


Abstract
We have determined the complete mitochondrial DNA (mtDNA) sequences of three chytridiomycete fungi, Monoblepharella15, Harpochytrium94 and Harpochytrium105. Our phylogenetic analysis based on concatenated mitochondrial protein sequences confirms the placement of Mono blepharella15 together with Harpochytrium spp. and Hyaloraphidium curvatum within the taxonomic order Monoblepharidales, with overwhelming support. These four mtDNA sequences encode the standard fungal mitochondrial gene complement and, like certain other chytridiomycete fungi, encode a reduced complement of 7–9 tRNAs, some of which require 5′-tRNA editing to be functional. Highly conserved sequence elements were identified upstream of almost all protein-coding genes in the mtDNAs of Monoblepharella15 and both Harpochytrium species. Finally, a guanosine residue is conserved upstream of the predicted ATG or GTG start codons of almost every protein-coding gene in these genomes. The appearance of this G residue correlates with the presence of a non-canonical cytosine residue at position 37 in the anticodon loop of the mitochondrial initiator tRNAs. Based on the unorthodox features in these four genomes, we propose that a 4 bp interaction between the CAUC anticodon of these tRNAs and GAUG/GGUG codons is involved in translation initiation in monoblepharidalean mitochondria. Intriguingly, a similar interaction may also be involved in mitochondrial translation initiation in the sea anemone Metridium senile.
INTRODUCTION
Although members of the ‘higher’ fungi (ascomycetes and basidiomycetes) have received the vast majority of scientific study, the ‘lower’ fungi (chytridiomycetes and zygomycetes) contain most of the genetic diversity within the fungal kingdom. Unfortunately, few molecular, and even fewer genomic data, are available currently from lower fungi. In addition, the evolutionary relationships of lower fungi to each other and to the higher fungi are poorly understood.
The most widely used molecular data for inferring fungal phylogeny, rDNA sequence data, have not been able to resolve the fungal phylogeny with significant support (1–5), and thus cannot be used alone to confirm or refine the taxonomic classification of the fungi. In fact, much more sequence information than is contained in rRNA is required to recover the phylogenetic signal needed to resolve a phylogeny as deep as that of the fungi, which are believed to have diverged from a common ancestor with the animals ∼1 billion years ago (6). Further, the taxon sampling in other data sets, such as tubulins (7), RPB1 [the largest subunit of RNA polymerase II (8)] and concatenated nuclear protein sequences (9) currently is too poor to be of use in the prediction of the global fungal phylogeny. In contrast, a large amount of complete mitochondrial genome data is available from a broad selection of species from all major lineages. Concatenated mitochondrial protein data presently are the best way of classifying and understanding the evolution of Chytridiomycota and other fungi (e.g. 10–12). A robust phylogenetic tree, such as that inferred using mitochondrial sequence data, can then serve as a framework for comparative mitochondrial genome analysis.
In recent years, considerable progress has been made in understanding mitochondrial genome structure and evolution in a broad selection of eukaryotes using a comparative genomics approach. For example, sequences from closely related organisms have been compared in order to identify promoters or other little conserved sequence motifs (12), and more substantial sequence conservation across larger evolutionary distances has been used to identify previously unrecognized genes [e.g. Rps3 (13) or the fungal mitochondrial RNase P gene rnpB (E.Seif and B.F.Lang, unpublished)]. In addition, sequence variation in gene homologs from organisms with well defined relationships has aided in RNA secondary structure prediction (e.g. the above-mentioned RNase P RNA). This approach has also allowed the time of emergence of traits to be pinpointed [e.g. tRNA editing in chytridiomycetes (14); M.-J.Laforest and B.F.Lang, unpublished results].
The Fungal Mitochondrial Genome Project (FMGP) has as its primary goal to sequence mitochondrial DNAs (mtDNAs) from representatives of all fungal lineages, with a particular focus on the lower fungi (15) (see also http://megasun.bch.umontreal.ca/People/lang/FMGP/progress.html). These new sequence data have been used to study genome structure and content in the fungi, trace the evolution of gene expression in these systems and reconstruct fungal phylogeny. To date, the FMGP has sequenced mtDNAs from members of Ascomycota, Basidiomycota and Zygomycota, as well as from members of four of the five known taxonomic orders of chytridiomycetes: Allomyces macrogynus (Blastocladiales), Rhizophydium136 (Chytridiales), Spizellomyces punctatus (Spizellomycetales) and Hyaloraphidium curvatum (Mono blepharidales). Members of the Neocallimastigales, the remaining order in Chytridiomycota, have not been investigated as all known representatives are amitochondriate.
In continuation of the FMGP’s aims, we have determined the mitochondrial genome sequences of three chytridiomycetes, Monoblepharella15, Harpochytrium94 and Harpo chytrium105, and compared these sequences with that of H.curvatum. In this study, we (i) present a phylogeny based on mitochondrial protein sequences which further clarifies relationships within the lower fungi; (ii) address issues of genome organization, gene content, gene order and gene expression in these mtDNAs; and (iii) propose that a 4 bp anticodon–codon interaction is involved in mitochondrial translation initiation in this fungal order.
MATERIALS AND METHODS
Strains and culture conditions
The strains Monoblepharella15, Harpochytrium94 and Harpochytrium105 were collected, identified and generously provided by M. R. N. Mollicone and J. E. Longcore (University of Maine). Cells were grown using TYG medium, consisting of 0.25% tryptone, 0.125% yeast extract and 0.3% glucose. Liquid cultures of 300 ml in 2 l Erlenmeyer flasks were inoculated with growing cells and placed at room temperature either with shaking (Harpochytrium spp., 70 r.p.m.) or without shaking (Monoblepharella15) for 2–4 weeks. Purification of mitochondria was performed from ∼10 g (wet weight) of cells.
Purification of mtDNAs
Due to their differing growth forms, cells of Harpochytrium spp. and Monoblepharella15 were disrupted differently. Harpochytrium spp. cells (unicellular) were harvested by centrifugation. After resuspension in a sorbitol buffer (0.6 M sorbitol, 5 mM EDTA, 50 mM Tris pH 8), the cells were broken by shaking in the presence of glass beads. Monoblepharella15 cells (filamentous) were harvested by filtration through cheesecloth, then ground using a pestle and mortar in the presence of glass beads. A sorbitol buffer was used to wash the beads after cell disruption in both cases.
Crude mitochondrial fractions were recovered by differential centrifugation, and these fractions were lysed in the presence of 1% SDS and 100 µg/ml proteinase K at 50°C for 1 h. SDS was eliminated from the lysate by addition of 1 M NaCl. After 1 h on ice, the SDS–protein complex was removed by centrifugation. Nucleic acids were fractionated on a CsCl gradient (1.1 g/ml, 40 000 r.p.m. for 48 h) in the presence of 10 µg/ml bis-benzimide (Hoechst dye 33258). The prominent upper band (AT-rich mtDNA) was extracted and recentrifuged in a second CsCl gradient.
Cloning, sequencing and sequence analysis of mtDNAs
MtDNA was physically sheared by nebulization (16), and a size fraction of 500–3000 bp was recovered after agarose gel electrophoresis. DNAs were incubated in the presence of dNTPs, the Klenow fragment of DNA polymerase I and T7 DNA polymerase to generate blunt ends, and cloned into the SmaI site of a modified Bluescript II KS+ vector with a shortened multicloning site (pFBS; B.F.Lang, unpublished). Recombinant plasmids containing mtDNA inserts were identified by colony hybridization using mtDNA as a probe.
DNA sequencing, sequence assembly and sequences analysis were performed as described (17).
The mtDNA sequences of Monoblepharella15, Harpo chytrium94 and Harpochytrium105 have been deposited in GenBank (accession numbers AY182007, AY182005 and AY182006, respectively).
Purification of mtRNA
A crude mitochondrial pellet was recovered from cell lysates by differential centrifugation (see above). Then 8 M guanidinium chloride was added to lyse mitochondria. Ethanol-precipitated material from this lysate was incubated in the presence of 1% SDS and 100 µg/ml proteinase K at room temperature for 30 min. Following phenol extraction, SDS was eliminated from the lysate by addition of 1 M NaCl. After 20 min on ice, the SDS–protein complex was removed by centrifugation and 2 M LiCl was added for 1 h on ice. After centrifugation, the pellet was enriched in salt-insoluble large RNAs such as rRNAs and mRNAs, whereas the supernatant contained predominantly salt-soluble tRNAs and DNA.
Primer extension analysis
Oligonucleotides used in this study were: atp6, 5′-(G/A) AA(A/T)GGGAACCAAGCACC(G/A)GC-3′; atp8, 5′-AAAGTATAGCGAGAAAGATGAG-3′; nad3, 5′-AAGACGAGCATCACTATAAGG-3′; nad4L, 5′-ACAGCAGATTCAGC ACCAGC-3′; nad5, 5′-GAAGTACAACTTCATAGTAACC-3′; nad6, 5′-CCACCAGCTAGGTAAAGACC-3′; and cox3, 5′-CACGGGTAATATCACGGAACCA-3′.
Oligonucleotides were phosphorylated by incubation in the presence of T4 polynucleotide kinase and [γ-32P]ATP. A fraction of the radiolabeled oligonucleotide was annealed to mRNA in the salt-insoluble mitochondrial RNA fraction. Reverse transcription was performed using AMV reverse transcriptase in the presence of dNTPs. The resulting 5′-radiolabeled cDNAs were subjected to electrophoresis in 4% polyacrylamide sequencing gels. The length of the reverse-transcribed cDNA was determined by comparison with a DNA sequencing ladder from a mtDNA clone containing the gene of interest, using the same oligonucleotide (5′-phosphorylated prior to use) as was used for the primer extension.
RT–PCR of tRNAs
Salt-soluble mitochondrial RNAs (enriched in tRNAs) were treated with DNase I. A DNA oligonucleotide (see below) was annealed to the tRNAfMet, and this mixture was incubated in the presence of AMV reverse transcriptase and dNTPs. cDNAs were amplified by PCR using Taq polymerase I and a second primer. Products were separated on 2% agarose gels. A negative control (DNase-treated tRNAs, no reverse transcription) did not produce an amplification product in the PCR, confirming the cDNA (and not the mitochondrial or nuclear DNA) origin of the PCR product. The PCR products were cloned and sequenced as described (see above).
Primers used were: first strand synthesis, 5′-GCAGAA GAAGGATTCGAACC(T/C)C-3′; second strand synthesis, 5′-TAGAATGGAGTAAGGGTAACTCG-3′.
Phylogenetic inference
For phylogenetic analyses, we used a total of 2895 amino acid positions (termed ‘complete’ data set) from 13 concatenated, well conserved protein sequences (Cox1, 2 and 3, Cob, Atp6 and 9, and Nad1, 2, 3, 4, 4L, 5 and 6) that are encoded in most fungal mtDNAs. Multiple sequence alignments were performed with CLUSTALW (18), and regions of uncertain alignment were eliminated by comparing alignments inferred with various parameters, using SOAP (19), thus eliminating the need for further, disputable user interventions. The data used for the phylogenetic analysis will be made available on request.
Many fungal mtDNA sequences have a high rate of evolution. Therefore, certain artifacts of phylogenetic analyses, notably long branch attraction (20) and/or combinations of mutational oversaturation of sites, bias introduced by variation in nucleotide composition, strong codon preferences (10) and non-standard translation codes (14), may obscure true phylogenetic relationships. To minimize the effect of these artifacts, we have chosen to infer phylogenies with protein sequences and to base our conclusions on maximum likelihood (ML) methods, which are known to be least sensitive to long branch attraction phenomena. In addition, we have inferred trees from a ‘filtered’ data set containing only highly conserved amino acid positions (at least 60% amino acid identity, a total of 1636 positions) in order to recognize potential artifacts due to mutational oversaturation of sites. Phylogenetic inferences employed either the ML method as implemented in PROML (21) or a distance approach using ML distance tables calculated with TREE-PUZZLE (22), and BIONJ (23) for tree construction. In all instances, a Γ-distribution model of site variation was used [α = 1.0, calculated with PAML (24)]. To assess the level of confidence in tree selection, statistical tests were performed employing CONSEL (25), which provides the least biased and most rigorous statistical tests available to date (26,27). The standard AU and WKH tests (results not shown) confirm the topology of the monoblepharidalean lineage shown in Figure 1, i.e. they reject all alternative scenarios at a significance level of 0.05.
Figure 1.

Phylogenetic analysis based on concatenated mitochondrial proteins. The topology shown was inferred using ProML and the JTT model of protein evolution. It was constructed from the ‘complete’ data set of the unambiguously aligned portions of 13 concatenated protein sequences. Bootstrap support for this tree (%, upper number) was calculated from 100 replicates. A similar topology was obtained using ProtDist and BIONJ (%, second number; ‘–’ in cases of differences in topology compared with the ML tree). Bootstrap support for the distance tree was calculated from 1000 replicates. In order to explore the impact of oversaturated amino acid positions in the alignment, an alternative ProML analysis was performed based on the ‘filtered’ data set (see Materials and Methods). Bootstrap support for this ML tree, calculated from 100 replicates, is indicated only for the fungi (%, lower number). The postulated events marking the origin of quartet GAUG/GGUG initiation codons (I), fragmentation of the gene encoding the small subunit rRNA (F), the introduction of 5′ tRNA editing (E), and the drastic reduction in mtDNA-encoded tRNAs (T) are indicated with arrows. Sequences obtained from GenBank: A.castellanii (NC_001637); Allomyces macrogynus (U41288); Amoebidium parasiticum (AF538042-AF538052); Arabidopsis thaliana (NC_001284); Aspergillus nidulans (ODAS1, CAA33481, AAA99207, AAA31737, CAA25707, AAA31736, CAA23994, X15442, P15956, CAA23995, CAA33116X00790, X15441, X06960, J01387, X01507); Chondrus crispus (NC_001677); Chrysodidymus synuroideus (NC_002174); Dictyostelium discoideum (NC_000895); H.curvatum (AF402142); Marchantia polymorpha (NC_001660); M.senile (AF000023); Monosiga brevicollis (AF538053); Mesostigma viride (AF353999); Mesorhizobium loti (NC_002678); Nephroselmis olivacea (AF110138); Phytophthora infestans (NC_002387); Podospora anserina (X55026); Porphyra purpurea (NC_002007); R.americana (AF007261); Rhizophydium136 (NC_003053); Rickettsia prowazekii (NC_000963); Sarcophyton glaucum (AF064823, AF063191); Schizophyllum commune (AF402141); Sinorhizobium meliloti (NC_003047); Spizellomyces punctatus (AF402142); Yarrowia lipolytica (AJ307410). Protein sequences of Rhizopus stolonifer can be downloaded from http://megasun.bch.umontreal.ca/People/lang/FMGP/proteins/.
Taxon sampling was given particular consideration in these analyses. We excluded fungal taxa that completely lack mitochondrial nad genes [Saccharomyces cerevisiae, as well as the three available Schizosaccharomyces species (12)], limited the number of ascomycetes to the three species with the shortest relative branch lengths, and chose a variety of non-fungal species among protists, animals, plants and bacteria that have relatively low evolutionary rates. Finally, the number of taxa was limited to 30 due to the computational demands of the ML method when using evolutionary models that permit change of rate at all amino acid positions of the protein sequences, and when applying subsequent bootstrap analyses (28).
RESULTS AND DISCUSSION
Molecular phylogeny of the chytridiomycetes
Our molecular phylogenetic analyses (Fig. 1) using 13 concatenated mitochondrial protein sequences, and including an established member of the chytridiomycete order Mono blepharidales Monoblepharella15, support several features of fungal evolution with overwhelming support (95–100% bootstrap support both with ML and distance-based methods). In particular, these analyses support the specific relationship of fungi and animals [Opisthokonts; see for example Wainright et al. (29) and Paquin et al. (30)] and the deep divergence separating Chytridiomycota from Zygomycota, Basidiomycota and Ascomycota (but note that the phylogenetic affiliation of the blastacladialean Allomyces macrogynus remains unresolved; see below). Within Chytridiomycota, S.punctatus (Spizellomyces in Fig. 1) and Rhizophydium136 (from the taxonomic orders Spizello mycetales and Chytridiales, respectively), branch separately from the Monoblepharidales group, as expected. Mono blepharella15 and the two Harpochytrium species are sister lineages within the Monoblepharidales to the exclusion of H.curvatum (Hyaloraphidium in Fig. 1). The close relationship of Monoblepharella15 and the two Harpochytrium species is also strongly supported by the high level of gene order conservation in these three genomes (Fig. 2). This grouping of Harpochytrium spp. within the Mono blepharidales is contrary to the assignment of this genus to the Chytridiales (31), but consistent with recent phylogenies using ribosomal sequence data (5). However, it should be noted that studies based on rRNA data have been unable to recover the Monoblepharidales within Chytridiomycota, with significant support (5,32). In contrast, our analyses not only reveal the branching order within the Monoblepharidales, but also confirm the specific affiliation of H.curvatum with this taxonomic order (Fig. 1) (10).
Figure 2.

Gene and physical map of the Monoblepharella15, Harpochytrium94, Harpochytrium105 and H.curvatum mitochondrial genomes. Black blocks on the outer circle represent genes and ORFs, which are all transcribed clockwise in this representation. Green blocks indicate intron-encoded ORFs. tRNA gene names are indicated by the amino acid (one-letter code) they specify. The inner circle gives the size scale (in kb); colors on the intermediate circle of the Monoblepharella15 and Harpochytrium94 mtDNA indicate gene order conservation.
The branching position of A.macrogynus remains uncertain [Allomyces in Fig. 1; see also Leigh et al. (11)] despite the large array of fungal mitochondrial data currently available. In fact, it has proven difficult to confirm or disprove the hypothesis of chytridiomycete paraphyly (15). By using ML inference methods with among-site rate variation on our mitochondrial data set, A.macrogynus has migrated away from a previously well supported affiliation with the Zygomycota + Basidiomycota + Ascomycota clade to a basal position within Chytridiomycota. However, the bootstrap support for the monophyly of Chytridiomycota is only ∼60% with both the ‘complete’ and ‘filtered’ data sets (see Materials and Methods). The inconsistency of results using distance and ML methods may be due to a long branch attraction phenomenon, which is known to be more predominant in analyses employing distance methods. We are confident that addition of data from more chytridiomycetes will stabilize the position of A.macrogynus within Chytridiomycota.
Genome organization, gene content and gene order
The mitochondrial genomes of the monoblepharidalean fungi Monoblepharella15, Harpochytrium94 and Harpo chytrium105 were completely sequenced. In contrast to the linear conformation of the mtDNA of H.curvatum (10), the mtDNAs of Monoblepharella15, Harpochytrium94 and Harpochytrium105 map as circular molecules (Fig. 2), although they are probably multimeric concatemers in vivo as are most other mtDNAs (33). All four of these mtDNAs encode the basic fungal set of mitochondrial genes: the large and small subunit rRNAs (SSU rRNAs; rnl and rns, respectively), three subunits of the cytochrome oxidoreductase complex (cox1, 2 and 3), apocytochrome b (cob), three subunits of the ATP synthase complex (atp6, 8 and 9) and seven subunits of the NADH:ubiquinone oxidoreductase complex (nad1, 2, 3, 4, 4L, 5 and 6) (Table 1). As they are in the mtDNA of H.curvatum, genes are encoded on only one strand in the other three mtDNAs. The gene order in the two Harpochytrium species is identical, and highly similar to that in Monoblepharella15 (refer to color code in Fig. 2). However, there is no gene order conservation between these genomes and that of H.curvatum. This agrees with our understanding of the phylogenetic distances between these organisms (Fig. 1).
Table 1. Gene and intron content in fungal mtDNAsa.
| Genes | Monoblepharella15 | Harpochytrium94 | Harpochytrium105 | H.curvatum | S.punctatus | A.macrogynus | S.pombe |
|---|---|---|---|---|---|---|---|
| rnl, rns | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
| atp6,8,9 | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
| cob | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
| cox1,2,3 | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
| nad1-6b | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ○ |
| trnA-W | 9 | 8 | 8 | 7 | 8 | 25 | 25 |
| rnpB | ○ | ○ | ○ | ○ | ○ | ○ | ▪ |
| rps3 | ○ | ○ | ○ | ○ | ○ | ▪ | ▪ |
| ORFsc | 5 | 0 | 0 | 3 | 5 | 3 | 0 |
| IntronId | 8 | 0 | 0 | 1 | 12 | 26 | 2 |
| IntronIId | 0 | 0 | 0 | 0 | 0 | 2 | 1 |
aFilled squares indicate the presence, open circles the absence, of a gene or genes.
bIncludes nad1, nad2, nad3, nad4, nad4L, nad5 and nad6.
cOnly ORFs greater than 100 amino acids in length are listed.
dIntronI and IntronII denote introns of group I and group II, respectively.
The mtDNAs of H.curvatum, Harpochytrium94 and Harpochytrium105 are relatively compact, at 29.97, 19.473 and 24.169 kb, respectively. In contrast, the mtDNA of Monoblepharella15 is 60.440 kb. This is partly due to additional coding regions in the larger genome. Monoblepharella15 encodes five open reading frames (ORFs) with no significant similarity to known proteins, whereas H.curvatum encodes three, and both Harpochytrium species encode none; Monoblepharella15 encodes nine tRNAs whereas H.curvatum encodes seven and the Harpochytrium genomes each encode eight (see below); and Monoblepharella15 encodes eight introns [all of group I, seven of which encode ORFs with either LAGLIDADG or GIY-YIG motifs, characteristic of group I intron ORFs (34)], whereas H.curvatum encodes only one and the Harpochytrium genomes are devoid of introns. Intergenic (non-coding) regions account for the majority of mitochondrial genome size variation in Monoblepharidales.
G + C content is relatively high in these genomes compared with other fungi, at 39% in both H.curvatum and Monoblepharella15 and 36% in Harpochytrium spp. This is also true in intergenic regions, as Harpochytrium94, Harpochytrium105, H.curvatum and Monoblepharella15 have intergenic G + C contents of 32, 38, 43 and 53%, respectively. The remarkably high value in Mono blepharella15 intergenic regions is due predominantly to the presence of many G + C-rich repeats, possibly the vestiges of double hairpin elements [DHEs (35)]. The intergenic regions of Harpochytrium105 contain several canonical DHE structures (15), whereas those of Harpochytrium94 and H.curvatum do not contain any of these elements. The biology of DHEs is not well understood, but they have been proposed to be mobile elements and contribute to mitochondrial genome plasticity (12,35).
Conserved sequence elements upstream of protein-coding genes
All genes are encoded on the same DNA strand in the four mtDNAs compared here. It is therefore possible that few, or even a single promoter is responsible for the transcription of all genes in each genome. However, nothing is known about promoter structure in chytridiomycete mtDNAs. Certain sequences in the H.curvatum mtDNA have been suggested as possible promoters based on their location (10), but this possibility has not been verified experimentally. Similar sequences to those in H.curvatum were not identified in the other three Monoblepharidales mtDNAs.
We identified a highly conserved sequence motif specific to the Monoblepharella15, Harpochytrium94 and Harpo chytrium105 genomes (consensus: 5′-TTATAGGAAAT-3′; Fig. 3A) between 15 and 26 nt upstream of the start codons of the atp6, atp8, nad3, nad4L, nad5 and nad6 genes. Additional conservation (consensus: 5′-CTAGGT-3′; Fig. 3A) was identified immediately 3′ to this conserved sequence upstream of the atp6 and atp8 genes in all three species. Another conserved sequence (consensus: AGAGTGTANTNNAATT; Fig. 3B) was identified 8–13 nt upstream of the cox3 gene in these three species. We speculated that some or all of these sequences might serve as promoters in these mtDNAs, like the nonanucleotide sequences which promote transcription in yeast mitochondria (36,37). If these sequences were truly functionally analogous to those in yeast, one would expect that RNA transcripts would begin near the last nucleotide in the conserved sequence block. To test this hypothesis, we performed primer extension experiments to map the 5′ ends of seven mitochondrial transcripts (not shown), which have conserved sequences upstream of their coding regions in the mtDNA of Monoblepharella15. In all cases, however, the 5′ end of the RNA mapped 1–6 nt upstream of the 5′ end of the conserved sequence (Fig. 3). These experiments indicate that the conserved sequences are completely included within RNA transcripts (at least in Monoblepharella15), arguing against the role of these sequences as promoters.
Figure 3.

Sequences immediately preceding selected genes in the mtDNAs of Monoblepharella15, Harpochytrium94 and Harpochytrium105. (A) Widespread conserved sequence element. (B) cox3-specific conserved sequence element. Blocks conserved in all genes are indicated in yellow. Additional blocks conserved in the atp6 and atp8 genes are indicated in purple. Inferred translation initiation codons are indicated in red. RNA 5′ ends determined by primer extension analysis are underlined.
The conservation of these sequence motifs close to the end of these RNAs may suggest their implication in 5′-RNA processing. However, we currently favor the alternative hypothesis that these sequences function in translation regulation. Poorly conserved structures and/or sequences [possibly a conserved octanucleotide (38)] in the 5′-untranslated regions of mitochondrial RNA transcripts have been implicated in translation regulation in yeast (39–41). The conserved sequences probably do not serve as Shine–Dalgarno-like sequences [as observed in the minimally derived mtDNA of Reclinomonas americana (42)] as no sequences can be identified at the 3′ end of monoblepharidalean SSU rRNAs with which they might interact.
A reduced set of tRNAs, codon bias and tRNA editing
A reduced set of tRNA genes is found in these four monoblepharidalean mitochondrial genomes, insufficient to decode all codons (Table 2). This is also the case in the mtDNAs of two other chytridiomycete fungi, S.punctatus and Rhizophydium136, which encode eight and seven tRNAs, respectively. Monoblepharella15 encodes nine mitochondrial tRNAs (tRNAAsp, tRNAGlu, tRNAGly, tRNALys, tRNAfMet, tRNAPro, tRNAGln, tRNATrp and tRNATyr), both Harpo chytrium species encode eight (the same set as in Mono blepharella15, except that tRNAGly is absent) and H.curvatum encodes seven (the same set as in Harpochytrium spp. except that tRNALys is absent). Additional tRNAs required for mitochondrial translation presumably are encoded in the nucleus and imported into mitochondria from the cytoplasm. Interestingly, tRNAfMet, tRNAPro, tRNAGln, tRNATrp and tRNATyr are found in all six of these chytridiomycete mtDNAs, testifying to a common history of tRNA gene loss in this lineage. Retention of these particular tRNAs perhaps is due to a difficulty in replacing features which are not found in their cytoplasmic tRNA counterparts. For example, tRNAfMet must be recognized and formylated in order to serve as the initiator tRNA for mitochondrial protein synthesis. It might therefore be retained because the mitochondrial methionyl-tRNAfMet transformylase cannot recognize the cytoplasmic initiator tRNA. As the chytridiomycete A.macrogynus encodes a tRNA set capable of decoding all codons in its mtDNA (43), we assume that the majority of tRNAs were lost deep within the chytridiomycete lineage after the divergence of A.macrogynus (Fig. 1).
Table 2. Partially shared, reduced complement of tRNAs encoded by chytridiomycete mtDNAsa,b.
| Species | D (GUC) | E (UUC) | G (UCC) | K (UUU) | Lc (CUA) | M (CAU) | P (UGG) | Q (UUG) | W (CCA) | Y (GUA) |
| Monoblepharella15 | ▪ | ▪ | ▪ | ▪ | ○ | ▪ | ▪ | ▪ | ▪ | ▪ |
| Harpochytrium94 | ▪ | ▪ | ○ | ▪ | ○ | ▪ | ▪ | ▪ | ▪ | ▪ |
| Harpochytrium105 | ▪ | ▪ | ○ | ▪ | ○ | ▪ | ▪ | ▪ | ▪ | ▪ |
| H.curvatum | ▪ | ▪ | ○ | ○ | ○ | ▪ | ▪ | ▪ | ▪ | ▪ |
| S.punctatus | ▪ | ○ | ○ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
| Rhizophydium136 | ○ | ○ | ○ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
atRNA species are identified by their corresponding amino acid (in one-letter code) and anticodon sequence.
bFilled squares indicate the presence, open circles the absence, of a gene.
cAn unusual tRNA decodes UAG ‘stop’ codons as leucine in S.punctatus and Rhizophydium136.
Several codons are not used in the standard protein-coding genes encoded in these genomes (i.e. not including intronic or free-standing ORFs), although the universal translation code is otherwise used. Of 64 possible codons, H.curvatum does not use seven (TGA, TCA, ATA, GCA, GCG, AGA and AGG), Monoblepharella15 does not use 10 (TGA, CTG, TCA, TCG, ACA, ACG, CAG, AAG, GAG and CGG), and the two Harpochytrium species do not use 14 (the same as those in Monoblepharella15, as well as ATA, GCG, AGG and GGA). This marked non-use of codons is in contrast to other fungal mtDNAs, including those of S.punctatus and Rhizo phydium136, which use all but two and three codons, respectively. Interestingly, all of the unused codons in the four monoblepharidalean genomes have a purine residue in the third position. The significance of this observation is unknown.
Finally, the majority of tRNAs in these genomes contain mismatches in the first three base pairs of their acceptor stems, potentially rendering them non-functional (10). The acceptor stem mismatches in Monoblepharella15, Harpochytrium94 and Harpochytrium105 have been show to be corrected at the RNA level to create standard Watson–Crick base pairs, by removal of the first 3 nt from the 5′-half of the acceptor stem followed by nucleotide addition (M.-J.Laforest and B.F.Lang, unpublished). This type of tRNA editing was first identified in the amoeboid protist Acanthamoeba castellanii (44), and has also been described in the chytridiomycete S.punctatus (14). The origin, evolution and mechanism of this form of RNA editing in chytridiomycete mitochondria will be discussed in detail elsewhere (M.-J.Laforest and B.F.Lang, unpublished).
SSU rRNA in pieces
Fragmented and scrambled mitochondrial rRNA genes were first observed in Tetrahymena pyriformis (45). They also occur in the mitochondria of certain green algae (46–48) and in alveolates such as Plasmodium falciparum (49) and Theileria parva (50). In Monoblepharella15 and both Harpochytrium species, the mitochondrial SSU rRNA is encoded in two pieces, exactly as it is in H.curvatum (10). The 3′-fragment is found upstream of the 5′-fragment in all four cases, separated by a gene (either nad2 or rnl; see Fig. 2). The secondary structures of these rRNAs, derived from the gene sequences, indicate structures consistent with the eubacterial model. The inferred break point in the SSU rRNA is located within the same variable region in all four cases, corresponding to nucleotides 590–649 of the Escherichia coli SSU rRNA, and the two rRNA fragments have the potential to assemble by intermolecular base pairing. It should be noted that there may be break points in other variable regions of these genes, and determination of rRNA ends would be necessary to establish the true fragmentation pattern. The rns gene is not fragmented in the mtDNAs of the three other characterized chytridiomycetes, A.macrogynus, S.punctatus and Rhizophydium136. This suggests that fragmentation of the rns gene occurred within the Monoblepharidales lineage prior to the divergence of H.curvatum (Fig. 1).
A 4 bp interaction is potentially involved in translation initiation
Comparison of the four Monoblepharidales mitochondrial genome sequences revealed an unusual feature: a guanosine residue is found immediately 5′ to the ATG and GTG start codons (inferred by protein sequence alignment) of almost all protein-coding genes. The only protein-coding genes for which a G does not precede the start codon are the cox1 genes of Monoblepharella15 and Harpochytrium spp. (whereas it is present in the cox1 gene of H.curvatum) and the nad1 gene of H.curvatum (whereas it is present in the nad1 genes of Mono blepharella15, Harpochytrium94 and Harpochytrium105). No alternative in-frame GATG or GGTG codons are present in either of these two cases. Further analysis revealed that the initiator tRNAsfMet encoded by these mtDNAs also contain unusual features (Fig. 4). Most tRNAs adhere to a well defined secondary structure, which includes a 5 bp anticodon stem and a 7 nt anticodon loop. The tRNAsfMet of Monoblepharella15, Harpochytrium94 and Harpochytrium105, on the other hand, have mismatches at the base of the anticodon stem (CxC in Monoblepharella15, UxU in Harpochytrium spp.). In fact, non-Watson–Crick base pairs are observed at this position in all four Monoblepharidales tRNAsfMet (a U–G pair is present at this position in H.curvatum). Further, the nucleotide at position 37 (immediately 3′ to the anticodon) of all four of these tRNAs is occupied by a cytidine residue (Fig. 4). This feature is particularly surprising, as a purine residue, usually modified, is found in the corresponding position of the vast majority of tRNAs.
Figure 4.
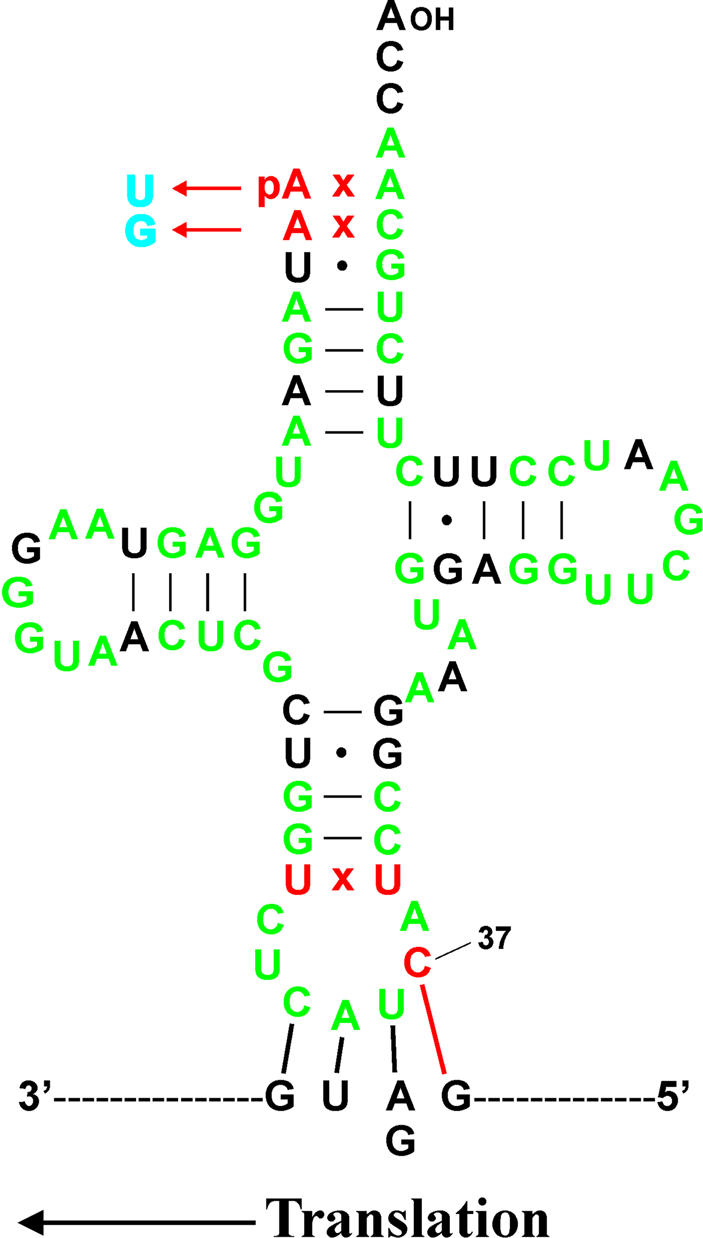
Conserved unorthodox features in Monoblepharidales mitochondrial initiator tRNAs, and proposed 4 bp interaction involved in translation initiation. The sequence of tRNAfMet from Harpochytrium105 is shown. Nucleotides identical in all four Monoblepharidales mitochondria are indicated in green. Nucleotide replacement 5′-tRNA editing is indicated in blue. Conserved, unorthodox features are indicated in red, including the non-Watson–Crick base pair at the base of the anticodon stem and the proposed base pair between the cytosine at position 37 of tRNAfMet and the guanosine residue immediately 5′ to AUG and GUG start codons. Note that the size of the dihydrouridine (D) loop is one nucleotide longer in H.curvatum compared with the other Monoblepharidalean tRNAsfMet.
As editing of tRNA acceptor stems is observed in the mitochondria of these organisms, it was formally possible that another type of tRNA editing altered the sequence of the anticodon stem and/or loop. Therefore, we reverse-transcribed and PCR-amplified the anticodon stem–loop region of tRNAfMet from both Monoblepharella15 and Harpo chytrium105. In both cases, cDNA sequencing demonstrated that these unusual tRNAs are expressed, and that the RNA sequence of the amplified regions was identical to that encoded by the mtDNA (not shown). Therefore, we assume that these are functional mitochondrial tRNAs.
The unorthodox features of the initiator tRNAs encoded by these mtDNAs, in conjunction with those of initiation codons, strongly suggest that a 4 bp interaction between the CAUC anticodon and quartet GAUG/GGUG initiation codons is involved in translation initiation in Monoblepharidales mitochondria (Fig. 4), in contrast to the standard 3 bp interaction. Extended anticodon–codon interactions occur in other contexts; in fact, selection experiments have shown that the most efficient engineered suppressor tRNAs for frameshifting at four-base codons have Watson–Crick complementarity at all four bases of the codon, supporting direct interaction of a four-base anticodon and codon (51). More recently, evidence has been presented supporting an extended interaction in translation initiation in Chlamydomonas reinhardtii chloroplasts (52). In this case, additional base pairing is between the unmodified adenosine at position 37 of tRNAfMet and the uridine residue immediately 5′ to an initiation codon (uridine is found at this position in 75% of initiation codons in this system). The absence of a modified purine residue at position 37 of monoblepharidalean mitochondrial tRNAsfMet (as well as C.reinhardtii chloroplast tRNAfMet) may be important for this type of interaction to occur, as the presence of a modified purine residue would be expected to impede interaction with the mRNA during translation (53–55). Finally, the lack of a Watson–Crick base pair at the base of the anticodon stem of Mono blepharidalean tRNAsfMet may be required for increased flexibility of the anticodon loop, allowing the presentation of the extended anticodon in a conformation which makes it possible for the interaction to occur. It is of great interest to understand the function of this unorthodox feature of mitochondrial translation. One attractive possibility is that the additional anticodon–codon complementarity allows more accurate selection of true initiation codons.
Intriguingly, the mitochondrial initiator tRNA in the sea anemone Metridium senile also encodes a cytosine at position 37 (56). Inspection of the M.senile mtDNA reveals that 5 of the 13 protein-coding genes in this genome (cox1, cox2, atp6, nad2 and nad3) have a guanosine residue immediately preceding the ATG initiation codon predicted by the authors (56). Three other genes have in-frame GATG codons in close proximity to the predicted initiation codon (one codon downstream in atp8; seven codons downstream in nad1; 12 codons downstream in cob). The remaining five genes, cox3, nad4, nad4L, nad5 and nad6, do not have a GATG codon in close proximity to the predicted initiation codon, although an in-frame GGTG codon is located five codons upstream of the predicted initiation codon of the nad4L gene. There is therefore a strong tendency for a G immediately 5′ to the likely ATG initiation codon (8 out of 13), whereas the overall G + C content of this mtDNA is only 37.5%. Based on the correlation of these features of initiation codons and tRNAfMet, it is probable that a 4 bp interaction is involved in translation initiation in M.senile mitochondria, as we propose here for monoblepharidalean mitochondria. As there is no evidence to support a specific relationship between Monoblepharidales and the cnidarian M.senile (see Fig. 1), this extended anticodon–codon interaction must have evolved independently in each lineage. Furthermore, it does not appear to be widespread even among cnidarians, as the mitochondrial initiator tRNAs of two other distant members of this group, Acropora tenuis and Sarcophyton glaucum, have purine residues at position 37 (57,58). It is possible nevertheless that evidence of further 4 bp anticodon–codon interactions involved in translation initiation will come to light as mitochondrial genome sequences from close relatives of M.senile become available.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank J. E. Longcore and M. R. N. Mollicone (both of the University of Maine) for identification and supply of lower fungal strains. We also acknowledge the contributions of H. Hamel and M.-J. Laforest in DNA sequencing, and the excellent technical assistance of Z. Wang. This research was supported by the Canadian Institutes of Health Research (CIHR grants MT-14028 and MOP42475 to B.F.L.), an academic equipment grant by SUN microsystems (Palo Alto, CA, to B.F.L.), the donation of an automatic sequencer by Licor (Lincoln, NB, to B.F.L.) and a Master’s Research Scholarship from Fonds F.C.A.R. (C.E.B.). Salary and interaction support from the Canadian Institute for Advanced Research is gratefully acknowledged.
DDBJ/EMBL/GenBank accession nos+ AY182005–AY182007
REFERENCES
- 1.Bruns T.D., Vilgalys,R., Barns,S.M., Gonzalez,D., Hibbett,D.S., Lane,D.J., Simon,L., Stickel,S., Szaro,T.M., Weisburg,W.G. and Sogin,M.L. (1992) Evolutionary relationships within the fungi: analyses of nuclear small subunit rRNA sequences. Mol. Phylogenet. Evol., 1, 231–241. [DOI] [PubMed] [Google Scholar]
- 2.Nagahama T., Sato,H., Shimazu,M. and Sugiyama,J. (1995) Phylogenetic divergence of the Entomophthoralean fungi: evidence from nuclear 18S ribosomal RNA gene sequence. Mycologia, 87, 203–209. [Google Scholar]
- 3.Jensen A.B., Gargas,A., Eilenberg,J. and Rosendahl,S. (1998) Relationships of the insect-pathogenic order Entomophthorales (Zygomycota, Fungi) based on phylogenetic analyses of nuclear small subunit ribosomal DNA sequences (SSU rDNA). Fungal Genet. Biol., 24, 325–334. [DOI] [PubMed] [Google Scholar]
- 4.Berbee M.L., Carmean,D.A. and Winka,K. (2000) Ribosomal DNA and resolution of branching order among the Ascomycota: how many nucleotides are enough? Mol. Phylogenet. Evol., 17, 337–344. [DOI] [PubMed] [Google Scholar]
- 5.James T.Y., Porter,D., Leander,C.A., Vilgalys,R. and Longcore,J.E. (2000) Molecular phylogenetics of the Chytridiomycota supports the utility of ultrastructural data in chytrid systematics. Can. J. Bot., 78, 336–350. [Google Scholar]
- 6.Doolittle R.F., Feng,D.-F., Tsang,S., Cho,G. and Little,E. (1996) Determination of divergence times of the major kingdoms of living organisms with a protein clock. Science, 271, 470–477. [DOI] [PubMed] [Google Scholar]
- 7.Keeling P.J., Luker,M.A. and Palmer,J.D. (2000) Evidence from beta-tubulin phylogeny that microsporidia evolved from within the fungi. Mol. Biol. Evol., 17, 23–31. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y.J., Whelen,S. and Hall,B.D. (1999) Phylogenetic relationships among ascomycetes: evidence from an RNA polymerase II subunit. Mol. Biol. Evol., 16, 1799–1808. [DOI] [PubMed] [Google Scholar]
- 9.Baldauf S.L., Roger,A.J., Wenk-Siefert,I. and Doolittle,W.F. (2000) A kingdom-level phylogeny of eukaryotes based on combined protein data. Science, 290, 972–977. [DOI] [PubMed] [Google Scholar]
- 10.Forget L., Ustinova,J., Wang,Z., Huss,V.A.R. and Lang,B.F. (2002) Hyaloraphidium curvatum: a linear mitochondrial genome, tRNA editing and an evolutionary link to lower fungi. Mol. Biol. Evol., 19, 310–319. [DOI] [PubMed] [Google Scholar]
- 11.Leigh J., Seif,E., Rodriguez,N., Jacob,Y. and Lang,B.F. (2003) Fungal evolution meets fungal genomics. In Arora D. (ed.), Handbook of Fungal Biotechnology, 2nd Edn. Marcel Dekker Inc., New York, in press.
- 12.Bullerwell C.E., Leigh,J., Forget,L. and Lang,B.F. (2003) A comparison of three fission yeast mitochondrial genomes. Nucleic Acids Res., 31, 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bullerwell C.E., Burger,G. and Lang,B.F. (2000) A novel motif for identifying Rps3 homologs in fungal mitochondrial genomes. Trends Biochem. Sci., 25, 363–365. [DOI] [PubMed] [Google Scholar]
- 14.Laforest M.J., Roewer,I. and Lang,B.F. (1997) Mitochondrial tRNAs in the lower fungus Spizellomyces punctatus: tRNA editing and UAG ‘stop’ codons recognized as leucine. Nucleic Acids Res., 25, 626–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paquin B., Laforest,M.-J., Forget,L., Roewer,I., Wang,Z., Longcore,J. and Lang,B.F. (1997) The fungal mitochondrial genome project: evolution of fungal mitochondrial genomes and their gene expression. Curr. Genet., 31, 380–395. [DOI] [PubMed] [Google Scholar]
- 16.Okpodu C.M., Robertson,D., Boss,W.F., Togasaki,R.K. and Surzycki,S.J. (1994) Rapid isolation of nuclei from carrot suspension culture cells using a BioNebulizer. Biotechniques, 16, 154–159. [PubMed] [Google Scholar]
- 17.Burger G., Saint-Louis,D., Gray,M.W. and Lang,B.F. (1999) Complete sequence of the mitochondrial DNA of the red alga Porphyra purpurea: cyanobacterial introns and shared ancestry of red and green algae. Plant Cell, 11, 1675–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Löytynoja A. and Milinkovitch,M.C. (2001) SOAP, cleaning multiple alignments from unstable blocks. Bioinformatics, 17, 573–574. [DOI] [PubMed] [Google Scholar]
- 20.Felsenstein J. (1978) Cases in which parsimony and compatibility methods will be positively misleading. Syst. Zool., 27, 401–410. [Google Scholar]
- 21.Felsenstein J. (2002) Phylip (Phylogeny Inference Package) Version 3.6. Distributed by the author. University of Washington, Seattle.
- 22.Strimmer K. and von Haeseler,A. (1996) Quartet puzzling: a quartet maximum-likelihood method for reconstructing tree topologies. Mol. Biol. Evol., 13, 964–969. [Google Scholar]
- 23.Gascuel O. (1997) BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol., 14, 685–695. [DOI] [PubMed] [Google Scholar]
- 24.Yang Z. (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. CABIOS, 13, 555–556. [DOI] [PubMed] [Google Scholar]
- 25.Shimodaira H. and Hasegawa,M. (2000) CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics, 17, 1246–1247. [DOI] [PubMed] [Google Scholar]
- 26.Shimodaira H. (2002) An approximately unbiased test of phylogenetic tree selection. Syst. Biol., 51, 492–508. [DOI] [PubMed] [Google Scholar]
- 27.Whelan S., Lio,P. and Goldman,N. (2001) Molecular phylogenetics: state of the art methods for looking into the past. Trends Genet., 17, 262–272. [DOI] [PubMed] [Google Scholar]
- 28.Felsenstein J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution, 39, 783–791. [DOI] [PubMed] [Google Scholar]
- 29.Wainright P.O., Hinkle,G., Sogin,M.L. and Stickel,S.K. (1993) Monophyletic origins of the metazoa: an evolutionary link with fungi. Science, 260, 340–342. [DOI] [PubMed] [Google Scholar]
- 30.Paquin B., Forget,L., Roewer,I. and Lang B.F. (1995) Molecular phylogeny of Allomyces macrogynus: congruency between nuclear ribosomal RNA- and mitochondrial protein-based trees. J. Mol. Evol., 41, 657–665. [DOI] [PubMed] [Google Scholar]
- 31.Barr D.J.S. (1990) Phylum Chytridiomycota. In Margulis,L., Corliss,J.O., Melkonian,M. and Chapman,D.J. (eds), Handbook of Protoctista. Jones and Bartlett, Boston, pp. 454–466.
- 32.Ustinova I., Krienitz,L. and Huss,V.A.R. (2000) Hyaloraphidium curvatum is not a green alga, but a lower fungus; Amoebidium parasiticum is not a fungus, but a member of the DRIPs. Protist, 151, 253–262. [DOI] [PubMed] [Google Scholar]
- 33.Bendich A.J. (1996) Structural analysis of mitochondrial DNA molecules from fungi and plants using moving pictures and pulsed-field gel electrophoresis. J. Mol. Biol., 255, 564–588. [DOI] [PubMed] [Google Scholar]
- 34.Michel F., Jacquier,A. and Dujon,B. (1982) Comparisons of fungal mitochondrial introns reveals extensive homologies in RNA secondary structure. Biochimie, 64, 867–881. [DOI] [PubMed] [Google Scholar]
- 35.Paquin B., Laforest,M.-J. and Lang,B.F. (2000) Double-hairpin elements in the mitochondrial DNA of Allomyces: evidence for mobility. Mol. Biol. Evol., 17, 1760–1768. [DOI] [PubMed] [Google Scholar]
- 36.Osinga K.A., De Haan,M., Christianson,T. and Tabak,H.F. (1982) A nonanucleotide sequence involved in promotion of ribosomal RNA synthesis and RNA priming of DNA replication in yeast mitochondria. Nucleic Acids Res., 10, 7993–8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christianson T. and Rabinowitz,M. (1983) Identification of multiple transcriptional initiation sites on the yeast mitochondrial genome by in vitro capping with guanylyltransferase. J. Biol. Chem., 258, 14025–14033. [PubMed] [Google Scholar]
- 38.Dunstan H.M., Green-Williams,N.S. and Fox,T.D. (1997) In vivo analysis of Saccharomyces cerevisiae cox2 mRNA 5′-untranslated leader functions in mitochondrial translation initiation and translational activation. Genetics, 147, 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Costanzo M.C. and Fox,T.D. (1988) Specific translational activation by nuclear gene products occurs in the 5′-untranslated leader of a yeast mitochondrial mRNA. Proc. Natl Acad. Sci. USA, 85, 2677–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mulero J.J. and Fox,T.D. (1993) PET111 acts in the 5′-leader of the Saccharomyces cerevisiae mitochondrial cox2 mRNA to promote its translation. Genetics, 133, 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steele D.F., Butler,C.A. and Fox,T.D. (1996) Expression of a recoded nuclear gene inserted into yeast mitochondrial DNA is limited by mRNA-specific translational activation. Proc. Natl Acad. Sci. USA, 93, 5253–5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lang B.F., Burger,G., O’Kelly,C.J., Cedergren,R.J., Golding,G.B., Lemieux,C., Sankoff,D., Turmel,M. and Gray,M.W. (1997) An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature, 387, 493–497. [DOI] [PubMed] [Google Scholar]
- 43.Paquin B. and Lang,B.F. (1996) The mitochondrial DNA of Allomyces macrogynus: the complete genomic sequence from an ancestral fungus. J. Mol. Biol., 255, 688–701. [DOI] [PubMed] [Google Scholar]
- 44.Lonergan K.M. and Gray,M.W. (1993) Editing of transfer RNAs in Acanthamoeba castellanii mitochondria. Science, 259, 812–816. [DOI] [PubMed] [Google Scholar]
- 45.Schnare M.N., Heinonen,T.Y., Young,P.G. and Gray,M.W. (1986) A discontinuous small subunit ribosomal RNA in Tetrahymena pyriformis mitochondria. J. Biol. Chem., 261, 5187–5193. [PubMed] [Google Scholar]
- 46.Boer P.H. and Gray,M.W. (1988) Scrambled ribosomal RNA gene pieces in Chlamydomonas reinhardtii mitochondrial DNA. Cell, 55, 399–411. [DOI] [PubMed] [Google Scholar]
- 47.Turmel M., Lemieux,C., Burger,G., Lang,B.F., Otis,C., Plante,I. and Gray,M.W. (1999) The complete mitochondrial DNA sequences of Nephroselmis olivacea and Pedinomonas minor: two radically different evolutionary patterns within green algae. Plant Cell, 11, 1717–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nedelcu A.M., Lee,R.W., Lemieux,C., Gray,M.W. and Burger,G. (2000) The complete mitochondrial DNA sequence of Scenedesmus obliquus reflects an intermediate stage in the evolution of the green algal mitochondrial genome. Genome Res., 10, 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gillespie D.E., Salazar,N.A., Rehkopf,D.H. and Feagin,J.E. (1999) The fragmented mitochondrial ribosomal RNAs of Plasmodium falciparum have short A tails. Nucleic Acids Res., 27, 2416–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kairo A., Fairlamb,A.H., Gobright,E. and Nene,V. (1994) A 7.1 kb linear DNA molecule of Theileria parva has scrambled rDNA sequences and open reading frames for mitochondrially encoded proteins. EMBO J., 13, 898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Magliery T.J., Anderson,J.C. and Schultz,P.G. (2001) Expanding the genetic code: selection of efficient suppressors of four-base codons and identification of ‘shifty’ four-base codons with a library approach in E.coli. J. Mol. Biol., 307, 755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Esposito D., Fey,J.P., Eberhard,S., Hicks,A.J. and Stern,D.B. (2003) In vivo evidence for the prokaryotic model of extended codon–anticodon interaction in translation initiation. EMBO J., 22, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pieczenik G. (1980) Predicting coding function from nucleotide sequence or survival of ‘fitness’ of tRNA. Proc. Natl Acad. Sci. USA, 77, 3539–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Björk G.R., Wilkström,P.M. and Byström,A.S. (1989) Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science, 244, 986–989. [DOI] [PubMed] [Google Scholar]
- 55.Hagervall T.G., Tuohy,T.M.F., Atkins,J.F. and Björk,G.R. (1993) Deficiency of 1-methylguanosine in tRNA from Salmonella typhimurium induces frameshifting by quadruplet translocation. J. Mol. Biol., 232, 756–765. [DOI] [PubMed] [Google Scholar]
- 56.Beagley C.T., Okimoto,R. and Wolstenholme,D.R. (1998) The mitochondrial genome of the sea anemone Metridium senile (Cnidaria): introns, a paucity of tRNA genes and a near-standard genetic code. Genetics, 148, 1091–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Oppen M.J.H., Catmull,J., McDonald,B.J., Hislop,N.R., Hagerman,P.J. and Miller,D.J. (2002) The mitochondrial genome of Acropora tenuis (Cnidaria; Scleractina) contains a large group I intron and a candidate control region. J. Mol. Evol., 55, 1–13. [DOI] [PubMed] [Google Scholar]
- 58.Pont-Kingdon G., Okada,N.A., Macfarlane,J.L., Beagley,C.T., Watkins-Sims,C.D., Cavalier-Smith,T., Clark-Walker,G.D. and Wolstenholme,D.R. (1998) Mitochondrial DNA of the coral Sarcophyton glaucum contains a gene for a homologue of bacterial MutS: a possible case of gene transfer from the nucleus to the mitochondrion. J. Mol. Evol., 46, 419–431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.