

Abstract
The Escherichia coli DsbA protein is the major oxidative catalyst in the periplasm. Dartigalongue et al. (EMBO J., 19, 5980–5988, 2000) reported that null mutations in the ompL gene of E.coli fully suppress all phenotypes associated with dsbA mutants, i.e. sensitivity to the reducing agent dithiothreitol (DTT) and the antibiotic benzylpenicillin, lack of motility, reduced alkaline phosphatase activity and mucoidy. They showed that OmpL is a porin and hypothesized that ompL null mutations exert their suppressive effect by preventing efflux of a putative oxidizing–reducing compound into the medium. We have repeated these experiments using two different ompL null alleles in at least three different E.coli K-12 genetic backgrounds and have failed to reproduce any of the ompL suppressive effects noted above. Also, we show that, contrary to earlier results, ompL null mutations alone do not result in partial DTT sensitivity or partial motility, nor do they appreciably affect bacterial growth rates or block propagation of the male-specific bacteriophage M13. Thus, our findings clearly demonstrate that ompL plays no perceptible role in modulating redox potential in the periplasm of E.coli.
Keywords: alkaline phosphatase/bacterial motility/disulfide bond formation/DsbA/DTT resistance
Introduction
During the past 10 years, the cumulative efforts of various laboratories have demonstrated the existence of complex oxidative–reductive pathways in the periplasm of Escherichia coli. It is known that the product of the dsbA gene is the major oxidant in the periplasm of E.coli. As a consequence, dsbA null mutants exhibit highly pleiotropic phenotypes, most likely due to defects in periplasmic disulfide bond formation (Bardwell et al., 1991; Kamitani et al., 1992; Jacob-Dubuisson et al., 1994; Genevaux et al., 1999). These phenotypes include reduction in levels of activity of the periplasmic enzyme alkaline phosphatase (AP), sensitivity to the reducing agent dithiothreitol (DTT), mucoidy, resistance to male-specific bacteriophages such as M13 and lack of both cell motility and biofilm formation. The DsbA protein has been shown to be reoxidized by the integral membrane protein DsbB (Bardwell et al., 1993; Dailey and Berg, 1993; Missiakas et al., 1993; Raina and Missiakas, 1997; Ritz and Beckwith, 2001; Collet and Bardwell, 2002). Hence, null mutations in dsbB result in pleiotropic phenotypes similar to those exhibited by dsbA single mutants. In turn, reduced DsbB is oxidized by the respiratory chain (Kobayashi et al., 1997; Bader et al., 1999).
In addition to the DsbA–DsbB oxidation pathway, there are also disulfide bond isomerization pathways carried out by the DsbC and DsbG proteins. The integral membrane protein DsbD is responsible for reducing the DsbC and DsbG isomerases (Raina and Missiakas, 1997; Ritz and Beckwith, 2001; Collet and Bardwell, 2002).
Null mutations in the dsbD gene were identified as exhibiting partial sensitivity to DTT and as partial suppressors of dsbA mutations (Missiakas et al., 1995). In an effort to identify better suppressors of dsbA mutations, the same group reported the isolation of null mutations in the ompL gene of E.coli using two separate genetic approaches (Dartigalongue et al., 2000). The first approach selected directly for insertional mutations in the chromosome of E.coli dsbA null mutants that restore wild-type levels of resistance to the reducing agent DTT. Upon subsequent testing, these dsbA ompL double null mutant strains were shown to possess full AP activity and to be non-mucoid, resistant to benzylpenicillin and fully motile. The second genetic approach involved screening for insertional mutations in an otherwise wild-type chromosome of E.coli that alone confer moderate sensitivity to DTT but do not inactivate any of the previously identified dsb genes. One such insertion, mapping in ompL, was characterized and also shown to result in a bacterial slow growth phenotype and resistance to the male-specific filamentous bacteriophage M13 (Dartigalongue et al., 2000). The ompL gene product was overexpressed, purified and shown to encode a porin protein. In contrast with their suppressive effect on dsbA null mutants, ompL null mutations did not suppress any of the examined defects in the dsbB gene. To explain their findings, the authors proposed that, in addition to DsbA, there may be an as yet unidentified low molecular weight compound(s) which is also oxidized by DsbB in the periplasm. It was suggested that this putative powerful oxidized agent(s) completely substitutes for DsbA provided that its leakage into the medium is not facilitated by the OmpL porin. Thus, in this model, the OmpL porin was postulated to be the major efflux mediator of this putative low molecular weight redox agent into the medium.
Attempts by one of us (A.A.S.) to identify the putative redox compound(s) predicted by the above model led to preliminary observations suggesting that the ompL null allele does not suppress a dsbA null mutant, forcing us to re-examine this in greater detail. Here, we report a complete failure to reproduce any of the numerous physiological phenotypes previously assigned to ompL null mutations.
Results and discussion
Construction and testing of mutant bacterial strains
Throughout this study we used the dsbA::mini-Tn10TetR (SR2262) insertion mutation of Dartigalongue et al. (2000). We verified that it is indeed a null mutation of dsbA by demonstrating that there is no full-length DsbA antigen in extracts prepared from SR2262 mutant bacteria, using standard western blot methodology (data not shown). We transferred the dsbA::mini-Tn10TetR allele by bacteriophage P1-mediated transduction into three standard, but different, E.coli K-12 genetic backgrounds, namely CA8000, MG1655 and MC4100. In all cases, the introduction of the dsbA::mini-Tn10TetR allele was associated with a complete lack of DsbA-specific antigen. Furthermore, as expected, the introduction of the dsbA::mini-Tn10TetR mutation resulted in the previously reported phenotypes of high sensitivity to DTT, lack of bacterial motility, mucoidy, reduced expression of AP activity and resistance to the male-specific bacteriophage M13.
In this work, we also employed the ompL::mini-Tn10KanR (SR1791) mutation used by Dartigalongue et al. (2000). We verified that the ompL::mini-Tn10KanR and the dsbA::mini-Tn10TetR mutations are closely linked (∼50%) by P1-mediated transduction (data not shown), in agreement with the previously reported findings of Dartigalongue et al. (2000). In addition, we constructed our own ompL deletion/substitution mutation (ΔompL::KanR) by taking advantage of the rapid gene substitution methodology of Yu et al. (2000), as described in Materials and methods. We have verified the authenticity of both the ompL::mini-Tn10KanR and the ΔompL::KanR alleles by carrying out PCR and DNA sequencing experiments using the appropriate DNA primers (see Materials and methods for details). We showed that the site of insertion of the mini-Tn10KanR cassette in strain SR1791 is indeed at nucleotide position 553 of the region surrounding ompL, as reported by Dartigalongue et al. (2000).
Reconstruction of dsbA ompL double mutants
To reconstruct the double mutants, the dsbA mutation was first transduced from SR2262 by bacteriophage P1 into the three E.coli genetic backgrounds listed above. Sub sequently, we introduced either the ompL::mini- Tn10KanR (SR1791) or the ΔompL::KanR allele that we constructed into both the dsbA single mutants and their corresponding wild-type parents. This resulted in the construction of the following set of isogenic mutants in the three genetic backgrounds: (1) dsbA::mini-Tn10TetR; (2) ompL::mini-Tn10KanR; (3) ΔompL::KanR, (4) dsbA:: mini-Tn10TetR ompL::mini-Tn10KanR; (5) dsbA::mini- Tn10TetR ΔompL::KanR. Again, the ompL mutant alleles were confirmed by PCR and DNA sequence analysis and, in the case of dsbA, by the lack of DsbA-specific antigen. As detailed below, none of the genetic and biochemical experiments that we carried out revealed any phenotypes associated with the ompL null mutations, either by themselves or in combination with the dsbA null allele.
ompL null mutations do not suppress the DTT sensitivity of dsbA null mutants
Having verified the authenticity of the various dsbA and ompL mutants that we constructed, we proceeded to test their physiological properties. Figure 1 shows that, as expected, the mutant dsbA allele alone results in hypersensitivity to the reducing agent DTT, i.e. dsbA mutant bacteria are completely unable to form colonies on LB agar containing 10 mM DTT. In contrast, both the wild-type and ompL single mutants form normal sized colonies under these conditions (Figure 1; data not shown). At higher concentrations of DTT, we were still unable to observe any differential behavior between the ompL null mutants and their corresponding isogenic wild-type parents (data not shown). Surprisingly, in our hands, neither of the two different ompL null mutations suppressed the DTT sensitivity of dsbA mutants in any of the three different backgrounds used (Figure 1; data not shown). This contrasts sharply with the results of Dartigalongue et al. (2000), who identified two out of their three ompL null mutations on the basis of complete suppression of the DTT sensitivity exhibited by dsbA null mutants, finding that the dsbA ompL double mutant is as resistant to DTT as the wild-type strain, if not more so.

Fig. 1. An ompL null mutation does not suppress DTT sensitivity of a dsbA null mutant. Ten-fold serial dilutions of exponentially growing bacterial cultures were spotted on LB agar plates containing various DTT concentrations and incubated at 37°C for ∼18 h. A representative set of mutants derived from the wild-type strain MG1655 (WT) spotted on LB agar at 37°C with or without 10 mM DTT is shown. The corresponding mutations are indicated at the top of the figure. The ompL allele shown here is the ompL::mini-Tn10KanR allele (Dartigalongue et al., 2000).
ompL null mutations do not suppress the motility defect of dsbA mutant bacteria
Owing to the defective formation of disulfide bonds, dsbA mutants fail to assemble the P ring of the flagellar hook-basal-body and are therefore non-motile (Dailey and Berg, 1993). Dartigalongue et al. (2000) showed that the introduction of any one of their three ompL mutations into a dsbA null mutant restores full motility. However, when we tested the motility of our MG1655 mutant strains, we clearly found that the dsbA ompL double mutants are as defective in motility as the isogenic dsbA single mutant (Figure 2; data not shown). This experiment was repeated in the CA8000 motile background and again the dsbA ompL double mutants were as non-motile as the dsbA single mutant (data not shown). Our conclusion from these motility experiments is that ompL null mutations play no detectable role in bacterial motility, either alone or in combination with the dsbA null mutation.
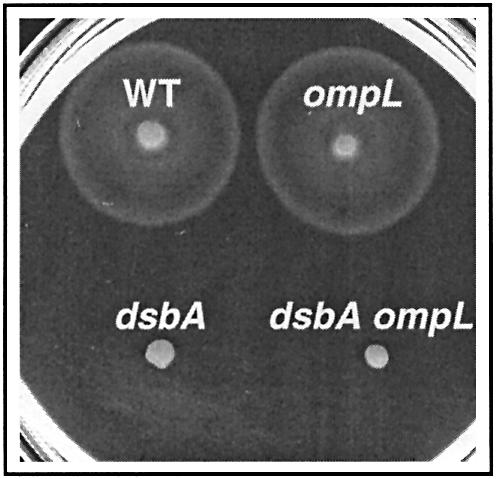
Fig. 2. An ompL null mutation does not restore the motility defect of a dsbA null mutant. Bacterial motility was monitored after 8 h of incubation at 37°C on soft agar plates. A typical set of results showing the radial growth of the dsbA and ompL single mutants and of the dsbA ompL double mutant compared with the parental strain MG1655 (WT) is presented. The ompL allele shown here is the ompL::mini-Tn10KanR allele (Dartigalongue et al., 2000).
ompL null mutations do not influence the levels of AP activity in a dsbA null mutant
Next, we measured the levels of AP activity in our isogenic constructs, both in LB broth (‘repressing’ conditions for AP activity) and in phosphate-limited minimal medium (‘de-repressing’ conditions for AP activity). In both cases, neither of the two ompL null mutations had any demonstrable effect in modulating the much lower levels of AP activity exhibited by the dsbA null mutant (Figure 3; data not shown). Thus, in contrast with the reported results of Dartigalongue et al. (2000), an ompL null mutation does not restore full or even partial levels of AP activity in the dsbA null mutant.

Fig. 3. The defect in AP folding exhibited by a dsbA mutation is not suppressed by mutations in ompL. AP assays were performed in rich and minimal media as described in Materials and methods. A representative set of results obtained in minimal medium with the wild-type strain MG1655 (WT) and its mutant derivatives is shown. The corresponding mutations are indicated at the bottom of the figure. The assays were done in triplicate and the standard deviation is represented by the error bars. The ompL allele shown here is the ompL::mini-Tn10KanR allele (Dartigalongue et al., 2000).
Suppression of other phenotypes associated with dsbA null mutations
Dartigalongue et al. (2000) reported that null mutations in the ompL gene completely eliminate the mucoid phenotype associated with dsbA null mutants. Again, in contrast with their findings, we observed that introduction of an ompL null mutation into a dsbA null mutant does not influence to any detectable extent the mucoidy exhibited by dsbA mutant bacteria in any of the three genetic backgrounds tested (data not shown).
We also examined the effect of an ompL null mutation on the sensitivity of a dsbA mutant to benzylpenicillin. Contrary to the findings of Dartigalongue et al. (2000), we surprisingly found that, in all three genetic backgrounds used, the dsbA null mutant was not more sensitive to benzylpenicillin than the isogenic wild-type parent or the ompL single mutants. In addition, introduction of an ompL null mutation in the dsbA mutant did not improve its growth on benzylpenicillin (data not shown).
Previously, it was reported that dsbA null mutants are defective in the formation of biofilms (Genevaux et al., 1999). This phenotype is related to the defective pili and flagellar biogenesis observed in a dsbA mutant (Dailey and Berg, 1993; Jacob-Dubuisson et al., 1994; Genevaux et al., 1999). As with motility, only the CA8000 and MG1655 parents are capable of biofilm formation. We tested both of these backgrounds for a potential effect of the ompL mutations on this dsbA-associated phenotype. As shown in Figure 4, the introduction of an ompL null mutation does not influence to any detectable extent the failure of dsbA mutants to form biofilms.
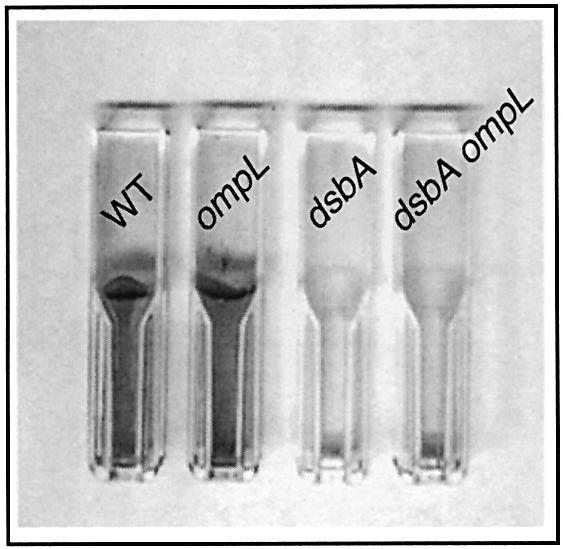
Fig. 4. The altered biofilm formation of a dsbA null mutant is not suppressed by an ompL null mutation. Biofilm formation was monitored after 12 h of incubation in LB medium at 37°C using polystyrene as the attachment surface. A representative set of experiments obtained with the wild-type strain MG1655 (WT), the dsbA and ompL single mutants and the dsbA ompL double mutant is presented. The ompL allele shown here is the ompL::mini-Tn10KanR allele (Dartigalongue et al., 2000).
Phenotypes of ompL single mutants
Dartigalongue et al. (2000) enumerated four phenotypes associated with all three of their independently isolated ompL null insertion mutations. The first is a partial sensitivity to the reducing agent DTT, which was originally employed as a screen to isolate one of their original ompL mutants. As stated above, we did not detect any difference between the ompL single mutants and the isogenic wild-type parents in this respect (Figure 1; data not shown). The second phenotype is slow bacterial growth, with an ompL single null mutant having a generation time of 70 min compared with 45 min for the wild-type parents at 37°C (the type of medium used was not stated). In contrast, we did not observe any significant difference in the growth rates between the wild-type parents and the ompL null mutants in either LB or minimal media (Figure 1; data not shown). The third phenotype associated with ompL null mutations is a block on the growth of the male-specific bacteriophage M13. Again, contrary to previous results (Dartigalongue et al., 2000), we did not observe any M13 resistance in the ompL null mutants in either the JM101 or CA8000 male backgrounds (Figure 5; data not shown). The failure to block bacteriophage M13 is particularly incomprehensible, given that complementation of this particular phenotype was used to originally clone and subclone the wild-type ompL gene (Dartigalongue et al., 2000). In addition, Figure 5 shows that the introduction of an ompL null mutation into a dsbA mutant does not suppress M13 resistance to any extent. Lastly, as stated above, we did not observe any reduction in the motility of single ompL mutants (Figure 2; data not shown), contrary to the reported results of Dartigalongue et al. (2000).
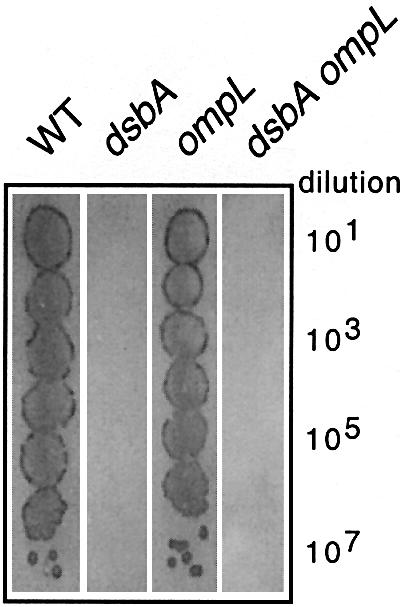
Fig. 5. The ompL null mutants are not resistant to bacteriophage M13 infection. Lawns of isogenic derivatives of JM101 were prepared on L-agar plates in the presence of IPTG and X-gal. Ten-fold serial dilutions of bacteriophage M13mp7 were spotted on the lawns followed by incubation of the plates at 37°C for ∼18 h. A representative set of results obtained with the parent JM101 (WT), the dsbA and ompL single mutants and the dsbA ompL double mutant is shown. The ompL allele shown here is the ompL::mini-Tn10KanR allele (Dartigalongue et al., 2000).
To explain their findings, Dartigalongue et al. (2000) suggested that a small, as yet unidentified, molecule(s), capable of substituting for DsbA, is oxidized by DsbB and that the oxidized and/or reduced forms of this hypothetical molecule are normally effluxed into the medium by the OmpL porin. Hence, in the absence of the OmpL porin, the molecule accumulates in the periplasmic space, thus completely complementing the lack of DsbA in a dsbA null mutant.
We have no experimental reason to doubt that the OmpL protein is a bona fide porin (established in the laboratory of Dr Hiroshi Nikaido; Dartigalongue et al., 2000). In addition, some of the features of the model proposed by Dartigalongue et al. (2000) may still turn out to be correct. However, taking all our genetic and biochemical results together, we conclude that null mutations in the ompL gene result in none of the remarkable phenotypes previously assigned to them, and that they play no demonstrable role in the suppression of the pleiotropic phenotypes associated with a dsbA null mutation.
The disparity between our results and those of Dartigalongue et al. (2000) may be due to several factors. Potential pitfalls associated with this type of genetic experiment may include closely linked uncharacterized suppressor mutations in some of the strains used, mix-ups or undetected contamination of strains. However, given the many discrepancies in the execution and reporting of the genetic experiments of Dartigalongue et al. (2000), including a serious lack of concordance between statements made in the Materials and methods section and the Results section, we are unable to offer a single logical scientific explanation for the failure of these authors to correctly assign any of the OmpL phenotypes to their various bacterial constructs.
Materials and methods
Bacterial and bacteriophage strains
We used the standard E.coli K-12 strains CA8000, MG1655 and MC4100 for this study. The MC4100 strain (Casadaban, 1976) is female and non-motile. The MG1655 strain, used in the E.coli genome sequencing project (Guyer et al., 1980; Blattner et al., 1997), is motile and female. The CA8000 strain (Hayes, 1953) is motile and male, and thus is the only one of the three capable of propagating bacteriophage M13 and its derivatives. Strain JM101 (Messing, 1979) (a kind gift from Dr Dominique Belin, University of Geneva) is male and was used primarily because it carries the chromosomally encoded lacZΔM15 mutation that is complemented by the LacZ α fragment encoded by bacteriophage M13mp7. The in vivo reconstituted LacZ activity enables the easy visualization of M13 plaques in the presence of IPTG (0.1 mM final) and X-gal (0.003% final). The strains SR2262 (MC4100 dsbA:: mini-Tn10TetR) and SR1791 (CA8000 ompL::mini-Tn10KanR) were obtained from Dr Satish Raina (University of Geneva) (Dartigalongue et al., 2000). Construction of the deletion/substitution allele of ompL was performed in strain DY378 (Yu et al., 2000) and is described below. Bacteriophage P1L4 (originally from Dr Lucien Caro, University of Geneva) was used for transduction experiments. Bacteriophage M13mp7 (Messing et al., 1981) (the kind gift of Dr William Kelley, University of Geneva) was used to test strains for M13 resistance.
Media and chemicals
LB broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl pH 7) and LB agar (LB broth containing 1% agar) were supplemented when necessary with either kanamycin (50 µg/ml) or tetracycline (15 µg/ml). Other chemicals and media are described under the appropriate methods.
Construction of an ompL deletion/substitution mutation
We took advantage of the methodology described by Yu et al. (2000) to create a complete deletion of the ompL gene. The 693 nucleotide long ompL (also named yshA) open reading frame was substituted by the kanamycin-resistance (KanR) cassette from plasmid pKD4 (Datsenko and Wanner, 2000) to score its inheritance easily. To do this, we generated the following two primers: primer LKOR, 5′-ctggtgggtggcggcctccctacgtt taaaaaatggacttattcatatgaatatcctccttag, whose 5′ end carries homology to the nucleotide region immediately upstream of the ompL start codon (in bold type) and whose 3′ end carries the appropriate homology to one end of the KanR cassette of the plasmid pKD4; and primer LKOF, 5′-gtaggccggataaggcgtttacgccgcatccggcagtttcacagtgtaggctgga gctgcttcg, whose 5′ end carries homology to the nucleotide region immediately downstream of the ompL stop codon (in bold type) and whose 3′ end carries the appropriate homology to the other end of the KanR cassette of the plasmid pKD4.
PCR amplification of pKD4 with these two primers resulted in a DNA fragment encoding the kanamycin-resistance cassette flanked by sequences homologous to the regions immediately upstream and downstream of ompL. The purified DNA fragment was crossed into the chromosome of DY378 using the protocol of Yu et al. (2000). The ompL deletion/substitution on the chromosome was verified by PCR amplification of the ompL region using external primers located 88 nucleotides upstream (primer LCR: 5′-cgggcttccgtaacgttataaatg) and 112 nucleotides downstream (primer LCF: 5′-gatgctggcggctagagctta) of the ompL gene. The PCR products were subsequently sequence-verified using the same primers. The deletion was further confirmed by Southern blot analysis using full-length ompL as a DNA probe (Sambrook et al., 1989). This ΔompL::KanR strain was used as a donor in subsequent bacteriophage P1 transduction experiments.
P1 transduction experiments
The various mutant alleles used in this study were moved from strain to strain by employing bacteriophage P1-mediated transduction, essentially as described by Miller (1992), and selecting at 37°C for the appropriate drug resistance on LB agar plates supplemented with 5 × 10–3 M sodium citrate.
AP activity measurements
Assays were performed with mid-log phase bacterial cell cultures grown at 37°C in either LB broth (repressing conditions for the phoA operon) or minimal 121 salts (Miller, 1992) supplemented with 0.2% glucose and limiting for PO4 (de-repressing conditions for the phoA operon) as described previously (Brickman and Beckwith, 1975). AP activity was measured as described previously (Brickman and Beckwith, 1975; Michaelis et al., 1983).
DTT sensitivity experiments
A freshly prepared 1 M stock solution of DTT (Eurobio) in H2O was diluted appropriately into molten LB agar to give the desired final concentrations. The solidified DTT agar plates were used within 1 h of solidification to minimize DTT oxidation by air. Exponentially growing cultures were serially diluted 10-fold, and 5 µl of each dilution was deposited on the plates. The dilutions were allowed to dry and the plates were then incubated at 37°C for ∼18 h.
Benzylpenicillin sensitivity experiments
A 30 mg/ml stock solution of benzylpenicillin (potassium salt; Sigma PEN-K, 1596 units penicillin-G base per mg) in H2O was diluted appropriately into molten LB agar to give the desired final concentration. The same dilutions of exponentially growing cultures used for the DTT sensitivity experiments were tested on the benzylpenicillin plates at the same time. The plates were incubated at 37°C for ∼18 h.
Growth rate, motility and biofilm measurements
Growth rates were assayed in LB broth and in 121 salts supplemented with 0.2% glucose (Miller, 1992) at 37°C under constant shaking. At regular time intervals, the optical density (OD) of the cultures was measured at a wavelength of 600 nm. Motility assays were performed essentially as described by Macnab (1986). Two microliters of each culture grown to an OD600 nm of ∼1.0 were applied to freshly prepared soft agar plates (1% tryptone, 0.5% NaCl, 0.3% agar). Plates were incubated at 37°C for 8 h. Under these conditions, dsbA null mutants exhibit no significant motility as judged by the diameter of the bacterial growth zone. Biofilm experiments were carried out for 12 h at 37°C in LB broth using polystyrene surfaces as described previously (Genevaux et al., 1999).
Immunoblot analysis
Whole-cell extracts were prepared as described previously (Kelley and Georgopoulos, 1997), and proteins were separated in 12% (w/v) polyacrylamide–SDS gels, transferred to nitrocellulose membrane (Schleicher and Schuell) and probed with rabbit monoclonal anti-DsbA antibodies (the kind gift of Dr Joen Luirink, Department of Molecular Microbiology, Free University of Amsterdam). Goat anti-rabbit horseradish peroxidase conjugate (Santa Cruz Biotechnology Inc.) was used as the secondary antibody. Blots were developed by enhanced chemiluminescence according to the manufacturer (Amersham Pharmacia Biotech).
Acknowledgments
Acknowledgements
Anti-DsbA antibodies, bacteriophage M13mp7 and strain JM101 were the generous gifts of Drs Joen Luirink, William Kelley and Dominique Belin, respectively. We thank Dr Colette Rossier for DNA sequencing services. One of the authors (A.A.S.) initiated this work as a postdoctoral fellow in the laboratory of Dr Satish Raina (supported by FN-59131) in an attempt to identify the nature of the putative small oxidizing–reducing agent in the periplasmic space of E.coli. The other authors were supported by FN-65403 and the Canton of Geneva.
References
- Bader M., Muse,W., Ballou,D.P., Gassner,C. and Bardwell,J.C. (1999) Oxidative protein folding is driven by the electron transport system. Cell, 98, 217–227. [DOI] [PubMed] [Google Scholar]
- Bardwell J.C.A., McGovern,K. and Beckwith,J. (1991) Identification of a protein required for disulfide bond formation in vivo. Cell, 67, 581–589. [DOI] [PubMed] [Google Scholar]
- Bardwell J.C.A., Lee,J.-O., Jander,G., Martin,N., Belin,D. and Beckwith,J. (1993) A pathway for disulphide bond formation in vivo. Proc. Natl Acad. Sci. USA, 90, 1038–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner F.R. et al. (1997) The complete genome sequence of Escherichia coli K-12. Science, 277, 1453–1462. [DOI] [PubMed] [Google Scholar]
- Brickman E. and Beckwith,J. (1975) Analysis of the regulation of Escherichia coli alkaline phosphatase synthesis using deletions and phi80 transducing phages. J. Mol. Biol., 96, 307–316. [DOI] [PubMed] [Google Scholar]
- Casadaban M.J. (1976) Transposition and fusion of the lac genes to selected promoters in Escherichia coli using phage λ and Mu. J. Mol. Biol., 104, 541–555. [DOI] [PubMed] [Google Scholar]
- Collet J.-F. and Bardwell,J.C.A. (2002) Oxidative protein folding in bacteria. Mol. Microbiol., 44, 1–8. [DOI] [PubMed] [Google Scholar]
- Dailey F.E. and Berg,H.C. (1993) Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. Proc. Natl Acad. Sci. USA, 90, 1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartigalongue C., Nikaido,H. and Raina,S. (2000) Protein folding in the periplasm in the absence of primary oxidant DsbA: modulation of redox potential in periplasmic space via OmpL porin. EMBO J., 19, 5980–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K.A. and Wanner,B.L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA, 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genevaux P., Bauda,P., DuBow,M.S. and Oudega,B. (1999) Identification of Tn10 insertions in the dsbA gene affecting Escherichia coli biofilm formation. FEMS Microbiol. Lett., 173, 403–409. [DOI] [PubMed] [Google Scholar]
- Guyer M.S., Reed,R.R., Steitz,J.A. and Low,K.B. (1980) Identification of a sex-factor-affinity site in Escherichia coli as γδ. Cold Spring Harb. Symp. Quant. Biol., 45, 135–140. [DOI] [PubMed] [Google Scholar]
- Hayes W. (1953) The mechanism of genetic recombination in Escherichia coli. Cold Spring Harb. Symp. Quant. Biol., 18, 75–93. [DOI] [PubMed] [Google Scholar]
- Jacob-Dubuisson F., Pinkner,J., Xu,Z., Striker,R., Padmanhaban,A. and Hultgren,S.J. (1994) PapD chaperone function in pilus biogenesis depends on oxidant and chaperone-like activities of DsbA. Proc. Natl Acad. Sci. USA, 91, 11552–11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani S., Akiyama,Y. and Ito,K. (1992) Identification and characterization of an Escherichia coli gene required for the formation of correctly folded alkaline phosphatase, a periplasmic enzyme. EMBO J., 11, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley W.L. and Georgopoulos,C. (1997) The T/t common exon of simian virus 40, JC and BK polyomavirus T antigens can functionally replace the J-domain of the Escherichia coli DnaJ molecular chaperone. Proc. Natl Acad. Sci. USA, 94, 3679–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Kishigami,S., Sone,M., Inokuchi,H., Mogi,T. and Ito,K. (1997) Respiratory chain is required to maintain oxidized states of the DsbA–DsbB disulphide bond formation system in aerobically growing Escherichia coli cells. Proc. Natl Acad. Sci. USA, 94, 11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macnab R.M. (1986) Proton-driven bacterial flagellar motor. Methods Enzymol., 125, 563–581. [DOI] [PubMed] [Google Scholar]
- Messing J. (1979) A multipurpose cloning system based on the single-stranded DNA bacteriophage M13. Recomb. DNA Tech. Bull., 2, 43–48. [Google Scholar]
- Messing J., Crea.R. and Seeburg,P.H. (1981) A system for shotgun DNA sequencing. Nucleic Acids Res., 9, 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis S., Inouye,H., Oliver,D. and Beckwith,J. (1983) Mutations that alter the signal sequence of alkaline phosphatase in Escherichia coli. J. Bacteriol., 154, 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. (1992) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Missiakas D., Georgopoulos,C. and Raina,S. (1993) Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc. Natl Acad. Sci. USA, 90, 7084–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiakas D., Schwager,F. and Raina,S. (1995) Identification and characterization of a new disulfide-isomerase like protein (DsbD) in Escherichia coli. EMBO J., 14, 3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina S. and Missiakas,D. (1997) Making and breaking of disulfide bonds. Annu. Rev. Microbiol., 51, 179–202. [DOI] [PubMed] [Google Scholar]
- Ritz D. and Beckwith,J. (2001) Roles of thiol-redox pathways in bacteria. Annu. Rev. Microbiol., 55, 21–48. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Yu D., Ellis,H.M., Lee,E.C., Jenkins,N.A., Copeland,N.G. and Court,D.L. (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl Acad. Sci. USA, 97, 5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]