

Abstract
The TGF-β superfamily of ligands and receptors stimulate cellular events in diverse processes ranging from cell fate specification in development to immune suppression. Activins define a major subgroup of TGF-β ligands that regulate cellular differentiation, proliferation, activation and apoptosis. Activins signal through complexes formed with type I and type II serine/threonine kinase receptors. We have solved the crystal structure of activin A bound to the extracellular domain of a type II receptor, ActRIIB, revealing the details of this interaction. ActRIIB binds to the outer edges of the activin finger regions, with the two receptors juxtaposed in close proximity, in a mode that differs from TGF-β3 binding to type II receptors. The dimeric activin A structure differs from other known TGF-β ligand structures, adopting a compact folded-back conformation. The crystal structure of the complex is consistent with recruitment of two type I receptors into a close packed arrangement at the cell surface and suggests that diversity in the conformational arrangements of TGF-β ligand dimers could influence cellular signaling processes.
Keywords: activin/ActRIIB/TGF-β/receptors/signaling/structure
Introduction
The transforming growth factor β (TGF-β) cytokines are a large superfamily with more than 30 mammalian family members divided into subgroups that include the TGF-βs, the bone morphogenetic proteins (BMPs), the activins and other more distant relatives (Massagué, 2000; Massagué and Chen, 2000; Miyazono et al., 2001). TGF-β cytokines can activate cell differentiation, apoptosis, hematopoiesis, angiogenesis, steroid synthesis, adhesion, migration and extracellular matrix production (Woodruff, 1998; Massagué and Wotton, 2000; Miyazono et al., 2001; Dennler et al., 2002). The specific response depends upon the types and levels of the TGF-β ligand and receptors as well as the cellular state and environment. TGF-β ligands act as potent inhibitors of cellular proliferation and disruption of the signaling pathway is associated with cellular transformation and oncogenesis (Massagué et al., 2000; Pasche, 2001).
TGF-β ligands interact with type I and type II signaling receptors (Massagué and Weis-Garcia, 1996; Massagué and Chen, 2000; Miyazono et al., 2001; Dennler et al., 2002). Both receptors contain a short extracellular domain that binds ligand, and a larger intracellular serine/threonine kinase domain. Type I and type II receptor kinase domains phosphorylate distinct cellular substrates. While type II receptor kinases are constitutively active, type I receptors are activated by type II receptor phosphorylation of a membrane-proximal GS-domain (Massagué et al., 2000). Ligands generally bind type II receptors with higher affinity followed by recruitment of lower affinity type I receptors into a complex, although certain BMP ligands can bind directly to type I receptors without requiring prior type II receptor interactions (Kirsch et al., 2000a; Miyazono et al., 2001). Activated type I receptor kinases phosphorylate receptor-regulated SMAD proteins (R-SMADS), which associate with a cytoplasmic common-mediator SMAD (Co-SMAD), forming a complex that translocates to the nucleus to mediate transcription (Massagué and Wotton, 2000).
In contrast to the larger number of TGF-β ligands, fewer receptors and SMAD proteins are involved in the signaling pathway (Miyazono et al., 2001). In mammals, only five type II receptors and seven type I receptors have been identified. Five R-SMADS, one Co-SMAD and two inhibitory SMAD proteins (I-SMADS) form the core of the signal transduction pathway. The SMAD1, SMAD5 and SMAD8 proteins are activated by BMP receptors, while the SMAD2 and SMAD3 proteins are activated by TGF-β and activin receptors. The diversity of the signaling pathway at the level of receptors and R-SMADs is limited in comparison with the diversity of physiological roles that the TGF-β superfamily exhibits in vivo. SMAD proteins are regulated by additional intracellular mechanisms that also provide links to other signaling pathways, contributing to the diversity of responses induced by TGF-β ligands (Massagué and Chen, 2000).
Both BMPs and activins act as classical morphogens (Gurdon et al., 1999; Miyazono et al., 2001; Bourillot et al., 2002), with cellular responses dependent upon the levels of ligand:receptor complexes. Activin concentration differences of as little as 3-fold are quantitatively correlated with steady-state levels of nuclear SMAD complexes that elicit distinct gene expression profiles in Xenopus blastula cells (Gurdon et al., 1999; Ryan et al., 2000; Bourillot et al., 2002). Access of ligand to receptors is blocked by extracellular ligand binding proteins and receptor inhibitors, and can also be enhanced by the presentation of ligand by type III receptors. The pleiotropic effects of TGF-β ligands on cellular activation likely relies on details of receptor:ligand complex formation related to the persistence and strength of signaling and the quantitative localization of SMAD complexes in the nucleus. Factors that may influence this include the kinetics and stabilities of receptor:ligand assemblies and the conformational arrangements of related receptor: ligand complexes.
A number of TGF-β ligand structures have been determined, revealing a common cysteine knot protein fold. TGF-β ligands are typically found as homodimers, although heterodimers can also form. The structures of TGF-β1 (Hinck et al., 1996), TGF-β2 (Daopin et al., 1992; Schlunegger and Grutter, 1992), BMP-2 (Scheufler et al., 1999; Kirsch et al., 2000b) and BMP-7 (Griffith et al., 1996) all show a common elongated dimer structure. The structures of TGF-β3 (Mittl et al., 1996) and GDNF (Eigenbrot and Gerber, 1997) revealed that certain family members deviate from the canonical dimer conformation. The structures of a type I receptor complex, BMP-2 bound to BRIA (Kirsch et al., 2000b), and a type II receptor complex, TGF-β3 bound to TβRII (Hart et al., 2002), have also been determined. We report here the structure of activin A bound to the type II receptor ActRIIB determined to 3.05 Å resolution. In two crystal forms, the activin A dimer adopts a compact conformation that is distinct from previously determined superfamily members. ActRIIB binding is observed on the outside of the activin A finger regions interacting with a surface site that differs from that occupied by TβRII bound to TGF-β3. The structure suggests that different binding modes and conformational arrangements of TGF-β ligand complexes could influence cellular signaling and responses.
Results
Overall description of the complex in P41 and P41212 crystal forms
The structure of the ActRIIB:activin A complex is shown in Figure 1A. Two receptors bind on the convex outer surface of the anti-parallel β sheets that form the finger or knuckle regions of the TGF-β fold. In both the P41 and P41212 crystal forms activin A adopts a highly bent conformation that differs from other known TGF-β ligand structures. Superposition of the two crystal form structures yields an overall root mean square (r.m.s.) deviation for Cα atoms of 0.69 Å2 (Figure 1B). In both forms, one of the activin A monomers and the associated ActRIIB is more ordered than the second, with the most pronounced difference in atomic temperature factors observed for the P41212 crystal form. Electron density for ActRIIB is observed for residues 25–117, but not for residues 118–120 and a C-terminal thrombin cleavage site. For activin A, residues 49–76 that form the canonical TGF-β ‘wrist’ region are disordered in both crystal forms, similar to the TGF-β3:TβRII complex. However, the ActRIIB: activin A interface is well-defined in electron density maps (Figure 1C and D).

Fig. 1. Structure of the ActRIIB:activin A complex. (A) Ribbon diagram of the complex. The activin A dimer is shown in light and dark blue for each monomer and the fingers are labeled F1–F4; ActRIIB models are represented in yellow and red CPK atoms with the hydrophobic cluster (Y60, W78, F101) are shown in grey. (B) Superposition of the P41 (black) and P41212 (red and blue) crystal forms. (C) Electron density showing the edge of the hydrophobic interface and the intramolecular salt bridge between K102 and D104 (activin A shown in red; ActRIIB, yellow). (D) Expanded stereo view of the electron density of the complex interface. Electron density is from a simulated annealing composite omit map contoured at 1.5 and 0.9 σ, respectively.
The ActRIIB:activin A complex contains a 2-fold symmetry axis between two nearly anti-parallel activin A monomers (Figure 1A). The two receptors are brought into closer proximity than observed in the TGF-β3:TβRII complex (Hart et al., 2002) or in complexes that would form with the canonical TGF-β dimer conformation. The two ActRIIB receptors are located on two sides of the activin A dimer, separated by a center-of-mass distance of 31 Å. This arrangement is consistent with the membrane anchoring of both receptors to the cell surface through a flexible linker to the transmembrane region.
Comparison of activin A with other TGF-β ligands
Activin A monomers in the complex maintain the canonical TGF-β fold with minor variations in the loops between β-strands forming the finger region of the fold, excluding the disordered wrist region. Superposition of the activin A monomers onto TGF-β2 (Daopin et al., 1992; Schlunegger and Grutter, 1992; Hart et al., 2002), TGF-β3 (Mittl et al., 1996), BMP-2 (Scheufler et al., 1999; Kirsch et al., 2000b) and BMP-7 (Griffith et al., 1996) monomers yields r.m.s. deviations in Cα positions ranging from 1.25 Å2 for BMP-2 to 1.70 Å2 for TGF-β3 (Figure 2A).

Fig. 2. Observed activin A monomer and dimer conformations. (A) Superposition of the activin A monomer with other TGF-β ligands. TGF-β2 (green), TGF-β3 (orange), BMP-2 (dark blue), BMP-7 (purple), TGF-β3 in complex with TβRII (crimson), BMP-2 in complex with BRIA (blue) and activin A (black) are shown. (B) Comparison of activin A (top), BMP-7 (middle) and TGF-β3 (bottom) dimers. Monomers are colored red and blue in each of the structures and aligned by a single monomer (blue, left). A schematic of the dimer configuration is shown to the right. (C) Superposition of the activin A (blue), BMP-7 (purple) and TGF-β3 (green) dimers using one of the monomers. The difference in monomer arrangement in activin A corresponds to a rotation of 75° from the BMP-7 configuration about the red axis and, for TGF-β3, a rotation of 110° around the yellow axis. (D) Activin A residues buried at the dimer interface. The ribbon of one chain is colored blue, the other red. Residues interact symmetrically across the dimer interface and come from two regions of the activin A fold (residues Cys80, Val82, Pro83, Leu86, Gln106 and Asn107).
The conformational arrangement of the activin A dimer differs from other observed TGF-β ligand conformations (Figure 2B). Most TGF-β dimers show an extended, symmetric arrangement like BMP-7 (Griffith et al., 1996), including TGF-β1 (Hinck et al., 1996), TGF-β2 (Daopin et al., 1992; Schlunegger and Grutter, 1992) and BMP-2 (Scheufler et al., 1999; Kirsch et al., 2000b). Another dimer arrangement was observed for TGF-β3 in complex with TβRII (Figure 2B), and NMR studies of TGF-β3 in solution are consistent with a flexible dimer interface (Bocharov et al., 2002). Superposition of activin A, TGF-β3 and BMP-7 aligned on one monomer highlights the large conformational differences in the second monomer position (Figure 2C), which can be described as domain movements relative to the BMP-7 structure. The structural change from the extended BMP-7 dimer to the compact activin A dimer involves a rotation of 75° of one monomer about a rotation axis nearly perpendicular to the long axis of the BMP-7 dimer. In contrast, structural changes in TGF-β3 involve a rotation of 110° about an axis along the length of the BMP-7 dimer. The rotations for activin A and TGF-β3 occur in opposite directions creating a ‘closed’ form for activin A and an ‘open’ form for TGF-β3.
The activin A dimer is linked by an interchain disulfide bond (Cys80) adjacent to the disordered wrist region. The finger regions of the activin A monomers bury 342 Å2 of surface area in their contact interface excluding the interchain disulfide. The dimer interface is primarily hydrophobic and involves two short segments, including residues Cys80, Val82, Pro83, Leu86, Gln106 and Asn107 symmetrically arranged between the monomers (Figure 2D). These residues are partially conserved across the TGF-β superfamily (Innis et al., 2000). Cys80 and Pro83 are completely conserved in activins B and C, while Val82 is replaced by Ile in activin B, Leu86 is replaced by Arg in activin C, Gln106 is replaced by Pro in both activins B and C, and Asn107 is replaced by Asp in activin C.
Residue differences between activin A and other family members at the canonical TGF-β dimer interface might also affect activin A conformational flexibility. Although most of these residues are disordered in the ActRIIB: activin A crystals, potential dimer contact residues can be identified from other structures. In BMP-7 and TGF-β2, there is good correspondence between the residues involved in the dimerization interfaces (Griffith et al., 1996). Out of 22 residues buried in the canonical dimer interface, 17 are identical or conservatively substituted in activin A. Only five residues are significantly different in size and/or polarity. Activin A Phe16 replaces Leu in both BMP-7 and TGF-β2, Phe58 replaces Asp and Gln, Gln65 replaces Leu in both, Asn76 replaces Val and Ser, and Leu77 replaces Pro and Ala. Leu86 is involved in both the canonical dimer interface and the activin A dimer interface. These residue changes may couple with other structural differences in activin A, such as positioning, stability or length of the α1 helix, enabling greater flexibility in the wrist region.
The activin A conformation may be affected by the crystal lattices. In both lattices, ActRIIB forms back-to-back interactions with symmetry-related receptors, burying ∼1400 Å2 of total surface area. Contacts between receptors across the activin A dimer axis may also stabilize the activin A conformation. The activin A dimer may be flexible in solution and adapt to these packing arrangements.
Comparison of ActRIIB with other type I/II receptors
ActRIIB exhibits a three-fingered toxin fold similar to ActRII (Greenwald et al., 1999). The r.m.s.deviations in Cα positions between ActRIIB and related receptors are 0.55 Å2 for ActRII, 2.29 Å2 for TβRII and 1.24 Å2 for BRIA. The location of the ligand-binding surface on ActRIIB differs significantly from that on TβRII (Hart et al., 2002) and correlates with large structural differences in the two proteins (Figure 3A). TGF-β3 binding by TβRII is almost exclusively through the first finger of the fold, burying only 515 Å2 (Figure 3A and B). In contrast, activin A binding by ActRIIB involves the concave surface at the base of fingers 2 and 3, with many contacts in the loop between fingers 2 and 3 (L23 in Figure 3A and B). The length and position of L23 differs in the two receptors (Figure 3A). In ActRIIB the L23 loop is short, forms part of the complex interface and allows access to hydrophobic residues important for activin binding. In TβRII, L23 is longer by six amino acids and extends 12 Å up towards the top of the fingers covering the surface analogous to the hydrophobic cluster of ActRIIB. Another major difference in the receptors is in the size and orientation of finger 1 (Figure 3B). Finger 1 proceeds in opposite directions at position G45 in ActRIIB (S49 in TβRII). In TβRII the finger wraps around the backside of the receptor, while in ActRIIB the finger points away from the core into the space occupied by TGF-β3 when bound to TΒRII. The large structural changes in finger 1 and the L23 loop, in addition to interface-specific sequence changes, contribute to the two modes of type II receptor binding.
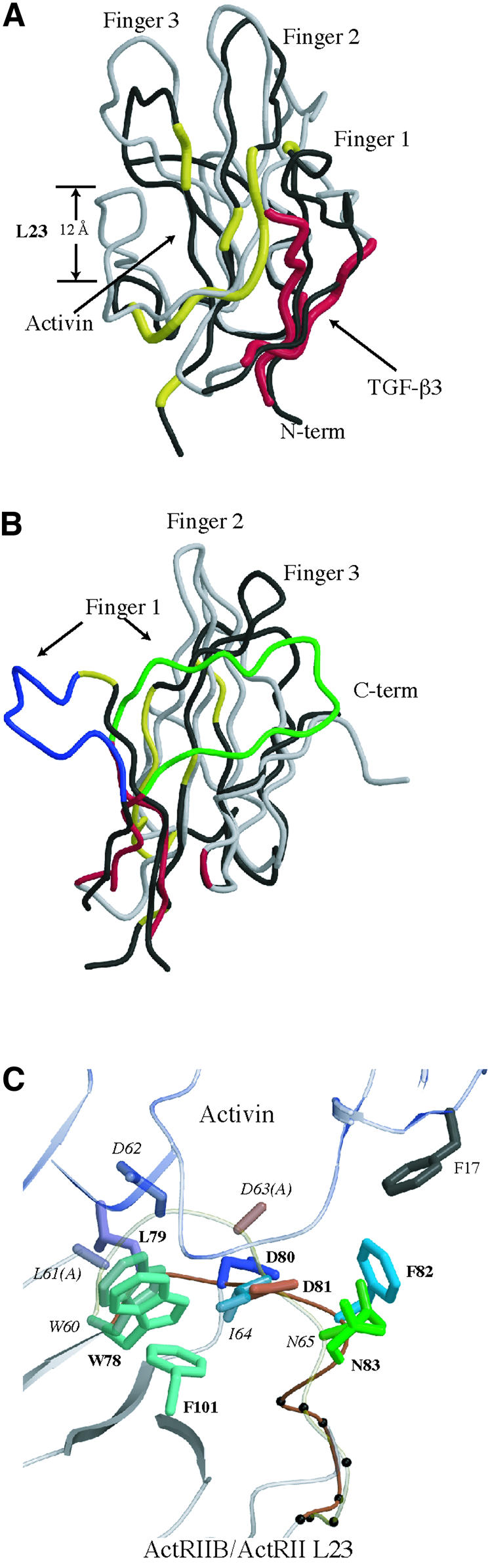
Fig. 3. Comparison of the ActRIIB structure with related TGF-β receptors. (A) Superposition of ActRIIB (black) and TβRII (grey) in complex with ligands. The activin A and TGF-β3 interaction surfaces are colored yellow and red, for the respective receptors. The loop joining fingers 2 and 3 of the three-finger toxin fold is labeled L23. (B) Finger 1 conformational differences in ActRIIB and TβRII. Finger 1 is colored blue for ActRIIB and green for TβRII. The remaining color code is identical to (A). (C) Superposition of ActRIIB and ActRII showing differences in the L23 loop. Residues are colored according to sequence alignment, with ActRIIB labels in bold and ActRII labels in italics. The L23 loop for ActRIIB is colored orange and ActRII is colored light yellow. Sequence numbers for ActRIIB are for the full-length protein, while those for ActRII are as in Greenwald et al. (1999). For example F82 in ActRIIB corresponds to Ile64 in ActRII.
Comparison of ActRIIB in the complex with the ligand-free structure of ActRII (Greenwald et al., 1999) highlights structural changes that may be due to either ligand binding or receptor sequence differences. ActRII and ActRIIB bind activin A with similar affinity (Kd <10 nM) and share ∼60% sequence identity, but exhibit different affinities for inhibin (Chapman et al., 2002). ActRIIB binding to activin A involves 14 residues and all but three are identical in ActRII (Tyr60, Phe82 and Glu39 are substituted to Phe, Ile and Asp, respectively). In ActRIIB, the L23 loop (residues 78–87) deviates significantly from that in ActRII and contributes over half the residues found at the activin A interface (Figure 3C). While the overall r.m.s. deviation for ActRII and ActRIIB Cα positions is 0.55 Å2, Cα positions in the loop deviate by an average of 3.4 Å2. In ActRII the loop protrudes out an additional 3 Å creating a potential steric block to activin A binding. The altered loop conformation may be affected by substitution of Ile64 in ActRII for Phe82 in ActRIIB. In ActRII Ile64 packs against a cluster of hydrophobic residues, while Phe82 in ActRIIB is solvent exposed and interacts with activin A. Activin A binding may shift Phe82 from intramolecular hydrophobic interactions, inducing a conformational change of the loop. Alternatively, this loop may represent a major static difference between ActRII and ActRIIB.
Overview of the ActRIIB:activin A binding interface
The activin A fingers form concave and convex surfaces implicated in binding type I and type II receptors, respectively. ActRIIB also exhibits a concave surface, generated by the three-finger toxin fold. The ActRIIB: activin A structure reveals that the convex surface at the second set of anti-parallel β-strands of the activin A fingers (the knuckles) bind into the concave groove of ActRIIB (Figure 4A). In activin A, the end of the fingers and loop joining fingers 1 and 2 form the interface. Complex formation buries a total of 1324 Å2 of solvent accessible surface area on the two proteins.

Fig. 4. The ActRIIB:activin A interface. (A) Surfaces of interaction between activin A and ActRIIB. Molecular surfaces showing contact residues at the complex interface for the activin A monomer (right) and ActRIIB (left). Positively and negatively charged residues are colored blue and red, respectively, while polar and hydrophobic residues are colored purple and green, respectively. (B) Hydrophobic interactions at the ActRIIB:activin A interface. (C) Hydrophilic interactions at the ActRIIB:activin A interface.
The ActRIIB:activin A interface involves hydrophobic, charged and polar residues in both ActRIIB and activin A (Figure 4A). In ActRIIB, hydrophobic amino acids on the concave surface generate the core of the interface that interacts with hydrophobic residues in the activin A knuckle region. Additional electrostatic interactions are formed at the edges of the interface. For clarity, the activin A and ActRIIB interface residues are subscripted with A and II, respectively.
The hydrophobic interface
Figure 4A and B and the schematic in Figure 5A show the hydrophobic interactions formed in the complex. On ActRIIB, a cluster of three aromatic amino acids, TyrII60, TrpII78 and PheII101 lie at the center of the interface. Together they contribute 20–25% of the total buried surface area on the receptor. In addition, residues ValII73, LeuII79 and PheII82 from L23, along with ValII99 of finger 3 contribute significant hydrophobic and van der Waals contacts.

Fig. 5. Binding schematic of the complex interface. Total buried surface area per residue is shown under the residue ID. Sequence comparison with the BMP and TGF-β subfamilies are shown to the right and BMP and TGF-β type II receptors to the left. (A) Hydrophobic contacts within 4.5 Å and (B) potential charge–charge and hydrogen bonding interactions. A negative sign indicates there is no comparable residue in the sequence alignment.
The complementary hydrophobic surface of activin A derives from four segments. A total of eight hydrophobic residues make receptor contacts, including PheA17 on finger 1, IleA30, AlaA31 and ProA32 on the loop between fingers 1 and 2, ProA88, LeuA92 and TyrA94 on finger 3 and IleA100 on finger 4. At the center of the hydrophobic interface, AlaA31, ProA32 and LeuA92 interact with TyrII60, TrpII78 and PheII101 (Figure 4B). Towards the tips of the activin A fingers, IleA30 and LeuA92 pack at the edge of the hydrophobic cluster with PheII101 on one side and ValII99 on the other. On the finger-tip edge of the interface IleA100 and TyrA94 interact with ValII73 and ValII99. Additional minor hydrophobic contacts are formed at the opposite end of the interface, towards the activin A wrist motif, where ProA88 packs with LeuII79, and PheA17 and HisA36 pack with PheII82. IleA100 and LeuA92 contribute >20% of the total buried surface area on activin A (Figure 5A).
The ionic/polar interface
Both inter- and intra-molecular ionic interactions are observed at the interface (Figures 4A, C and 5B). Potential inter-molecular electrostatic salt bridges and hydrogen bonds between ActRIIB and activin A surround the hydrophobic center of the interface on both sides (Figure 4A and C). LysII55 contacts GluA111, and AspII80 contacts ArgA87 near the edge of the interface towards the activin A wrist region. Intra-molecular interactions in both activin A and ActRIIB also contribute to the binding surfaces. In activin A, LysA102 interacts with AspA104 (Figures 4C and 5B) in addition to forming a potential hydrogen bond with the carbonyl oxygen of CysII77. The LysA102 interaction with AspA104 neutralizes the two charges and orients the aliphatic side chain atoms of LysA102 against the outside of the hydrophobic cluster at TyrII60. SerA90 may also stabilize the AspA104 conformation by interacting with its carboxylate group while forming a hydrogen bond across the interface to the amide hydrogen of LeuII79. In ActRIIB, LysII74 and GluII39 make intra-molecular contacts that position the aliphatic side chain of LysII74 between ValII73, TyrII60 and the aliphatic portion of LysA102.
The complex structure is consistent with mutagenesis experiments that identified residues in activin A and ActRII that are involved in binding. A cluster of three aromatic amino acids (TyrII60, TrpII78 and PheII101 in ActRIIB) were identified in the ActRII structure, and mutation of these residues to alanine decreases the affinity for activin A and inhibin (Greenwald et al., 1999; Gray et al., 2000;). Seven of 13 ActRII amino acids that were mutated correspond to ActRIIB residues in the complex interface, but only mutation of the three aromatic residues dramatically reduced binding and signal transduction. Other mutations at the interface (ActRII sequence numbers are in parentheses), including LysII55(37), LysII74(56), AspII80(62) and ValII99(81), had little or no effect, although for KII55(37) the IC50 value was higher than the others (0.7 versus 0.1–0.2 nM). The charged residues are more peripheral to the interface and may not contribute significantly to the binding of activin A or inhibin, while alanine substitution of other residues such as Val99 may be too conservative to perturb binding.
Charged activin A residues have also been mutated, based on a model of the ligand structure (Wuytens et al., 1999). Two residues, LysA102 and AspA27, were implicated in receptor binding. Substituting LysA102 with alanine or glutamate significantly reduced the affinity of receptor binding and signaling, whereas an arginine substitution had little effect. The structure suggests that the aliphatic portion of the LysA102 side chain is important for ligand binding and that the positive charge may stabilize the LysA102 conformation. Substitution of AspA27 to lysine increased the affinity while mutation to alanine had little effect. Although AspA27 does not interact directly with ActRIIB, a substitution to lysine may allow interactions with GlnII98, explaining the observed increase in affinity.
Specificity of interaction across the TGF-β superfamily
The ActRIIB:activin A and TGF-β3:TβRII crystal structures reveal two different binding modes for type II receptors, with TβRII shifted towards the tips of the TGF-β3 fingers (Figure 6A and B). For residues buried in the ActRIIB:activin A hydrophobic interface, amino acid substitutions are evident in TGF-β and activin/BMP sequences. The most striking differences occur at activin A positions IleA30 and AlaA31, which are His and Glu in TGF-β family members (Figure 5). ActRIIB residues TrpII78, PheII82 and ValII99 are substituted to His, Asp and Gly, respectively, in TβRII (Figure 5A). These major changes to the ActRIIB:activin A interaction surfaces may in part drive the two modes of receptor binding.

Fig. 6. Comparison of the ActRIIB:activin A structure with other type I and type II receptor complexes. (A) ActRIIB:activin A complex, with activin A in blue and ActRIIB in red. (B) TGF-β3 (blue) bound to TβRII (red). (C) BMP-2 (blue) bound to BRIA (red).
Most of the hydrophobic contacts are conserved in the BMP subfamily although subtle changes in the size of side chains could affect specificity. For example, on the receptor, LeuII79 and ValII99 are both isoleucine in BMP type II receptors. The largest differences can be found in the ionic interactions (Figure 5B), including residues LysA102 and AspA104. The Lys102–X–Asp104 motif is conserved in the activins but not in BMPs. Other differences in the inter-molecular ionic contacts include the interaction between AspII80 and ArgA87, which is replaced by Gly in the BMP type II receptors and Ser or Asn in the ligands. The electrostatic interaction of LysII55 with GluA111 would be altered by a deletion on the BMP receptor and a highly variable residue on the ligand. Specificity in the activin and BMP sub-families may reside in ionic/electrostatic interactions at the periphery of the interface.
Different isoforms of activin (βA, βB, βC and βE) also display different physiological effects (Lau et al., 2000; Mellor et al., 2000; Chapman et al., 2002; Vejda et al., 2002) and can assemble into a variety of heterodimers. The isoforms show distinct expression patterns, with some overlap in tissues such as the liver. Differences in isoform-dependent physiological effects may result from differences in type II receptor binding. For example, activin C does not stimulate the same cellular responses as activin A (Mellor et al., 2000). Knockout mice for activins C and E develop normally and show no reproductive axis defects, in contrast to activin A and B knockouts (Lau et al., 2000). Minor differences occur in the residues at the ActRIIB interface in the activin A and B isoforms. ArgA87 is replaced by Ser, which could disrupt the electrostatic interaction with AspII80 of ActRIIB. In addition, HisA36 is replaced by Tyr, which could enhance the binding affinity of the B isoform by interacting more favorably with PheII82. In contrast, activin C contains a significant substitution of Gln at AlaA31, which might alter the core hydrophobic interaction and affect receptor specificity and function.
The activin antagonist inhibin is a disulfide-linked heterodimer generated from the combination of an activin β subunit and a sequence divergent α subunit. Based on the arrangement of the cysteines in the sequence, inhibin is thought to adopt the cysteine knot fold, but binds to the activin type II receptors with a 10-fold lower Kd. Several critical activin A interface residues differ significantly in the α subunit. For example, AlaA31 is substituted with Tyr, and LeuA92 is substituted with Arg or Thr, depending upon the sequence alignment. In addition, the completely conserved activin motif Lys102–X–Asp104 is changed to Tyr–X–Thr in the α subunit. Differences in residues that generate the core of the interaction suggest that the α subunit would display significantly decreased affinity for ActRII or ActRIIB if it interacts at the same surface. Alternatively, the α subunit may display a different mode of binding or may not bind the type II receptor.
Implications for the formation of diverse signaling complexes
The compact conformation of the activin A dimer brings two type II receptors into closer proximity than observed in the TGF-β3:TβRII complex (Figure 6A and B). The distance between the centers of mass of the ActRIIB domains in the activin A complex is 31 Å, while the corresponding distance between TβRII monomers is 94 Å. Although the TGF-β3:TβRII complex deviates from the canonical ligand dimer symmetry, TGF-β3 retains an extended conformation. The BMP2:BRIA complex would also generate a larger distance between type II receptors, assuming binding of a type II receptor at the ends of the BMP2 dimer, similar to the ActRIIB or TβRII interactions (Figure 6C).
Figure 7A shows a hypothetical ternary complex between an activin A monomer, ActRIIB and a type I receptor, modeled with the BMP-2:BRIA complex. Assuming that activin A binds type I receptors similar to BMP-2, type I and type II receptors would be located on opposite sides of the activin A monomer. The activin A fingers are ‘sandwiched’ between the two receptors, with no direct contacts between the extracellular domains of ActRIIB and type I receptor ectodomains. The BRIA structure can also be accommodated in the dimeric activin A complex, with only minor steric clashes between BRIA and activin A. The two type I receptors are shifted to positions above the ActRIIB receptors on the sides of the complex (Figure 7B), and the C-terminal residues are oriented to the outside of the complex. Approximately 15 residues link the BRIA ectodomain structure to the transmembrane anchor, which would be consistent with the membrane anchoring of all four receptors in this complex to the same cellular surface (Figure 7B and C). This arrangement of type I and type II receptors would alter the proximity of the intracellular kinase domains compared with a canonical, extended TGF-β dimer, potentially resulting in quantitative differences in phosphorylation, SMAD activation and gene expression. Alternatively, flexibility in the activin A wrist structure may be reduced upon type I receptor binding, which might favor the canonical TGF-β conformation.

Fig. 7. Putative signaling complexes with the observed activin A dimer. (A) An activin A monomer (blue) is shown with ActRIIB and BRIA. The BRIA position is based on the superposition of activin A and BMP-2 monomers. (B) Side view of the putative ActRIIB: activin A:type I receptor complex. (C) Membrane-view of the putative ActRIIB:activin A:type I receptor complex.
Discussion
The diversity of cellular responses that can be induced by TGF-β ligands, their associated receptors and intracellular signaling components remains to be fully understood. In part, specificity of a cellular response must arise through interactions of multiple signaling pathways in cells and intra- and extracellular regulatory mechanisms that can block or enhance ligand:receptor interactions. However, the level of accumulation of activated SMAD complexes in the nucleus induced by TGF-β ligands is important in establishing morphogen gradients and associated cellular differentiation outcomes. These observations suggest that differences in affinity, kinetics or structures of related ligand:receptor complexes could quantitatively affect SMAD protein phosphorylation and thereby lead to distinct activation events.
The crystal structure of activin A bound to one of its type II receptors, ActRIIB, reveals the interactions that determine the specificity of ligand:receptor complex formation and how these differ across the broader TGF-β superfamily. Activin A binding to the ActRIIB receptor is significantly different from TGF-β3 binding to TβRII, consistent with previous structural and mutagenesis data. The specificity of these two distinct modes of interaction between type II receptors and TGF-β ligands is established at multiple levels. First, large structural differences are observed in ActRIIB as compared with TβRII in the loop connecting fingers 2 and 3 and in finger 1, which form the interaction surfaces in the two different binding modes. In the ligands, amino acid sequence differences between activin A and TGF-β3, rather than large structural changes in the monomer fold, result in distinct interactions with type II receptors. Activin A binds ActRIIB along the ‘knuckle’ region of the monomer structure while TGF-β3 binds TβRII at the tips of the monomer fingers. Based on sequence analysis and functional data, BMPs likely share a similar binding mode to type II receptors as observed in the ActRIIB:activin A complex, and the structure provides a basis for identifying key residues that may determine the specificity and affinity of these ligand:receptor interactions. The different TGF-β and activin A binding modes might affect signaling, through differential or cooperative recruitment of type I receptors or because of distinct spatial arrangements of type I and type II receptors in the holo complex.
The structure of the activin A dimer observed in the ActRIIB complex also differs significantly from other TGF-β ligand structures. The two activin A monomers are disordered in the ‘wrist’ region and form a compact arrangement that allows two ActRIIB receptors to interact across the ligand dimer axis. Although the two crystal forms reveal a similar arrangement for the activin A dimer, disorder in the wrist residues and in half of the dimer in the P41212 crystal form suggests that the activin A conformation may be more flexible in solution or at the cell surface. Superposition of a BMP type I receptor complex onto the ActRIIB:activin A complex suggests that the observed activin A conformation could interact with two type I receptors as part of a signaling complex. Alternatively, type I receptor binding may involve structural changes to the observed dimer.
Differences in the dimer conformation of activin A as compared with other TGF-β ligands could influence type I and type II receptor assembly and quantitative aspects of signaling and SMAD phosphorylation. Structural and functional studies of the EPO receptor suggest that the geometry of receptor dimerization may directly influence the efficiency of signal transduction (Syed et al., 1998). For example, preformed EPO receptor dimers are thought to exist in a conformational arrangement that prevents transphosphorylation (Livnah et al., 1999; Remy et al., 1999). Ligand binding may lead to a reorientation of receptors and subsequent signal activation. Ligand- independent complexes of other oligomerizing receptors have also been observed, including the EGF receptor (Gadella and Jovin, 1995; Sako et al., 2000), TNF receptor (Chan et al., 2000) and other members of the TGF-β superfamily (Wells et al., 1999; Gilboa et al., 2000; Nohe et al., 2002). TβRI and TβRII form homo- and heteromeric complexes in cells at low levels, requiring ligand binding for signal transduction (Chen and Derynck, 1994; Henis et al., 1994; Wells et al., 1999). The formation of TβRII homodimers stimulates intermolecular autophosphorylation of Ser409 and Ser416 (Luo and Lodish, 1997), in addition to the intramolecular autophosphorylation at Ser213. Phosphorylation at Ser213 and Ser409 are stimulatory, though by different mechanisms, while phosphorylation at Ser416 is inhibitory. BMP receptors have also been shown to form homo- and hetero-oligomers in the absence of ligand (Gilboa et al., 2000; Nohe et al., 2002) at levels higher than those observed for TβRI and TβRII. Signal transduction initiated by ligand binding to preformed receptor complexes may differ from ligand-induced complexes (Nohe et al., 2002), initiating SMAD and p38 MAPK pathways, respectively.
The activin A dimer observed in the crystal structure with ActRIIB may therefore profoundly influence type II receptor function in at least three distinct ways. First, the close clustering of type II receptors may directly influence the kinase domain activation state through autophosphorylation events, either before or after recruitment of type I receptors, leading to quantitative differences in the signal transduced. Secondly, distinct and close clustering of both type II and type I receptors bound to the compact activin A dimer may affect transphosphorylation of the type I receptor, altering the efficiency of SMAD activation. Thirdly, type I receptor binding to activin A may involve a folding transition in the activin A wrist region, which may affect the efficiency, stoichiometry or stability of complex formation and resultant intracellular phosphorylation events. Future structural and functional studies are required to address these possibilities and their potential impact on activin A signaling and associated cellular responses.
Materials and methods
Production and purification of ActRIIB and activin A
The extracellular domain (residues 1–120) of the rat ActRIIB gene, with a C-terminal thrombin cleavage site and His6 tag, was subcloned into the pvl1392 baculovirus vector. Two independent clones contained a single base substitution (C to G) at nucleotide 642, coding for Arg64 instead of Pro64. However, in most species residue 64 is an Arg, with exceptions of Pro in the rat and Ala in the human database sequences. Recombinant baculoviruses were generated using the Baculogold system (Pharmingen). Virus amplification and protein expression were carried out using standard protocols in SF+ insect cells (Protein Sciences Corp.). ActRIIB was purified from cell supernatants using Talon affinity resin (Clontech). ActRIIB was dialyzed into 50 mM Na2HPO4, 300 mM NaCl pH 7.5, digested with thrombin and repurified using the Talon resin. Final protein yields varied from 0.5 to 3 mg/l.
The production of recombinant human activin A has been described previously using transfected CHO cells (line BA83.6-02) and antibody affinity chromatography (Woodruff and Mather, 1995; Pangas and Woodruff, 2002). Briefly, activin A media was harvested, concentrated and dialyzed against 100 mM NaHCO3 pH 8.5. Activin A was bound to a monoclonal antibody (3D9) affinity column and eluted with 50 mM glycine, 0.1% Triton X-100 and 150 mM NaCl pH 2.5. The BA83.6-02 cell line produces ∼1 µg/ml activin A.
Complex formation and crystallization
Complex formation between recombinant ActRIIB and activin A was observed using gel filtration chromatography with a Superdex-75 gel filtration column. A peak with an apparent molecular weight of 65 kDa was observed using mixtures of the two proteins and contained both activin A and ActRIIB, based on SDS–PAGE and western blot analysis.
To establish appropriate mixing ratios of activin A and ActRIIB for crystallization, an ∼1:2 molar ratio mixture was prepared and analyzed by gel filtration chromotography. The ratio was optimized to minimize the amount of free ligand or receptor. The complex was concentrated to 7–10 mg/ml and dialyzed into 20 mM HEPES and 150 mM NaCl pH 7.5. Crystals were grown from hanging drops with a well solution containing 27% PEG 4000, 250 mM NaCl and 100 mM HEPES pH 7.0. Crystals appeared after 4–10 days with two distinct morphologies: a square-like crystal (‘base’) and a longer, pointed crystal form (‘monument’).
Data collection, structure determination and refinement
Crystals with the base morphology were transferred to a cryoprotectant solution of 32% PEG 4000, 400 mM NaCl and 5–10% ethylene glycol, 100 mM HEPES pH 7.0. Crystals with the monument morphology were transferred to a solution of 32% PEG 4000, 400 mM NaCl and 100 mM HEPES pH 7.0, and pulled through Paratone oil before flash cooling.
Data were collected at DND-CAT Beamline 5 ID of the Advanced Photon Source at Argonne National Laboratory. Diffraction data (Table I) were indexed and integrated with DENZO, and reflections merged with SCALEPACK (Otwinowski and Minor, 1997). The space groups for the two crystal forms were determined by diffraction symmetry, systematic absences and molecular replacement solutions. Initially, only diffraction data from a base crystal were available for molecular replacement analysis and these belong to space group P41212. Subsequent analysis of data from the monument crystals established the space group as P41.
Table I. Data collection and refinement statistics.
| Data collection | ||
|---|---|---|
| Space group | P41 | P41212 |
| Cell dimensions | a = b = 104.95, c = 46.20 | a = b = 141.33, c = 46.02 |
| Resolution (Å) | 25.0–3.05 | 15.0–3.10 |
| No. of observations (unique) | 44 194 (9789) | 128 261 (8815) |
| Average redundancy | 4.5 | 14.5 |
| Completeness | 93.5 (87.0)c | 97.6 (95.6)c |
| R-merge (%)a | 6.7 (43.5)c | 8.3 (43.7)c |
| <I/σ> |
22.14 (2.9)c |
31.1 (5.9)c |
| Refinement statistics |
|
|
| R-factorb | 25.1 | 26.8 |
| Rfree (5%)b | 29.8 | 29.1 |
| No. of protein atoms | 2637 | 2377 |
| R.m.s.d. bonds (Å) | 0.011 | 0.0096 |
| R.m.s.d. angles (°) | 1.84 | 2.1 |
| Average B-factor | ||
| ActRIIb chain A | 70.6 | 61.1 |
| ActRIIb chain C | 66.5 | 71.8 |
| Activin A chain B | 81.9 | 135.0 |
| Activin A chain D | 97.8 | 136.0 |
| Dihedral angles | ||
| Most favored (%) | 84.5 | 86.3 |
| Additionally allowed (%) | 13.2 | 13.3 |
| Generously allowed (%) | 2.0 | 0.0 |
| Disallowed (%) | 0.3 | 0.0 |
aR-merge = Σ|Ii – <I>|/Σ|I|, where Ii is the intensity of an individual reflection and <I> is the average intensity of that reflection.
bR-factor = Σ|Fo| – |Fc|/Σ|Fo|, where Fc is the calculated and Fo is the observed structure factor amplitude. Rfree is similarly calculated with a subset of the observed reflections.
cLast shell is 3.1–3.05 Å for P41 and 3.15–3.10 Å for P41212 crystal forms.
The structure of the complex was initially solved in the P41212 space group. A solution for one ActRIIB was found using a modified model of the crystal structure of ActRII (Greenwald et al., 1999). After rigid body refinement, minimization and grouped B-factor refinement with CNS (Brünger et al., 1998), the R-factor for this solution was 47.0% and the Rfree was 49.3%. Electron density maps revealed features near the receptor hydrophobic surface that were not interpretable. Several search models for activin A were also generated. A solution was found in a CNS phased translation search with a BMP-7 monomer (Griffith et al., 1996) that was altered to contain the minimal side chains in common with activin A. Inspection of the symmetry mates of the activin A monomer did not yield a disulfide-linked activin A dimer. An additional activin A monomer:receptor complex was identified using the program BEAST (Read, 2001). Since the observed activin A dimer differs from other TGF-β structures solved in the unbound state, the positions of the activin A monomer and the receptor were confirmed with a variety of molecular replacement searches using CNS (Brünger et al., 1998) and Bruteptf (http://zonker.bioc.aecom.yu.edu/cgi-bin/inhouse/bruteptf/bruteptf.cgi). Solutions were found at low σ levels for the more disordered set of activin A monomer and receptor, and the electron density is generally weaker throughout the more disordered half of the complex. Overall, 12 iterative cycles of manual building, minimization and grouped B-factor refinement with CNS were performed for the P41212 crystal form, providing the final model statistics collected in Table I. The cycle 7 model of a single ActRIIB and monomeric activin A complex was used to solve the P41 crystal form, providing two unambiguous solutions with initial R-factor and Rfree values of 44.4 and 44.7%, respectively. Refinement was initiated with the cycle 10 model from the P41212 crystal form, which showed significantly improved agreement with the P41 data, with R-factor and Rfree values of 37.9 and 36.9%, respectively. Six iterative cycles of building, minimization and B group refinement were carried out, along with a final round of B individual refinement, yielding the statistics collected in Table I. At all stages, overfitting of the data was avoided by monitoring Rfree values.
PDB codes
The PDB codes for the two complex structures are: P41, 1NYS access # RCSB018361 and P41212, 1NYU access # RCSB018363.
Acknowledgments
Acknowledgements
The authors would like to thank B.Joshi, D.Kozien and S.Pangas for technical support, and other members of the Jardetzky and Woodruff laboratories for helpful discussions. X-ray data were collected at beamline 5-ID (Dow-Northwestern-Dupont Collaborative Access Team) of the Advanced Photon Source and in the Keck Biophysics Facility at Northwestern University. This research has been supported by grants from the Cancer Research Institute (T.S.J.) and the National Institutes of Health (T.K.W.). T.S.J. is a Scholar of the Leukemia and Lymphoma Society of America and a recipient of a Cancer Research Institute Young Investigator Award. T.B.T. has been supported by an NIH Drug Discovery training fellowship (T32 AG00260) and National Research Service Award (F32 GM65717-01). T.K.W. and T.S.J. are members of the Northwestern University Robert H.Lurie Comprehensive Cancer Center. The Northwestern Structural Biology Center is supported by grants to the R.H.Lurie Cancer Center.
References
- Bocharov E.V., Korzhnev,D.M., Blommers,M.J., Arvinte,T., Orekhov,V.Y., Billeter,M. and Arseniev,A.S. (2002) Dynamics-modulated biological activity of transforming growth factor β3. J. Biol. Chem., 277, 46273–46279. [DOI] [PubMed] [Google Scholar]
- Bourillot P.Y., Garrett,N. and Gurdon,J.B. (2002) A changing morphogen gradient is interpreted by continuous transduction flow. Development, 129, 2167–2180. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Chan F.K., Chun,H.J., Zheng,L., Siegel,R.M., Bui,K.L. and Lenardo,M.J. (2000) A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science, 288, 2351–2354. [DOI] [PubMed] [Google Scholar]
- Chapman S.C., Bernard,D.J., Jelen,J. and Woodruff,T.K. (2002) Properties of inhibin binding to βglycan, InhBP/p120 and the activin type II receptors. Mol. Cell. Endocrinol., 196, 79. [DOI] [PubMed] [Google Scholar]
- Chen R.H. and Derynck,R. (1994) Homomeric interactions between type II transforming growth factor-β receptors. J. Biol. Chem., 269, 22868–22874. [PubMed] [Google Scholar]
- Daopin S., Piez,K.A., Ogawa,Y. and Davies,D.R. (1992) Crystal structure of transforming growth factor-β2: an unusual fold for the superfamily. Science, 257, 369–373. [DOI] [PubMed] [Google Scholar]
- Dennler S., Goumans,M.J. and ten Dijke,P. (2002) Transforming growth factor β signal transduction. J. Leukoc. Biol., 71, 731–740. [PubMed] [Google Scholar]
- Eigenbrot C. and Gerber,N. (1997) X-ray structure of glial cell-derived neurotrophic factor at 1.9 Å resolution and implications for receptor binding. Nat. Struct. Biol., 4, 435–438. [DOI] [PubMed] [Google Scholar]
- Gadella T.W. Jr, and Jovin,T.M. (1995) Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol., 129, 1543–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa L., Nohe,A., Geissendorfer,T., Sebald,W., Henis,Y.I. and Knaus,P. (2000) Bone morphogenetic protein receptor complexes on the surface of live cells: a new oligomerization mode for serine/threonine kinase receptors. Mol. Biol. Cell, 11, 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray P.C., Greenwald,J., Blount,A.L., Kunitake,K.S., Donaldson,C.J., Choe,S. and Vale,W. (2000) Identification of a binding site on the type II activin receptor for activin and inhibin. J. Biol. Chem., 275, 3206–3212. [DOI] [PubMed] [Google Scholar]
- Greenwald J., Fischer,W.H., Vale,W.W. and Choe,S. (1999) Three-finger toxin fold for the extracellular ligand-binding domain of the type II activin receptor serine kinase. Nat. Struct. Biol., 6, 18–22. [DOI] [PubMed] [Google Scholar]
- Griffith D.L., Keck,P.C., Sampath,T.K., Rueger,D.C. and Carlson,W.D. (1996) Three-dimensional structure of recombinant human osteogenic protein 1: structural paradigm for the transforming growth factor β superfamily. Proc. Natl Acad. Sci. USA, 93, 878–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdon J.B., Standley,H., Dyson,S., Butler,K., Langon,T., Ryan,K., Stennard,F., Shimizu,K. and Zorn,A. (1999) Single cells can sense their position in a morphogen gradient. Development, 126, 5309–5317. [DOI] [PubMed] [Google Scholar]
- Hart P.J., Deep,S., Taylor,A.B., Shu,Z., Hinck,C.S. and Hinck,A.P. (2002) Crystal structure of the human TβRII ectodomain–TGF-β3 complex. Nat. Struct. Biol., 9, 203–208. [DOI] [PubMed] [Google Scholar]
- Henis Y.I., Moustakas,A., Lin,H.Y. and Lodish,H.F. (1994) The types II and III transforming growth factor-β receptors form homo-oligomers. J. Cell Biol., 126, 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck A.P. et al. (1996) Transforming growth factor β1: three-dimensional structure in solution and comparison with the X-ray structure of transforming growth factor β2. Biochemistry, 35, 8517–8534. [DOI] [PubMed] [Google Scholar]
- Innis C.A., Shi,J. and Blundell,T.L. (2000) Evolutionary trace analysis of TGF-β and related growth factors: implications for site-directed mutagenesis. Protein Eng., 13, 839–847. [DOI] [PubMed] [Google Scholar]
- Kirsch T., Nickel,J. and Sebald,W. (2000a) BMP-2 antagonists emerge from alterations in the low-affinity binding epitope for receptor BMPR-II. EMBO J., 19, 3314–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch T., Sebald,W. and Dreyer,M.K. (2000b) Crystal structure of the BMP-2–BRIA ectodomain complex. Nat. Struct. Biol., 7, 492–496. [DOI] [PubMed] [Google Scholar]
- Lau A.L., Kumar,T.R., Nishimori,K., Bonadio,J. and Matzuk,M.M. (2000) Activin βC and βE genes are not essential for mouse liver growth, differentiation, and regeneration. Mol. Cell. Biol., 20, 6127–6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livnah O., Stura,E.A., Middleton,S.A., Johnson,D.L., Jolliffe,L.K. and Wilson,I.A. (1999) Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science, 283, 987–990. [DOI] [PubMed] [Google Scholar]
- Luo K. and Lodish,H.F. (1997) Positive and negative regulation of type II TGF-β receptor signal transduction by autophosphorylation on multiple serine residues. EMBO J., 16, 1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. (2000) How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol., 1, 169–178. [DOI] [PubMed] [Google Scholar]
- Massagué J. and Chen,Y.G. (2000) Controlling TGF-β signaling. Genes Dev., 14, 627–644. [PubMed] [Google Scholar]
- Massagué J. and Weis-Garcia,F. (1996) Serine/threonine kinase receptors: mediators of transforming growth factor β family signals. Cancer Surv., 27, 41–64. [PubMed] [Google Scholar]
- Massagué J. and Wotton,D. (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J., 19, 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J., Blain,S.W. and Lo,R.S. (2000) TGFβ signaling in growth control, cancer, and heritable disorders. Cell, 103, 295–309. [DOI] [PubMed] [Google Scholar]
- Mellor S.L., Cranfield,M., Ries,R., Pedersen,J., Cancilla,B., de Kretser,D., Groome,N.P., Mason,A.J. and Risbridger,G.P. (2000) Localization of activin β(A)-, β(B)-, and β(C)-subunits in humanprostate and evidence for formation of new activin heterodimers of β(C)-subunit. J. Clin. Endocrinol. Metab., 85, 4851–4858. [DOI] [PubMed] [Google Scholar]
- Mittl P.R., Priestle,J.P., Cox,D.A., McMaster,G., Cerletti,N. and Grutter,M.G. (1996) The crystal structure of TGF-β3 and comparison to TGF-β2: implications for receptor binding. Protein Sci., 5, 1261–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono K., Kusanagi,K. and Inoue,H. (2001) Divergence and convergence of TGF-β/BMP signaling. J. Cell Physiol., 187, 265–276. [DOI] [PubMed] [Google Scholar]
- Nohe A., Hassel,S., Ehrlich,M., Neubauer,F., Sebald,W., Henis,Y.I. and Knaus,P. (2002) The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J. Biol. Chem., 277, 5330–5338. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pangas S.A. and Woodruff,T.K. (2002) Production and purification of recombinant human inhibin and activin. J. Endocrinol., 172, 199–210. [DOI] [PubMed] [Google Scholar]
- Pasche B. (2001) Role of transforming growth factor β in cancer. J. Cell. Physiol., 186, 153–168. [DOI] [PubMed] [Google Scholar]
- Read R.J. (2001) Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. D, 57, 1373–1382. [DOI] [PubMed] [Google Scholar]
- Remy I., Wilson,I.A. and Michnick,S.W. (1999) Erythropoietin receptor activation by a ligand-induced conformation change. Science, 283, 990–993. [DOI] [PubMed] [Google Scholar]
- Ryan K., Garrett,N., Bourillot,P., Stennard,F. and Gurdon,J.B. (2000) The Xenopus eomesodermin promoter and its concentration-dependent response to activin. Mech. Dev., 94, 133–146. [DOI] [PubMed] [Google Scholar]
- Sako Y., Minoghchi,S. and Yanagida,T. (2000) Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol., 2, 168–172. [DOI] [PubMed] [Google Scholar]
- Scheufler C., Sebald,W. and Hulsmeyer,M. (1999) Crystal structure of human bone morphogenetic protein-2 at 2.7 Å resolution. J. Mol. Biol., 287, 103–115. [DOI] [PubMed] [Google Scholar]
- Schlunegger M.P. and Grutter,M.G. (1992) An unusual feature revealed by the crystal structure at 2.2 Å resolution of human transforming growth factor-β2. Nature, 358, 430–434. [DOI] [PubMed] [Google Scholar]
- Syed R.S. et al. (1998) Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature, 395, 511–516. [DOI] [PubMed] [Google Scholar]
- Vejda S., Cranfield,M., Peter,B., Mellor,S.L., Groome,N., Schulte-Hermann,R. and Rossmanith,W. (2002) Expression and dimerization of the rat activin subunits βC and βE: evidence for the formation of novel activin dimers. J. Mol. Endocrinol., 28, 137–148. [DOI] [PubMed] [Google Scholar]
- Wells R.G., Gilboa,L., Sun,Y., Liu,X., Henis,Y.I. and Lodish,H.F. (1999) Transforming growth factor-β induces formation of a dithiothreitol-resistant Type I/Type II receptor complex in live cells. J. Biol. Chem., 274, 5716–5722. [DOI] [PubMed] [Google Scholar]
- Woodruff T.K. (1998) Regulation of cellular and system function by activin. Biochem. Pharmacol., 55, 953–963. [DOI] [PubMed] [Google Scholar]
- Woodruff T.K. and Mather,J.P. (1995) Inhibin, activin and the female reproductive axis. Annu. Rev. Physiol., 57, 219–244. [DOI] [PubMed] [Google Scholar]
- Wuytens G. et al. (1999) Identification of two amino acids in activin A that are important for biological activity and binding to the activin type II receptors. J. Biol. Chem., 274, 9821–9827. [DOI] [PubMed] [Google Scholar]