecular mechanism involves an Sp1/Sp3 multiprotein complex. Endocrinology, 142, 3817–3827. [DOI] [PubMed] [Google Scholar]
- 32.Taniura S., Kamitani,H., Watanabe,T. and. Eling,T.E. (2002) Transcriptional regulation of cyclooxygenase-1 by histone deacetylase inhibitors in normal human astrocyte cells. J. Biol. Chem., 277, 16823–16830. [DOI] [PubMed] [Google Scholar]
- 33.Takakura M., Kyo,S., Sowa,Y., Wang,Z., Yatabe,N., Maida,Y., Tanaka,M. and Inoue,M. (2001) Telomerase activation by histone deacetylase inhibitor in normal cells. Nucleic Acids Res., 29, 3006–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y. and Dufau,M.L. (2002) Silencing of transcription of the human luteinizing hormone receptor gene by histone deacetylase-mSin3A complex. J. Biol. Chem., 277, 33431–33438. [DOI] [PubMed] [Google Scholar]
- 35.Xiao H., Hasegawa,T. and Isobe,K. (1999) Both Sp1 and Sp3 are responsible for p21waf1 promoter activity induced by histone deacetylase inhibitor in NIH3T3 cells. J. Cell. Biochem., 73, 291–302. [PubMed] [Google Scholar]
- 36.Tsubaki J., Hwa,V., Twigg,S.M. and Rosenfeld,R.G. (2002) Differential activation of the IGF binding protein-3 promoter by butyrate in prostate cancer cells. Endocrinology, 143, 1778–1788 [DOI] [PubMed] [Google Scholar]
- 37.Billon N., Carlisi,D., Datto,M.B., van Grunsven,L.A., Watt,A., Wang,X.F. and Rudkin,B.B. (1999) Cooperation of Sp1 and p300 in the induction of the CDK inhibitor p21WAF1/CIP1 during NGF-mediated neuronal differentiation. Oncogene, 6, 2872–2882. [DOI] [PubMed] [Google Scholar]
- 38.Xiao H., Hasegawa,T. and Isobe,K. (2000) p300 collaborates with Sp1 and Sp3 in p21(waf1/cip1) promoter activation induced by histone deacetylase inhibitor. J. Biol. Chem., 275, 1371–1376. [DOI] [PubMed] [Google Scholar]
- 39.Sun J.M., Chen,H.Y., Moniwa,M., Litchfield,D.W., Seto,E. and Davie,J.R. (2002) The transcriptional repressor Sp3 is associated with CK2-phosphorylated histone deacetylase 2. J. Biol. Chem., 277, 35783–35786. [DOI] [PubMed] [Google Scholar]
- 40.Won J., Yim,J. and Kim,T.K. (2002) Sp1 and Sp3 recruit histone deacetylase to repress transcription of human telomerase reverse transcriptase (hTERT) promoter in normal human somatic cells. J. Biol. Chem., 277, 38230–38238. [DOI] [PubMed] [Google Scholar]
- 41.Braun H., Koop,R., Ertmer,A., Nacht.S. and Suske,G. (2001) Transcription factor Sp3 is regulated by acetylation. Nucleic Acids Res., 29, 4994–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jass J.R. (1985) Diet, butyric acid and differentiation of gastrointestinal tract tumours. Med. Hypotheses, 18, 113–118. [DOI] [PubMed] [Google Scholar]
- 43.Mariadason J.M., Velcich,A., WilsonA.J., Augenlicht,L.H. and Gibson,P.R. (2001) Resistance to butyrate-induced cell differentiation and apoptosis during spontaneous Caco-2 cell differentiation. Gastroenterology, 120, 889–899. [DOI] [PubMed] [Google Scholar]