

Abstract
The highly conserved, ubiquitously expressed, zinc finger protein CTCF is involved in enhancer blocking, a mechanism crucial for shielding genes from illegitimate enhancer effects. Interestingly, CTCF-binding sites are often flanked by thyroid hormone response elements (TREs), as at the chicken lysozyme upstream silencer. Here we identify a similar composite site positioned upstream of the human c-myc gene. For both elements, we demonstrate that thyroid hormone abrogates enhancer blocking. Relief of enhancer blocking occurs even though CTCF remains bound to the lysozyme chromatin. Furthermore, chromatin immunoprecipitation analysis of the lysozyme upstream region revealed that histone H4 is acetylated at the CTCF-binding site. Loss of enhancer blocking by the addition of T3 led to increased histone acetylation, not only at the CTCF site, but also at the enhancer and the promoter. Thus, when TREs are adjacent to CTCF-binding sites, thyroid hormone can regulate enhancer blocking, thereby providing a new property for what was previously thought to be constitutive enhancer shielding by CTCF.
Keywords: chromatin/CTCF/enhancer blocking/histone acetylation/thyroid hormone
Introduction
CTCF is a highly conserved, ubiquitously expressed, 11-zinc finger phospho-protein that was first identified as a transcription factor that binds to silencer elements in the chicken c-myc and lysozyme genes (Baniahmad et al., 1990; Lobanenkov et al., 1990). The conservation of CTCF between man, mouse, rat, chicken and frog is such that the 11-zinc finger DNA-binding domain is almost identical, and the overall amino acid identity of the full length protein is in the range of 83–84% (Burke et al., 2002). Its ubiquitous expression profile and the above conservation argue for important functions at the cellular level. In addition to CTCF-mediated repression (Baniahmad et al., 1990; Lobanenkov et al., 1990) and activation (Quitschke et al., 1996) of promoters, enhancer blocking activity of CTCF on insulator elements has also been demonstrated. In fact, CTCF is the only mammalian protein so far identified that exhibits enhancer blocking activity (Bell et al., 1999). Enhancers can communicate over long distances with promoter elements, both from upstream and downstream sites. Specific mechanisms therefore had to evolve to control enhancer activity so that only the appropriate promoters are activated. Insulator elements with enhancer blocking activity fulfil this purpose (for reviews, see Mueller, 2000; Wolffe, 2000; Ohlsson et al., 2001; West et al., 2002). Constitutive enhancer blocking function has been found, for example, in the locus control region of β-globin genes (Bell et al., 1999). In contrast, CpG containing CTCF-binding sites have been shown to lose binding activity when methylated (Bell and Felsenfeld, 2000; Hark et al., 2000; Kanduri et al., 2000; Szabo et al., 2000; Filippova et al., 2001; Takai et al., 2001; Chao et al., 2002; Kanduri et al., 2002a). This activity is crucial for the proper function of the imprinting control region at the Igf2/H19 locus.
Functional regulatory elements in eukaryotic genomes are often built from modules consisting of individual sites for DNA-binding factors. Such a modular structure has been shown for the lysozyme upstream silencer that consists of a CTCF-binding site together with a thyroid hormone response element (TRE) (Arnold et al., 1996; Lutz et al., 2000a). The thyroid hormone receptor (TR) binds as a homodimer or as a heterodimer with the retinoid-X-receptor (RXR) to the two TRE half sites, which are arranged as an inverted palindrome. For this composite CTCF/TR element, synergy in repression in the absence of thyroid hormone (T3), as well as in gene activation in the presence of T3, has been demonstrated (Lutz et al., 2000a). Similarly, a TRE-containing rat genomic DNA segment (element 144) is found next to a CTCF-binding site (Awad et al., 1999). This TRE is a so-called negative response element, mediating transcriptional repression in the presence of T3. This repression is critically dependent on CTCF binding next to the TRE. Another example of a composite CTCF/TR-binding site is site A, downstream of the mouse c-myc gene, which is flanked by a TRE (Perez-Juste et al., 2000). This element also confers transcriptional repression in the presence of T3.
Here we addressed the question of whether composite CTCF/TR-binding sites confer their repressive effect directly on promoters, or whether the enhancer blocking activity of CTCF on insulator elements can also be modulated by TR and T3. We tested the composite binding site that lies upstream of the chicken lysozyme gene and a newly identified composite element upstream of the human c-myc gene. For both composite elements we demonstrate that enhancer blocking activity can be regulated, since in the presence of T3, enhancer blocking is abrogated. This is the first demonstration that enhancer blocking in vertebrates is not only a constitutive property or an activity regulated by long-term changes in DNA methylation, but, in the case of CTCF/TR composite elements, can also be regulated by ligand.
Results
CTCF sites are often found next to binding sites for the thyroid hormone receptor
Thyroid hormone response elements can be grouped into so-called positive TREs and negative TREs (for a review, see Dotzlaw and Baniahmad, 2002). In several cases, binding sites for CTCF and for TR have been found in close proximity (Figure 1A and B). For the 144 element, T3-dependent repression of this negative TRE is strictly dependent on the presence of a functional CTCF-binding site (Awad et al., 1999). The F2 sequence in the F1/F2 silencer element 2.4 kb upstream of the chicken lysozyme gene is a positive TRE and synergy with CTCF in repression in the absence of T3 has been seen (Lutz et al., 2000a). Encouraged by the frequent occurrence of adjacent TR- and CTCF-binding sites, we wondered whether a new CTCF site at position –2.1 kb upstream of the human c-myc gene (N-site; G.Filippova, D.Loukinov, Y.Hu, T.Awad, B.Penn, S.Tapscott, E.Klenova, P.Neiman, S.Collins, H.Morse III, R.Ohlsson, R.Renkawitz and V.Lobanenkov, submitted for publication) is also flanked by a TR-binding site. Sequence inspection indeed revealed a potential TR-binding site (Figure 1). This sequence element, TRE(myc-N), is a direct repeat element spaced by four nucleotides (DR4), the distance between the DR4 element and the border of the CTCF-binding site N being 10 bp (Figure 1). This arrangement is similar to the lysozyme F1 and F2 sequences, which show a 13 bp spacing between elements. TR binding to TRE(myc-N) was analysed by electrophoretic mobility shift experiments (EMSA). Addition of TR and RXR proteins resulted in a prominent shift in mobility of the TRE, demonstrating protein binding to this sequence (Figure 1C). This binding was specific, since competition with a bona fide TRE (DR4) efficiently removed protein binding, whereas competition with a CTCF-binding site (F1) did not. Furthermore, EMSAs in the presence of T3 displayed a slight increase in the mobility of DNA-bound TR/RXR, which is characteristic for the hormone-loaded receptor–DNA complex (Arnold et al., 1996). The direct repeat arrangement of the TRE(myc-N) half sites spaced by 4 nucleotides, as shown for other TREs, allows only heterodimeric TR/RXR binding, and not homodimeric TR/TR or RXR/RXR binding. Addition of CTCF induced the formation of a specific complex, which is abrogated by excess F1 DNA, but not by a DR4 element. Simultaneous addition of all binding components (TR, RXR, CTCF) generates all the possible shifts: TR/RXR, CTCF and TR/RXR plus CTCF. Inspection of the binding efficiency in the absence of DNA competitors indicates that TR/RXR and CTCF binding occur independently without any positive or negative cooperativity. Thus the myc-N/TRE, 2 kb upstream of the human c-myc gene, is another example of adjacent TRE and CTCF sites (Figure 1A and B).


Fig. 1. CTCF sites are often found close to binding sites for TR. Five different cases of CTCF/TR composite elements are listed: the human c-myc upstream elements N and TRE(myc-N) (see text), the chicken lysozyme upstream F1 and F2 sequences (Baniahmad et al., 1990), the human 144 CTCF- and TR-binding sites (Bigler and Eisenman, 1995; Awad et al., 1999), the promoter region of the human APP gene (Quitschke et al., 1996; Belandia et al., 1998) and the mouse c-myc site with the CTCF site A and the TRE (Filippova et al., 1996; Perez-Juste et al., 2000). (A) Alignment of the 10 different TRE half sites from the five different CTCF/TR composite elements and their arrangement as direct repeats spaced by 4 or 5 nucleotides (DR4 and DR5), or as inverted palindromes spaced by 3, 6 or 7 nucleotides (IP3, IP6 and IP7) is indicated. (B) Alignment of the five different CTCF-binding sites with their spacing (in bp) from their respective TREs, upstream (left hand side) or downstream (right hand side) of the CTCF binding sequence. (C) Gel retardation with the c-myc upstream composite element N and TRE(myc-N) as probe. TRα, RXRα or CTCF were added as indicated, in the absence or presence of T3. Competitors, DR4 to compete specifically for TR/RXR binding and F1 to compete CTCF binding, were added as indicated.
CTCF/TR-mediated enhancer blocking by the chicken lysozyme F1/F2 element is abrogated by thyroid hormone
Based on the occurrence of CTCF/TR composite elements and the functional interaction of CTCF and TR in regulation of transcription, we asked whether T3 has an effect on the enhancer blocking activity of CTCF. In order to functionally analyse the possible enhancer blocking activity of CTCF/TR composite elements, we used a colony assay, previously developed in K562 cells to analyze the insulator function of sequences from the chicken β-globin locus (Chung et al., 1993, 1997). This assay has been widely used for investigation of the enhancer blocking potential of DNA elements from diverse genes and species (Bell et al., 1999; Bell and Felsenfeld, 2000; Filippova et al., 2001; Chao et al., 2002). In this assay, any enhancer blocking element placed between an enhancer and the promoter of a neomycin resistance (Neor) gene reduces gene activity and thereby decreases the number of G418-resistant colonies. Since we were interested in the effect of T3 in this system, we first analysed whether K562 cells respond to induction by T3. Transient transfection of a reporter gene driven by a TRE-tk promoter showed a 10-fold T3-dependent induction (data not shown). In order to evaluate possible indirect hormone effects on colony numbers, we controlled whether the growth of the K562 cells is influenced by the presence of T3, but found no effect of the hormone on cell growth (data not shown).
In the absence of an enhancer blocker, the Neor gene is fully active and therefore induces a large number of resistant colonies (Figure 2A). The number of colonies is substantially reduced by inserting the β-globin insulator between the enhancer and the Neor gene, whereas a multimerized F1/F2 unit in neither the sense nor antisense orientation had any effect on colony numbers when placed outside of the enhancer/Neor gene unit. Since the enhancer blocking activity of the β-globin insulator is also mediated by CTCF, these results confirm previous observations that CTCF-binding sites block enhancer activity when placed between the enhancer and the promoter (Bell et al., 1999). We therefore carried out the colony assay in the presence or absence of T3. Independent of the presence of an enhancer blocking element (Figure 2A), addition of T3 reduces somewhat the observed number of colonies, indicating a general inhibitory effect on colony formation. Importantly, there was no increase in colony number and therefore no loss of enhancer blocking by the β-globin insulator, a segment containing a CTCF site that is not associated with a TRE. No increase in colony numbers was observed after T3 addition when the F1/F2 element was placed in a position outside of the promoter/enhancer unit, demonstrating that the TR-bound F2 element was unable to regulate gene activity in this downstream position. These controls allowed testing of the F1/F2 composite elements in a position which should mediate enhancer blocking (Figure 2B). In the absence of T3, the duplicate β-globin insulator and the F1/F2 element as dimer or hexamer, in both the sense or antisense orientation, mediate enhancer blocking with similar strength. Addition of T3 again had no effect on enhancer blocking activity after transfection of the constructs that lack a TRE. In sharp contrast, for all constructs containing F1/F2 sequences in an enhancer blocking position and in both orientations, T3 addition led to loss of enhancer blocking activity. In order to test whether T3-mediated relief of enhancer blocking is dependent on the gene-specific F1/F2 sequences of the CTCF- and TR-binding sites, we also tested the myc upstream composite element myc-N/TRE. Although this element is similar to the F1/F2 element with respect to its ability to bind TR/RXR heterodimers and CTCF, it is different with respect to the spacing and orientation of individual sequences within the TRE (Figure 1). We placed this composite element as a monomeric site at the enhancer blocking position and compared the observed number of colonies with the hexameric F1/F2 element (Figure 3). The single composite element of the myc gene caused a similar degree of enhancer blocking as the F1/F2 hexamer, and addition of T3 almost fully relieved the enhancer blocking activity of the myc composite element. Importantly, no T3 effect was seen with a mutated sequence, which had two nucleotides exchanged within the CTCF-binding site of the myc composite element. This mutation caused a loss of in vitro CTCF binding (Filippova et al., submitted for publication), but had no effect on the TR-binding site. The weak enhancer blocking activity remaining could result from residual CTCF binding and/or possibly from a weak blocking activity mediated by TR itself.


Fig. 2. CTCF/TR-mediated enhancer blocking by the chicken lysozyme F1/F2 element is abrogated by thyroid hormone. K562 cells were transfected with the indicated DNA constructs and after neomycin selection in the absence (open bars) or presence of thyroid hormone (filled bars) the number of colonies was determined. (A) The F1/F2 sequences outside of the enhancer (E)/promoter unit in the sense (6xsF1/F2E) or antisense orientation (6xaF1/F2E) do not effect colony numbers. Addition of thyroid hormone causes a general reduction in colony numbers irrespective of the presence of a TR-binding site (F2). (B) Relative enhancer blocking activity (determined by dividing the colony numbers obtained with the control plasmid pNI-MCS by the colony number from the respective DNA constructs) for four constructs. F1/F2 elements placed between the enhancer and promoter in the sense (E6xsF1/F2) or antisense orientation (E6xaF1/F2) mediate enhancer blocking in the absence, but not in the presence of thyroid hormone.

Fig. 3. CTCF/TR-mediated enhancer blocking by the c-myc upstream composite element N and TRE(myc-N) is abrogated by thyroid hormone. Relative enhancer blocking activity in the absence (open bars) or presence of thyroid hormone (filled bars) was determined as in Figure 2. The composite element was tested between the enhancer and the promoter as a single wild-type sequence (Emyc-N/TRE) or as a single mutated sequence with a mutation in the CTCF-binding site (Emyc-Nmut/TRE).
In summary, both the lysozyme F1/F2 and the myc-N/TRE composite CTCF/TR-binding sites mediate enhancer blocking. In contrast to previously analysed CTCF-binding sites, enhancer blocking by these composite elements is relieved by T3, thereby demonstrating a new feature of hormone regulated enhancer blocking.
CTCF stays bound on the chromatin even after relief of enhancer blocking by thyroid hormone
A simple explanation of the effects of T3 on CTCF insulation would be that CTCF binding to the composite elements is lost after hormone treatment. We therefore analysed the in vivo binding of CTCF on the F1/F2 element in chicken erythroblasts (HD37) and chicken monocytic cells (HD11), both of which show a DNase I hypersensitive site in the chromatin at the F1/F2 element at position –2.4 kb (Huber et al., 1995). HD37 cells are inactive for lysozyme expression whereas HD11 cells express the lysozyme gene, which can be further induced (Huber et al., 1995). Based on the DNase I responsiveness of the chromatin, a scenario was proposed in which the silencer (F1/F2) element at –2.4 kb is active in erythroblasts as well as in monocytes, whereas the enhancer at position –2.7 kb is only active in the monocytes (Huber et al., 1995). A further increase of lysozyme gene activity upon induction was envisaged as a loss of silencer function. In vivo DMS footprinting was therefore carried out using HD37 and HD11 cells in the absence or presence of thyroid hormone (Figure 4A and B). The large footprint typical of CTCF-binding sites covers ∼50 nucleotides in both HD37 and HD11 cells and is seen both in the presence as well as the absence of T3. Since in vivo footprints prove the occupation of specific sites but do not allow identification of the binding protein and do not allow quantitative analyses of changes in binding, we carried out chromatin immunoprecipitation (ChIP) assays. Formaldehyde cross-linked chromatin from both cell types, grown in the absence or presence of thyroid hormone, was sheared and precipitated with an antibody against CTCF. Different regions upstream of the lysozyme gene were detected by real-time PCR. Specifically, the region around the CTCF-binding site at –2.4 kb was tested and compared with CTCF negative controls at –3.9 kb, –4.8 kb and –6.1 kb. As seen in Figure 4C and D, the sequences enriched in the CTCF ChIP are those of the CTCF-binding site at –2.38 kb and the adjacent region at –2.54 kb, which although not harbouring a CTCF-binding site, is also precipitated since the sonicated chromatin fragments, average size ∼500 bp, represent an overlapping set of sequences. All of the other sequences tested showed a low ratio of precipitate to input chromatin. Furthermore, the presence or absence of thyroid hormone did not dramatically change the precipitation of the CTCF-binding site, in particular at position –2.38 kb. Therefore, it can be concluded from the in vivo footprinting and the ChIP experiments that loss of CTCF binding is not the cause of thyroid hormone-mediated relief of enhancer blocking by CTCF/TR composite elements.
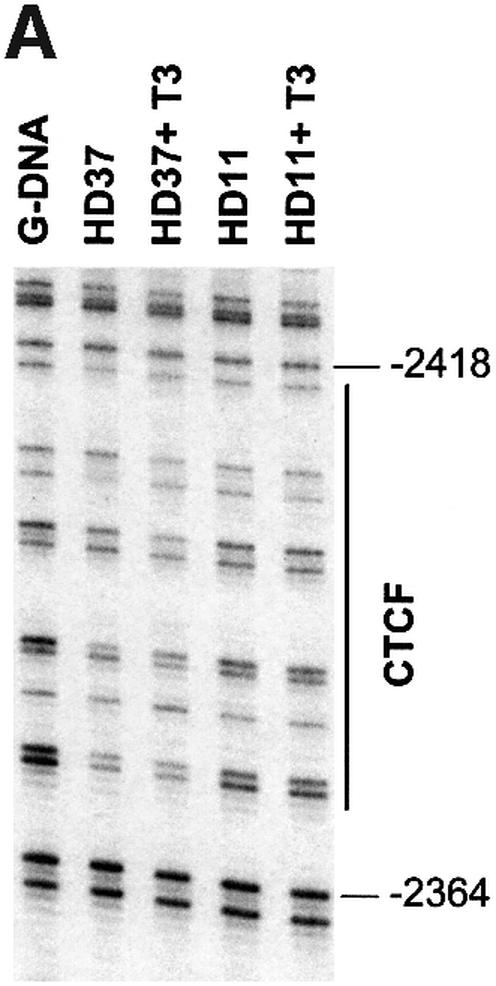



Fig. 4. CTCF remains bound to the chromatin upstream of the chicken lysozyme gene even after thyroid hormone-induced relief of enhancer blocking. HD37 cells (non-expressing) and HD11 cells (lysozyme expressing) were grown in the absence or presence of thyroid hormone. (A) DMS in vivo footprinting shows protection over the CTCF-binding site under all conditions when compared with the G-ladder (G-DNA) prepared from purified DNA. (B) The scan of the autoradiograph shown in (A) demonstrates that the footprint is not changed upon incubation with T3. (C and D) Chromatin immunoprecipitation with antibodies against CTCF shows CTCF binding on the endogeneous F1 element (–2.38kb) in HD37 cells and HD11 cells. Absence (–T3) or presence of thyroid hormone (+T3) does not change CTCF binding on the –2.38kb sequence. Fold enrichment or fold depletion is plotted relative to non-specific precipitation at CTCF free sites (–3.9 kb, –4.8 kb and –6.1 kb; see Materials and methods).
Relief of enhancer blocking by thyroid hormone is associated with enhanced acetylation of histone H4 in the enhancer blocking region as well as in the enhancer itself
Previously, we showed that CTCF binds histone deacetylase activity from HeLa nuclear extracts (Lutz et al., 2000b). Since deacetylation of histones is often associated with transcriptional repression (Aranda and Pascual, 2001; Forsberg and Bresnick, 2001; Wade, 2001), we wondered whether such deacetylation is associated with the ability of CTCF to repress promoter activities (Lutz et al., 2000a) and, specifically, whether enhancer blocking is also associated with histone deacetylation. ChIP experiments with antibodies against acetylated histones H3 and H4 across the chicken β-globin locus have revealed that the CTCF-binding site in the locus control region is associated with a strong constitutive focus of hyperacetylation (Litt et al., 2001b). We therefore analysed acetylation of histone H4 in the lysozyme non-expressing and expressing cell lines, HD37 and HD11, respectively, in the presence or absence of T3. Since the CTCF-binding site within the enhancer blocking sequence at position –2.4 kb is in close proximity to the enhancer element at –2.7 kb, we analysed acetylation of mono-nucleosomes from this region. The positions of nucleosomes in this region have previously been established (Huber et al., 1996) and are summarized in Figure 5A. Chromatin fragments were generated from nuclei by micrococcal nuclease digestion and fractionation on sucrose gradients, only the mono-nucleosomal fraction being used for the ChIP assays. Input chromatin, as well as precipitated chromatin was analysed by real-time PCR using Taqman probes for the amplicons indicated in Figure 5A. Amplicon 4 is specific for the CTCF region, whereas amplicons 2 and 3 are specific for the adjacent enhancer. In addition, we analysed a region further upstream (amplicon 1), as well as one much further downstream (amplicon 5). A 6th amplicon monitored the promoter region adjacent to the start of transcription. The signals from the four input mono-nucleosomal fractions (HD37 and HD11 cells, in the presence or absence of thyroid hormone) show a similar level of sequence representation at all six amplicons (data not shown), indicating that there is no under-representation of nucleosomes at any of the points investigated, i.e. that none of the amplicons is devoid of nucleosomes. Furthermore, the level of representation of these six sequences (15–20% of genomic DNA) is that found typically for MNase-generated mono-nucleosomes derived from genic regions. The lysozyme non-expressing HD37 cell line shows only a very low level of acetylated H4 histone, both in thyroid hormone-treated as well as untreated cells (Figure 5B). In sharp contrast, lysozyme-expressing HD11 cells, in the absence of thyroid hormone, show a substantial level of histone H4 acetylation in all of the regions tested, including the CTCF site (Figure 5C). After addition of thyroid hormone, acetylation levels at all six amplicons were further increased. This was true for the enhancer blocking region (CTCF/TR sites) at –2.4 kb and for both the enhancer (E) and the promoter.



Fig. 5. H4 acetylation at the enhancer blocker, the enhancer and the promoter is present only in lysozyme-expressing cells and is further induced by T3 additon. (A) Nucleosomal structure in the upstream lysozyme chromatin (Huber et al., 1996) is indicated by the position of hypersensitive sites for micrococcal nuclease digestion (arrows). Enhancer factor binding sites for PU.1 and C/EBP (Faust et al., 1999) and binding sites for CTCF and TR on the F1/F2 element are indicated. Nucleotide positions for the amplicons 1 to 5 used for the ChIP assay in (B) and (C) are indicated. Amplicon 6 covers the promoter region (–182/–77; data not shown). Chromatin immunoprecipitation of acetylated histone H4 from HD37 cells and HD11 cells are given in (B) and (C), respectively. Fold enrichment or fold depletion is the ratio of the PCR signal from the Ab-precipitated chromatin relative to that from the input chromatin (see Materials and methods). The x-axis shows the amplicon positions relative to the lysozyme upstream region, as indicated at the bottom of the graph.
There is thus no evidence for the involvement of unacetylated nucleosomes in CTCF-mediated enhancer blocking activity. Even in case of the HD37 cells, which show no significant H4 acetylation in the enhancer or the promoter regions, a small but significant acetylation over the CTCF site was observed. Lysozyme-expressing HD11 cells in the absence of thyroid hormone show enhancer blocking activity as well as significant histone H4 acetylation at the CTCF site. Relief of enhancer blocking by T3 is associated with a further increase in histone H4 acetylation.
Discussion
Thyroid hormones play important roles in metabolism, growth and differentiation. Almost every cell type has one or other isoform of TR. Brain, liver, heart, bone and blood cells have been used as model systems to study thyroid hormone action in differentiation. It is therefore not surprising that many thyroid hormone responsive genes and their corresponding TREs have been described in these tissues (for a review, see Yen, 2001). For both genes studied in this work, functional TREs have been identified (Baniahmad et al., 1990; Lutz et al., 2000a; Perez-Juste et al., 2000). In addition to the composite CTCF/TR-binding site in the promoter region of the c-myc gene, we identified another composite element located >2 kb upstream of the human c-myc gene, a position similar to a CTCF/TR composite element in the chicken lysozyme gene (Lutz et al., 2000a). For both composite elements, we demonstrate that in the absence of thyroid hormone, enhancer blocking activity can be seen, whereas the addition of thyroid hormone abrogates this enhancer blocking. The functional interaction between CTCF and TR is such that in the case of the myc composite element, a mutation in the CTCF-binding site prevents any T3 effects in the enhancer blocking assay. This finding offers an explanation for the function and arrangement of the regulatory elements upstream of the lysozyme gene (Huber et al., 1995). In myeloid precursor cells, when the upstream enhancer at –2.7 kb is active, the enhancer blocker at position –2.4 kb only allows a low level of lysozyme expression. However, after induction towards mature monocytes, maximal transcriptional activity is seen, most probably due to the fact that the upstream enhancer is no longer shielded by the enhancer blocker.
In order to understand the molecular mechanism of release of the blocking function of an insulator, one has to understand the mechanism of enhancer blocking. Several models on how insulators confer enhancer blocking have been postulated, based on convincing evidence for each mechanism (Pirrotta, 1998; Gause et al., 2001; Burgess-Beusse et al., 2002; Labrador and Corces, 2002). Most of these models require some change in chromatin conformation but are unable to fully explain enhancer blocking. For CTCF-binding sites, specific chromatin modifications have been found. These modifications include histone H3 and H4 acetylation, as well as H3 methylation at lysine 4 in case of the insulator at the locus control region of the chicken β-globin gene locus (Litt et al., 2001a). In this example, histone hyperacetylation has been seen at CTCF sites accompanied by a sharp drop of the modification in the neighbouring sequences. In the study presented here, we find that at the F1 site in the lysozyme gene, histone H4 is more strongly acetylated as compared with the flanking regions. Loss of enhancer blocking by the addition of T3 increases acetylation several fold, not only at the CTCF site, but also at the enhancer and other regions. The lower acetylation in the absence of T3 could be either explained by an inefficient recruitment of histone acetyl transferases or by a combined action of histone deacetylases and acetyl transferases, a possible scenario since CTCF has been shown to bind histone deacetylases (Lutz et al., 2000b). Subsequent action of thyroid hormone might lead to loss of deacetylases or to recruitment of acetylases. Alternatively, binding to histone deacetylase-containing complexes may only play a role in CTCF-mediated promoter repression and may not be involved in enhancer blocking. One can then ask how an increase in histone acetylation can explain the loss of enhancer blocking. Several types of conformational change are possible, for example the overall acetylation of nucleosomes could facilitate looping of chromatin to allow interaction of enhancer regions with the promoter.
Whatever the precise mechanisms involved, the present data show that the relief of enhancer blocking by thyroid hormone is not caused by a loss of CTCF from the chromatin. It has been shown for the imprinting control region of the Igf2 and H19 genes that CTCF binds in the linker region between nucleosomes (Kanduri et al., 2002b). In case of the lysozyme –2.4 kb region, a nucleosome is present since we find that the mono-nucleosomal fraction contains a normal representation of the CTCF site. Chromatin digestion with micrococcal nuclease showed hypersensitive regions flanking the CTCF/TR-binding site (Figure 5A; Huber et al., 1996), suggesting that CTCF binds at the centre of the nucleosome. Differences in CTCF zinc finger usage and/or different rotational phasing at the lysozyme F1 and the Igf2/H19 ICR sequence may explain why CTCF cannot bind to a nucleosome at the ICR, whereas at the F1 element nucleosome binding is possible. Similarly, TR binding to nucleosomes has also been demonstrated (Urnov and Wolffe, 2001), so it is possible that binding of TR facilitates the binding of CTCF to a nucleosome that includes a composite CTCF/TR-binding site. Most importantly, CTCF stays bound to a nucleosome even after addition of T3 and loss of enhancer blocking activity.
Enhancer blocking has been demonstrated for several DNA elements that shield a regulatory gene unit from enhancer action coming from a neighbouring regulatory gene unit (for reviews, see Mueller, 2000; Bell et al., 2001; Burgess-Beusse et al., 2002; Labrador and Corces, 2002). Such an activity has to be constitutive to provide permanent shielding. DNA methylation has been shown in some cases to regulate CTCF binding, thereby allowing enhancer function to be regulated. Such regulation is expected to turn genes on or off over a more long-term period, as shown for the imprinted Igf2/H19 and Kcnq1 loci and for the DM1 locus (Bell and Felsenfeld, 2000; Hark et al., 2000; Kanduri et al., 2000; Szabo et al., 2000; Filippova et al., 2001; Kanduri et al., 2002a). Here we show a new, short-term feature of enhancer blocking, namely regulation by thyroid hormone, which expands the biological role of enhancer blockers from constitutive or long-term regulators to fine-tuning switches.
Materials and methods
Plasmid construction
pNI-MCS was generated by inserting a multiple cloning site between the enhancer and promoter of the plasmid pNI, by cutting with AscI and ligating with a double-stranded oligonucleotide containing unique recognition sequences for NotI, EcoRV, Eco47III, KpnI, SacI and XhoI.
The construct pE6xsF1/F2 with the F1/F2 sequence in sense orientation was generated by isolating the hexamerized binding site of F1/F2 from an intermediary clone with EcoRI and NotI followed by Klenow fill in. This fragment was ligated into the EcoRV site of pNI-MCS. The plasmid pE6xaF1/F2 with the F1/F2 sequence in antisense orientation was generated by isolating the hexamerized binding site of F1/F2 from an intermediary clone with EcoRV and NotI and ligating this fragment to pNI-MCS cut with the same enzymes. For generating reporter constructs that contain the enhancer between the promoter and the hexamerized F1/F2 site, pNIΔLCR was first produced by eliminating the enhancer with EcoRI digestion and religating the vector. The plasmid p6xaF1/F2E was cloned by isolating the NotI/Eco47III fragment from pE6xaF1/F2 and ligating it with pNIΔLCR digested with the same enzymes. This intermediate then was cut with XhoI and filled with Klenow. The enhancer, isolated as an EcoRI fragment from pNI-MCS, was filled with Klenow and inserted into this site. The same strategy was used to obtain p6xsF1/F2E. In this case, the hexameric binding cassette was isolated from pE6xsF1/F2.
To generate the reporter construct pE2xaF1/F2, the BamHI/BglII DNA fragment of ptkCATsF1/F2 (B.Kaiser, unpublished data) containing the F1/F2 sequence was dimerized with T4 DNA ligase and the ends filled with Klenow. This insert was ligated to pNI, which had been cut with AscI and filled in with Klenow. To generate pEmyc-N/TRE and pEmyc-Nmut/TRE that carries a mutation in the CTCF site, the inserts were excised from pBend2-TR-N and pBend2-TR-N(Sac) with XbaI, treated with T4 polymerase to recess the ends and then gel purified. The globin inserts were excised from the pJC-3–4 using SacI and the resulting vector fragment was blunt ended with T4 polymerase and ligated to the N/TRE inserts.
Electrophoretic mobility shift assay
The myc probe containing the N site and the TRE(mycN) was generated from the c-myc-2050 region after EcoRI and SalI digestion and labelling radioactively with Klenow. DNA–protein binding reactions were carried out with an affinity purified CTCF fraction from HeLa cells (Burcin et al., 1997), bacterially expressed human TRα and in vitro translated human RXRα, as described previously (Arnold et al., 1996).
Colony formation assay in K562 cells and cell culture
Reporter plasmids were linearized with NdeI, phenol/chloroform extracted, precipitated with ethanol and 2 µg used to electroporate 1 × 107 K562 cells cultured in RPMI medium supplemented with 10% hormone-depleted fetal calf serum (FCS) and 1% PenStrep, essentially as described previously (Chung et al., 1993, 1997). After 2–3 weeks of growth in agar containing G418, in the absence or presence of thyroid hormone (10–6 M TRIAC), relative enhancer blocking was calculated by normalizing the number of colonies to that obtained with the control plasmid pNI-MCS.
The chicken cell lines HD37 and HD11 (Beug et al., 1979; Metz and Graf, 1991) were grown in Iscove’s modified DMEM medium containing GLUTAMAX-I (Life Technologies) supplemented with 8% FCS and 2% chicken serum, 100 U/ml penicillin, 100 µg/ml streptomycin and 0.15 mM monothioglycerol, as described previously (Kontaraki et al., 2000). T3 (10–6 M) was added to the medium for 48 h before harvesting cells when appropriate.
In vivo footprinting
Preparation of DMS-treated DNA and LM-PCR was carried out essentially as described previously (Hershkovitz and Riggs, 1997; Kontaraki et al., 2000; Tagoh et al., 2002). The first primer extension was carried out using Vent polymerase (New England Biolabs) and a 5′-biotinylated primer (CCTATGATTTGCTACAAAAGTAC) and products ligated to the linker LP25–21, consisting of a 25mer annealed to a 5′ phosphorylated 21mer. Specific biotinylated products were isolated on streptavidin paramagnetic beads (DYNAL) and used for PCR amplification using the 25mer and the nested specific primer GCTACAAAAGTACTCATAAACCCTAAATC with Pfu Turbo DNA polymerase (Stratagene) in a buffer containing 1.4 M betaine and 5% DMSO. PCR-amplified products were labelled by primer extension also using Pfu Turbo DNA Polymerase and the 32P-labelled primer (GATTTGCTACAAAAGTACTCATAAACC) and were separated on 7% sequencing gels.
ChIP assays
ChIP assays for CTCF (Orlando et al., 1997) and for acetylated histone H4 (Hebbes et al., 1994) were performed as described previously with modifications and primer sequences described in the Supplementary data.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We would like to thank Leni Schäfer-Pfeiffer for technical assistance, Aria Baniahmad and Joerg Leers for critically reading the manuscript, and Adam C.Bell for kindly providing the plasmids pNI, pJC 3–4 and pJC 13–1. This work was supported by a grant from the Deutsche Forschungsgemeinschaft and by the Fonds der Chemischen Industrie.
References
- Aranda A. and Pascual,A. (2001) Nuclear hormone receptors and gene expression. Physiol. Rev., 81, 1269–1304. [DOI] [PubMed] [Google Scholar]
- Arnold R., Burcin,M., Kaiser,B., Muller,M. and Renkawitz,R. (1996) DNA bending by the silencer protein NeP1 is modulated by TR and RXR. Nucleic Acids Res., 24, 2640–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad T.A. et al. (1999) Negative transcriptional regulation mediated by thyroid hormone response element 144 requires binding of the multivalent factor CTCF to a novel target DNA sequence. J. Biol. Chem., 274, 27092–27098. [DOI] [PubMed] [Google Scholar]
- Baniahmad A., Steiner,C., Koehne,A.C. and Renkawitz,R. (1990) Modular structure of a chicken lysozyme silencer: involvement of an unusual thyroid hormone receptor binding site. Cell, 61, 505–514. [DOI] [PubMed] [Google Scholar]
- Belandia B., Latasa,M.J., Villa,A. and Pascual,A. (1998) Thyroid hormone negatively regulates the transcriptional activity of the β-amyloid precursor protein gene. J. Biol. Chem., 273, 30366–30371. [DOI] [PubMed] [Google Scholar]
- Bell A.C. and Felsenfeld,G. (2000) Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature, 405, 482–485. [DOI] [PubMed] [Google Scholar]
- Bell A.C., West,A.G. and Felsenfeld,G. (1999) The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell, 98, 387–396. [DOI] [PubMed] [Google Scholar]
- Bell A.C., West,A.G. and Felsenfeld,G. (2001) Insulators and boundaries: versatile regulatory elements in the eukaryotic. Science, 291, 447–450. [DOI] [PubMed] [Google Scholar]
- Beug H., von Kirchbach,A., Doederlein,G., Conscience,J.F. and Graf,T. (1979) Chicken hematopoietic cells transformed by seven strains of defective avian leukemia viruses display three distinct phenotypes of differentiation. Cell, 18, 375–390. [DOI] [PubMed] [Google Scholar]
- Bigler J. and Eisenman,R.N. (1995) Novel location and function of a thyroid hormone response element. EMBO J., 14, 5710–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burcin M., Arnold,R., Lutz,M., Kaiser,B., Runge,D., Lottspeich,F., Filippova,G.N., Lobanenkov,V.V. and Renkawitz,R. (1997) Negative protein 1, which is required for function of the chicken lysozyme gene silencer in conjunction with hormone receptors, is identical to the multivalent zinc finger repressor CTCF. Mol. Cell. Biol., 17, 1281–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess-Beusse B., Farrell,C., Gaszner,M., Litt,M., Mutskov,V., Recillas-Targa,F., Simpson,M., West,A. and Felsenfeld,G. (2002) The insulation of genes from external enhancers and silencing chromatin. Proc. Natl Acad. Sci. USA, 99, 16433–16437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke L.J., Hollemann,T., Pieler,T. and Renkawitz,R. (2002) Molecular cloning and expression of the chromatin insulator protein CTCF in Xenopus laevis. Mech. Dev., 113, 95–98. [DOI] [PubMed] [Google Scholar]
- Chao W., Huynh,K.D., Spencer,R.J., Davidow,L.S. and Lee,J.T. (2002) CTCF, a candidate trans-acting factor for X-inactivation choice. Science, 295, 345–347. [DOI] [PubMed] [Google Scholar]
- Chung J.H., Whiteley,M. and Felsenfeld,G. (1993) A 5′ element of the chicken β-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell, 74, 505–514. [DOI] [PubMed] [Google Scholar]
- Chung J.H., Bell,A.C. and Felsenfeld,G. (1997) Characterization of the chicken β-globin insulator. Proc. Natl Acad. Sci. USA, 94, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotzlaw H. and Baniahmad,A. (2002) Gene regulation by thyroid hormone receptors. Methods Mol. Biol., 202, 109–128. [DOI] [PubMed] [Google Scholar]
- Faust N., Bonifer,C. and Sippel,A.E. (1999) Differential activity of the –2.7 kb chicken lysozyme enhancer in macrophages of different ontogenic origins is regulated by C/EBP and PU.1 transcription factors. DNA Cell Biol., 18, 631–642. [DOI] [PubMed] [Google Scholar]
- Filippova G.N., Fagerlie,S., Klenova,E.M., Myers,C., Dehner,Y., Goodwin,G., Neiman,P.E., Collins,S.J. and Lobanenkov,V.V. (1996) An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol., 16, 2802–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippova G.N., Thienes,C.P., Penn,B.H., Cho,D.H., Hu,Y.J., Moore,J.M., Klesert,T.R., Lobanenkov,V.V. and Tapscott,S.J. (2001) CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet., 28, 335–343. [DOI] [PubMed] [Google Scholar]
- Forsberg E.C. and Bresnick,E.H. (2001) Histone acetylation beyond promoters: long-range acetylation patterns in the chromatin world. BioEssays, 23, 820–830. [DOI] [PubMed] [Google Scholar]
- Gause M., Morcillo,P. and Dorsett,D. (2001) Insulation of enhancer–promoter communication by a gypsy transposon insert in the Drosophila cut gene: cooperation between suppressor of hairy-wing and modifier of mdg4 proteins. Mol. Cell. Biol., 21, 4807–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark A.T., Schoenherr,C.J., Katz,D.J., Ingram,R.S., Levorse,J.M. and Tilghman,S.M. (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature, 405, 486–489. [DOI] [PubMed] [Google Scholar]
- Hebbes T.R., Clayton,A.L., Thorne,A.W. and Crane-Robinson,C. (1994) Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken β-globin chromosomal domain. EMBO J., 13, 1823–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershkovitz M. and Riggs,A.D. (1997) Ligation-mediated PCR for chromatin-structure analysis of interphase and metaphase chromatin. Methods, 11, 253–263. [DOI] [PubMed] [Google Scholar]
- Huber M.C., Graf,T., Sippel,A.E. and Bonifer,C. (1995) Dynamic changes in the chromatin of the chicken lysozyme gene domain during differentiation of multipotent progenitors to macrophages. DNA Cell Biol., 14, 397–402. [DOI] [PubMed] [Google Scholar]
- Huber M.C., Krueger,G. and Bonifer,C. (1996) Genomic position effects lead to an inefficient reorganization of nucleosomes in the 5′-regulatory region of the chicken lysozyme locus in transgenic mice. Nucleic Acids Res., 24, 1443–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanduri C., Pant,V., Loukinov,D., Pugacheva,E., Qi,C.F., Wolffe,A., Ohlsson,R. and Lobanenkov,V.V. (2000) Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr. Biol., 10, 853–856. [DOI] [PubMed] [Google Scholar]
- Kanduri C., Fitzpatrick,G., Mukhopadhyay,R., Kanduri,M., Lobanenkov,V., Higgins,M. and Ohlsson,R. (2002a) A differentially methylated imprinting control region within the Kcnq1 locus harbors a methylation-sensitive chromatin insulator. J. Biol. Chem., 277, 18106–18110. [DOI] [PubMed] [Google Scholar]
- Kanduri M., Kanduri,C., Mariano,P., Vostrov,A.A., Quitschke,W., Lobanenkov,V. and Ohlsson,R. (2002b) Multiple nucleosome positioning sites regulate the CTCF-mediated insulator function of the H19 imprinting control region. Mol. Cell. Biol., 22, 3339–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontaraki J., Chen,H.H., Riggs,A. and Bonifer,C. (2000) Chromatin fine structure profiles for a developmentally regulated gene: reorganization of the lysozyme locus before trans-activator binding and gene expression. Genes Dev., 14, 2106–2122. [PMC free article] [PubMed] [Google Scholar]
- Labrador M. and Corces,V.G. (2002) Setting the boundaries of chromatin domains and nuclear organization. Cell, 111, 151–154. [DOI] [PubMed] [Google Scholar]
- Litt M.D., Simpson,M., Gaszner,M., Allis,C.D. and Felsenfeld,G. (2001a) Correlation between histone lysine methylation and developmental changes at the chicken β-globin locus. Science, 293, 2453–2455. [DOI] [PubMed] [Google Scholar]
- Litt M.D., Simpson,M., Recillas-Targa,F., Prioleau,M.N. and Felsenfeld,G. (2001b) Transitions in histone acetylation reveal boundaries of three separately regulated neighboring loci. EMBO J., 20, 2224–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanenkov V.V., Nicolas,R.H., Adler,V.V., Paterson,H., Klenova,E.M., Polotskaja,A.V. and Goodwin,G.H. (1990) A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene, 5, 1743–1753. [PubMed] [Google Scholar]
- Lutz M., Baniahmad,A. and Renkawitz,R. (2000a) Modulation of thyroid hormone receptor silencing function by co-repressors and a synergizing transcription factor. Biochem. Soc. Trans, 28, 386–389. [PubMed] [Google Scholar]
- Lutz M. et al. (2000b) Transcriptional repression by the insulator protein CTCF involves histone deacetylases. Nucleic Acids Res., 28, 1707–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz T. and Graf,T. (1991) v-myb and v-ets transform chicken erythroid cells and cooperate both in trans and in cis to induce distinct differentiation phenotypes. Genes Dev., 5, 369–380. [DOI] [PubMed] [Google Scholar]
- Mueller J. (2000) Transcriptional control: the benefits of selective insulation. Curr. Biol., 10, R241–R244. [DOI] [PubMed] [Google Scholar]
- Ohlsson R., Renkawitz,R. and Lobanenkov,V. (2001) CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet., 17, 520–527. [DOI] [PubMed] [Google Scholar]
- Orlando V., Strutt,H. and Paro,R. (1997) Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods, 11, 205–214. [DOI] [PubMed] [Google Scholar]
- Perez-Juste G., Garcia-Silva,S. and Aranda,A. (2000) An element in the region responsible for premature termination of transcription mediates repression of c-myc gene expression by thyroid hormone in neuroblastoma cells. J. Biol. Chem., 275, 1307–1314. [DOI] [PubMed] [Google Scholar]
- Pirrotta V. (1998) Polycombing the genome: PcG, trxG and chromatin silencing. Cell, 93, 333–336. [DOI] [PubMed] [Google Scholar]
- Quitschke W.W., Matthews,J.P., Kraus,R.J. and Vostrov,A.A. (1996) The initiator element and proximal upstream sequences affect transcriptional activity and start site selection in the amyloid β-protein precursor promoter. J. Biol. Chem., 271, 22231–22239. [DOI] [PubMed] [Google Scholar]
- Szabo P., Tang,S.H., Rentsendorj,A., Pfeifer,G.P. and Mann,J.R. (2000) Maternal-specific footprints at putative CTCF sites in the H19 imprinting control region give evidence for insulator function. Curr. Biol., 10, 607–610. [DOI] [PubMed] [Google Scholar]
- Tagoh H., Himes,R., Clarke,D., Leenen,P.J., Riggs,A.D., Hume,D. and Bonifer,C. (2002) Transcription factor complex formation and chromatin fine structure alterations at the murine c-fms (CSF-1 receptor) locus during maturation of myeloid precursor cells. Genes Dev., 16, 1721–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai D., Gonzales,F.A., Tsai,Y.C., Thayer,M.J. and Jones,P.A. (2001) Large scale mapping of methylcytosines in CTCF-binding sites in the human H19 promoter and aberrant hypomethylation in human bladder cancer. Hum. Mol. Genet., 10, 2619–2626. [DOI] [PubMed] [Google Scholar]
- Urnov F.D. and Wolffe,A.P. (2001) An array of positioned nucleosomes potentiates thyroid hormone receptor action in vivo. J. Biol. Chem., 276, 19753–19761. [DOI] [PubMed] [Google Scholar]
- Wade P.A. (2001) Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum. Mol. Genet., 10, 693–698. [DOI] [PubMed] [Google Scholar]
- West A.G., Gaszner,M. and Felsenfeld,G. (2002) Insulators: many functions, many mechanisms. Genes Dev., 16, 271–288. [DOI] [PubMed] [Google Scholar]
- Wolffe A.P. (2000) Transcriptional control: imprinting insulation. Curr. Biol., 10, R463–R465. [DOI] [PubMed] [Google Scholar]
- Yen P.M. (2001) Physiological and molecular basis of thyroid hormone action. Physiol. Rev., 81, 1097–1142. [DOI] [PubMed] [Google Scholar]