

Abstract
The human opportunistic pathogen Serratia marcescens is a bacterium with a broad host range, and represents a growing problem for public health. Serratia marcescens kills Caenorhabditis elegans after colonizing the nematode’s intestine. We used C.elegans to screen a bank of transposon-induced S.marcescens mutants and isolated 23 clones with an attenuated virulence. Nine of the selected bacterial clones also showed a reduced virulence in an insect model of infection. Of these, three exhibited a reduced cytotoxicity in vitro, and among them one was also markedly attenuated in its virulence in a murine lung infection model. For 21 of the 23 mutants, the transposon insertion site was identified. This revealed that among the genes necessary for full in vivo virulence are those that function in lipopolysaccharide (LPS) biosynthesis, iron uptake and hemolysin produc tion. Using this system we also identified novel conserved virulence factors required for Pseudomonas aeruginosa pathogenicity. This study extends the utility of C.elegans as an in vivo model for the study of bacterial virulence and advances the molecular understanding of S.marcescens pathogenicity.
Keywords: Caenorhabditis elegans/cytotoxicity/insertional mutagenesis/iron transport/LPS
Introduction
There is a continuing need to characterize the strategies used by bacterial pathogens during infection. This has been highlighted in recent times by the increase in resistance to multiple antibiotics of certain bacteria, and by the use of pathogens as biowarfare agents. Since certain virulence factors are necessary for full pathogenicity regardless of the host, one approach to the study of the interaction between pathogen and host is the use of simple model systems. It has been clearly demonstrated that the nematode Caenorhabditis elegans is an appropriate model host for the study of the virulence mechanisms deployed by the human opportunistic pathogen Pseudomonas aeruginosa (reviewed in Tan and Ausubel, 2000). The use of C.elegans for such studies has now been extended to a range of Gram-positive and Gram-negative bacteria (reviewed in Aballay and Ausubel, 2002; Ewbank, 2002), including the potential biowarfare agents Burkholderia pseudomallei (O’Quinn et al., 2001; Gan et al., 2002) and Yersinia pestis (Darby et al., 2002).
Serratia marcescens is a Gram-negative bacterium that causes disease in plants and in a wide range of both invertebrate and vertebrate hosts (Grimont and Grimont, 1978). It is an opportunistic human pathogen and in the last three decades there has been a steady increase in nosocomial S.marcescens infections that can be life-threatening (Haddy et al., 1996; Hejazi and Falkiner, 1997; and references cited therein). While environmental S.marcescens strains are often red, due to the production of prodigiosin (Thomson et al., 2000), the strains associated with hospital outbreaks are mostly non-pigmented (Hejazi and Falkiner, 1997; Carbonell et al., 2000). As many S.marcescens strains are also resistant to multiple antibiotics (Hejazi and Falkiner, 1997; Alexandrakis et al., 2000), it represents a growing problem for public health. However, relatively little is known about the factors that contribute to S.marcescens pathogenesis within its host (Hejazi and Falkiner, 1997).
Serratia marcescens is capable of killing C.elegans following the establishment of an intestinal infection (Mallo et al., 2002). We have characterized this interaction and used C.elegans to screen a bank of S.marcescens mutants. We isolated bacterial clones with a reduced virulence against the nematode. Some of them were also attenuated in their pathogenicity in two other infection models. Three candidates were tested in a murine lung infection model, and one of them displayed a very marked reduction in its pathogenicity. To our knowledge, this represents the first systematic study of S.marcescens virulence in vivo and has permitted the identification of previously uncharacterized virulence factors. Further, using this S.marcescens–C.elegans model we have identified conserved virulence factors also required for the full virulence of P.aeruginosa.
Results
Characterization of the infection of C.elegans by S.marcescens
Several pigmented and non-pigmented strains of S.marcescens are capable of infecting and killing C.elegans (Table I). We chose the non-pigmented strain Db11 for further study, thereby eliminating any potential contribution of the pigment prodigiosin to the infectious process. This strain was first described as a pathogen of Drosophila melanogaster (Flyg et al., 1980). More recently it has been used as a model pathogen to investigate innate immunity in C.elegans (Pujol et al., 2001; Mallo et al., 2002). The steps in the infection of worms by Db11 have been outlined (Mallo et al., 2002) and its repulsive effect on worms has previously been described (Pujol et al., 2001). When wild-type (N2) worms were transferred as L4 larvae from the standard Escherichia coli strain OP50 to lawns of Db11 they became visibly sick after 2 days. They did not show any sign of starvation, but started to die 1 day later. All worms were dead after 7 days when grown on Db11 (Figure 1A).
Table I. Pathogenicity of different strains of S.marcescens during C.elegans infection.
| Strain | Pigmentation | Mean lifespana |
|---|---|---|
| Db11 | – | 4.2 ± 0.3 (145) |
| Db1140 | – | 6.2 ± 0.7 (132) |
| Sma 3 | – | 4.9 ± 0.1 (95) |
| Sma 12 | – | 5.0 ± 0.2 (96) |
| Sma 13 | – | 5.1 ± 0.2 (98) |
| ATCC 274 | + | 2.6 ± 0.1 (100) |
| Sm 365 | + | 5.3 ± 0.7 (91) |
| Sm 2170 | + | 1.7 ± 0.3 (99) |
aMean survival time ± SD in days for N2 worms infected at the L4 stage at 25°C. The total number of worms used in two independent tests is given in parentheses. Under these conditions, the mean post-L4 survival time for worms cultivated on E.coli OP50 is 12.6 ± 1 (n = 88).

Fig. 1. Characterization of the infection of C.elegans by S.marcescens. (A) The killing of C.elegans by S.marcescens requires live bacteria. Kinetics of killing of worms exposed to S.marcescens Db11 (closed squares), E.coli OP50 (open squares), heat-killed Db11 (open triangles) and heat-killed Db11 supplemented with culture supernatant (open circles). (B) A short contact with S.marcescens is sufficient to infect C.elegans. Kinetics of killing of C.elegans by S.marcescens after different periods of contact with Db11. Worms were exposed to S.marcescens Db11 permanently (closed squares), for 4 h (open diamonds), for 8 h (open triangles) or for 18 h (open squares) and were then surface-sterilized and deposited on OP50. (C) The time course of the infection of C.elegans by Db11 is age dependent. Worms were transferred from OP50 to Db11 lawns at the L1 stage (open squares), at the L4 stage (closed squares), as 1-day-old adults (open triangles), as 2-day-old adults (open diamonds) or as 3-day-old adults (open circles) and their post-transfer survival was scored. In all cases, worms were grown on NGM plates at 25°C, and between 40 and 50 N2 hermaphrodites were used in each test. The curves are representative of at least two independent trials.
Certain bacteria kill worms via toxin-mediated mechanisms (Aballay and Ausubel, 2002; Couillault and Ewbank, 2002; Ewbank, 2002). To address this possibility, L4 worms were transferred to heat-killed Db11 in the presence or absence of supernatants from saturated Db11 cultures. In these cases, the worms did not appear sick and their survival was at least as long as that of worms fed on OP50 (Figure 1A). This suggests that live bacteria are needed for the infection and that a stable toxin does not mediate the killing of the worm. To establish whether a permanent contact was necessary for bacterially mediated killing, worms were transferred to Db11 at the L4 stage, allowed to feed for a fixed time and then surface-sterilized and returned to OP50. A contact of 30 h was sufficient to give survival curves that were indistinguishable from those obtained when worms were in constant contact with Db11 (data not shown). With shorter periods of contact, worms died much faster than control worms that were kept on E.coli, but the time course of the infection was longer than that for worms kept permanently in contact with Db11 (Figure 1B). Older worms were more susceptible to infection and the latency period before the first observed deaths was diminished as a function of the age of the worms, presumably reflecting a decrease in the antibacterial capabilities of older worms. Conversely, the early larval stages of C.elegans were resistant to Db11 (Figure 1C and see below).
To follow the fate of the bacteria upon ingestion by the worm, we used strains of E.coli OP50 and S.marcescens Db11 that express the green fluorescent protein (GFP). Db11-GFP is as virulent as Db11 with regard to the killing of C.elegans (data not shown). When L4 worms are placed on OP50-GFP, intact bacteria are not found in the intestine, as they are broken down by the grinder located in the terminal bulb of the pharynx (Figure 2A and B) (Labrousse et al., 2000). On the other hand, when worms were transferred to Db11-GFP at the L4 stage, after as little as 2 h of contact, intact bacteria were seen to accumulate in the lumen of the intestine (Figure 2C and D). If, however, L4 worms were fed briefly on Db11 before being transferred to OP50-GFP, intact fluorescent bacteria were able to pass the grinder (Figure 2E and F). This indicates that Db11 is capable of disrupting the function of the grinder. In contrast with L4 worms, no intact bacteria were observed in the intestines of earlier larval stages fed with Db11-GFP even for periods of several hours (data not shown). This suggests that the resistance of pre-L4 larvae to Db11 (Figure 1C) is most likely due to the incapacity of the bacteria to enter the intestinal lumen. After 24 h of contact with Db11-GFP, the intestinal lumen appeared distended and full of fluorescent bacteria that remained extracellular during the infection. They were restricted to the intestinal lumen, except on rare occasions when bacteria were observed in the uterus between eggs. The increase in volume of the lumen was concurrent with exponential bacterial growth (see Supplementary data, available at http://www.ciml.univ-mrs.fr/EWBANK_ jonathan/SuppMat/Screen/Kurz.html). Apart from this progressive distension of the intestinal lumen, outwardly, worms showed relatively little sign of infection for the first 24 h. The muscular contractions usually associated with feeding and defecation continued and their rate of egg-laying was normal (Mallo et al., 2002). In clear contrast with worms grown on OP50 (Figure 3A), there was then a progressive vacuolation of the intestinal cells (Figure 3C) accompanied by a decrease in the volume of the worm’s intestinal epithelium. After infection with Db11 for 3 days, there was also an accumulation within the lumen of autofluorescent vesicles that appeared to be derived from the intestinal epithelium (Figure 3B). Similar vesicles were observed in worms after 5 days of contact with Salmonella typhimurium (C.L.Kurz, unpublished observations). The size and number of these vesicles, which moved in the lumen as the intestine contracted, increased to such a degree that defecation became impaired. The other tissues, including the germ-line, were also gradually destroyed (Figure 3B) before the worms died.

Fig. 2. Early entry of intact S.marcescens into the intestinal lumen of C.elegans. Fluorescence (A, C, E) and Nomarski (B, D, F) photomicrographs of L4 N2 hermaphrodite worms fed with OP50-GFP for 2 h (A and B), with Db11-GFP for 2 h (C and D) and with Db11 for 2 h followed by brief washing and feeding with OP50-GFP for 5 min (E and F). In (A), intact bacteria (indicated by white arrows) can only be seen in the pharyngeal isthmus anterior to the terminal bulb and the grinder (indicated by the asterisk). In (C), intact Db11-GFP are in the intestinal lumen. In (E), intact OP50-GFP can freely pass the grinder after the short contact with Db11 and are found intact in the intestinal lumen. In all cases the head of the worm is to the left; scale bar, 10 µm.

Fig. 3. Symptoms of the infection of C.elegans by S.marcescens. Nomarski photomicrographs of N2 worms fed for 5 days post-L4 stage with OP50 (A) or Db11 for 5 days post-L4 stage (B, C). (A) The intestine is healthy with large cells (arrows), the intestinal lumen is of normal size (arrowheads) and the germ-line is clearly visible (dotted lines). (B) The intestine of the Db11-infected worm is distended (arrowheads) and full of intact bacteria. The intestinal cells are partially lysed (arrows) and the germ-line is no longer apparent. A large round vesicle can also be observed in the intestinal lumen. (C) The vacuolation of one posterior intestinal cell is highlighted (dotted lines). In all cases the head of the worm is to the left; scale bar, 10 µm.
Isolation of Db11 mutants with a reduced virulence against C.elegans
Having characterized the infection of C.elegans by Db11, we designed a screen to identify bacterial mutants with a reduced virulence. Given that wild-type worms infected by Db11 are able to lay eggs and these eggs hatch to give larvae that are resistant to Db11, we decided to use the mutant worm strain fer-15 that is conditionally sterile at 25°C to avoid the potential confusion between generations. The time course of survival of fer-15 on Db11 is essentially identical with that of wild-type worms (see Supplementary data). Therefore we individually screened clones from a S.marcescens Db11 mini-Tn5Cm insertion library for mutants that supported the growth and survival of fer-15 worms beyond the time observed for Db11 (see Supplementary data). From 2300 bacterial clones tested, 23 attenuated mutants were selected for further study. Each contained a single transposon insertion (see Supplementary data). To determine whether the observed attenuation in virulence was due to a problem of general metabolism and/or growth, or was the result of the disruption of the function of a virulence gene, we followed the growth of each clone in Luria broth (LB) at 25°C and 37°C. No major difference in replication rate between mutant and parental strains was observed during exponential growth. However, two clones, 8E11 and 18D4, grew more slowly on LB agar plates (data not shown). The different mutants were then individually tested for their pathogenicity during the infection of N2 worms and were classed into three categories: weakly attenuated, attenuated and strongly attenuated (Figure 4; Table II). With the exception of 10E5, all mutants supported the growth and survival of N2 worms beyond 7 days, something that was never seen with Db11 (n > 5000). Worms grown on the most strongly attenuated clone, 20C2, lived roughly twice as long as worms on Db11 (Figure 4). In the case of 10E5, a statistically significant reduction of virulence was only observed during the infection of the fer-15 strain (see Supplementary data).

Fig. 4. The isolated S.marcescens mutants are less virulent during their infection of C.elegans. Kinetics of killing of C.elegans infected by Db11 (closed squares), and representative weakly attenuated mutant 3H5 (open circles), attenuated mutant 7F1 (open triangles) and strongly attenuated mutant 20C2 (open diamonds). The dotted line with open squares shows the survival curve for worms fed on E.coli OP50. In all cases, worms were grown on NGM plates at 25°C and 40–50 N2 hermaphrodites were used in each test. The curves are representative of at least two independent trials.
Table II. Characterization of attenuated mutants.
| Clone | Model |
Within/upstream of gene encoding conserved protein/domain (gene name) | Species (DDBJ/EMBL/GenBank accession No.) | E valueb | Function | ||
|---|---|---|---|---|---|---|---|
| Worma | Fly | Cells | |||||
| 3H5 | – | wt | wt | No | na | na | na |
| 8G1 | – | wt | wt | baeS homologue | X.axonopodis (AAM37649) | 8e-07 | Two-component system sensor |
| 8H1 | – | wt | wt | mgtB homologue | Y.pestis (NP_405238) | 9e-24 | Magnesium transport |
| 10E5 | – | wt | wt | Ferrisiderophore receptor (y3343) | Y.pestis (AAM86893) | 4e-82 | Iron transport |
| 7D1 | – – | wt | wt | Yes (STM0278; see 22D9) | S.typhimurium (NP_459276) | 2e-05 | Unknown |
| 7E7 | – – | wt | wt | galR homologue | Y.pestis (NP_670482) | 2e-56 | Galactose operon repressor |
| 7F1 | – – | wt | wt | Amino oxidase domain (PA3713) | P.aeruginosa (A83182) | 5e-57 | Unknown |
| 8C7 | – – | wt | wt | No | na | na | na |
| 8E2 | – – | wt | wt | Yes (YPO2856) | Y.pestis (NP_406362) | 5e-06 | Unknown |
| 23C11 | – – | wt | wt | Not cloned | na | na | na |
| 18F3 | – – – | wt | wt | wbeiT homologue | E.ictaluri (AAL25633) | 1e-39 | O-antigen biosynthesis |
| 22D4 | – – – | wt | wt | ibpB homologue | Y.pestis (NP_671393) | 5e-36 | Stress resistance |
| 22D9 | – – – | wt | wt | Yes (STM0278; see 7D1) | S.typhimurium (NP_459276) | 3e-07 | Unknown |
| 23E6 | – – – | wt | wt | yjcE homologue | Y.pestis (NP_407085) | 3e-23 | Unknown |
| 10F7 | – | – | wt | No | na | na | na |
| 18D4 | – | – | wt | No | na | na | na |
| 10H4 | – | – – | wt | Yes (yfdR) | E.coli (P76514) | 5e-29 | Unknown |
| 8E11 | – – | – | wt | Not cloned | na | na | na |
| 7A8 | – – | – – | wt | DJ-1/PfpI domain (CC2959) | C.crescentus (NP_421753) | 2e-11 | Unknown |
| 21C1 | – – | – – – | wt | ATPase domain | E.coli (S28007) | 0.008 | Unknown |
| 10H1 | – | – – | – – | vibC homologue | P.fluorescens (CAA70528) | 2e-17 | Iron transport |
| 21C4 | – – – | – – – | – – | shlB | S.marcescens (AAA50322) | na | Hemolysin production |
| 20C2 | – – – | – – – | – – | wzm homologue | E.coli (BAA28324) | 1e-115 | LPS biosynthesis |
wt, wild type; –, weakly attenuated; – –, attenuated; – – –, strongly attenuated; na, not applicable.
aN2 worms except for 10E5, for which fer-15 worms were used.
bExpected value of BLASTX; see Supplementary data for details.
Certain mutants exhibit reduced virulence in other infection models
To determine whether the bacterial genes necessary for full virulence during the natural infection of C.elegans were also necessary for pathogenesis in other hosts, the selected mutants were individually tested in a D.melanogaster infection model. At 20°C, almost all flies died within 24 h following injection of 50–100 Db11 bacteria into the thorax (Figure 5A). Nine of the 23 mutants showed a clear and reproducible reduction of their virulence in this insect model. This shows that certain factors are necessary for the full virulence of Db11 during infection of both the nematode and the fly, despite the different modes of infection (ingestion versus injection). Of the nine mutants, 20C2, 21C1 and 21C4 showed the strongest attenuation of virulence (Figure 5A; Supplementary data).

Fig. 5. The selected S.marcescens mutants are less pathogenic in other models. (A) Kinetics of killing of D.melanogaster injected in the thorax with LB medium (open squares), Db11 (closed squares), or mutants 10F7 (open diamonds), 10H4 (open triangles) or 21C1 (open circles). (B) Cytotoxic effect of Db11 and derived mutants against a polarized human epithelial cell line. The release of lactate dehydrogenase from the epithelial cells was measured after 2 h contact with the bacteria and a cytotoxicity index calculated. An index of 1 corresponds to 100% lysis. The results represent the mean and standard deviation obtained from four independent trials. (C) Kinetics of killing of 8- to 10-week-old mice (n = 10) infected intranasally with Db11 (closed squares) or the mutant 21C4 (open squares). Their survival was plotted using the Kaplan–Meier method.
Serratia marcescens has previously been shown to possess a strong cytotoxic effect in vitro against several cell types (Poole et al., 1988; Carbonell et al., 1997; Hertle et al., 1999) and is also associated with nosocomial lung infections (Haddy et al., 1996). Therefore we tested the cytotoxicity of Db11 and of the 23 mutants against a human polarized pulmonary epithelial cell-line (16HBE14o–). In this test, the bacteria are in direct contact with the target cells. Three of the mutants, 10H1, 20C2 and 21C4, showed a strong attenuation of their cytotoxic effect in vitro compared with Db11 (Figure 5B). These three mutants, which also showed a reduced virulence in D.melanogaster, were then tested in vivo in a murine lung infection model. The mutant 21C4 showed a marked reduction in its pathogenicity (p < 0.0013) (Figure 5C), while 10H1 and 20C2 were at least as virulent as Db11 (data not shown).
Molecular characterization of the attenuated mutants
For 21 of the 23 mutants, the inserted transposon and part of the flanking genomic region was cloned; the remaining two regions have so far been refractory. The respective transposon insertion sites were identified (see Supple mentary data), and analyses revealed that for 19 mutants the sites were unrelated, indicating that the screen is far from being saturated. The mutants 7D1 and 22D9 contained insertions separated by less than 250 bp (see below), confirming the role of this particular locus in virulence. Only in the case of the clone 21C4 was the transposon inserted in a region previously characterized in S.marcescens, being inserted within the hemolysin shl operon. For four mutants (3H5, 8C7, 10F7 and 18D4), no conserved open reading frame was identified in the vicinity of the transposon insertion site. On the other hand, for 16 mutants, the transposon was inserted within, or just upstream of, a gene potentially encoding a protein with a homologue in at least one other bacterial species (see Supplementary data).
Based on the site of transposon insertion, the three mutants 10H1, 20C2 and 21C4 were predicted to be affected in iron uptake, lipopolysaccharide (LPS) biosynthesis or hemolysin production, respectively. For 10H1, the gene downstream of the transposon insertion site potentially encodes a VibC/EntC homologue. These proteins are involved in the biosynthesis of the siderophores vibriobactin and enterobactin necessary for iron uptake in Vibrio cholerae (Wyckoff et al., 2001) and E.coli (Nahlik et al., 1987), respectively. As judged by chrome azurol S tests (Schwyn and Neilands, 1987), the mutant 10H1 produced less siderophore compared with Db11 (E.Pradel, personal communication). Increasing the iron concentration in the culture medium had no effect on either the survival of worms grown on OP50 or the virulence of Db11 (data not shown), but increased the rate of killing of C.elegans by 10H1. At 25°C, the difference was marginal (data not shown). We have observed that at 20°C the time course of infection of C.elegans with S.marcescens is slower than at 25°C, and that this can help accentuate differences in survival between different worm strains on the same bacterial strain or between the same worm strain on different bacteria (C.L.Kurz, unpublished results). Therefore the effect of increasing the iron concentration was also assayed at 20°C. At this temperature, the augmentation of the virulence of 10H1 was significant (p < 0.014) (Figure 6). Taken together, these results confirm that the mutant is defective in its capacity to capture iron. The mutant 20C2, the most attenuated in the nematode, fly and cell models (Figures 4 and 5B; Supplementary data), contains an insertion in a homologue of wzm that codes for the membrane component of an ABC-2 transporter specialized in the translocation of LPS O-antigen (Sugiyama et al., 1998). Consistent with this, analysis of the 20C2 LPS by Tricine–SDS–PAGE revealed the absence of O-antigen. A derivative of Db11, Db1140 (Flyg and Xanthopoulos, 1983), also lacked O-antigen, as previously predicted (Pujol et al., 2001), in contrast with the other mutants (Figure 7; data not shown). In 21C4, the transposon is inserted 61 bp upstream of the initiation codon of shlB, the first gene of the shlBA operon. ShlA is a well characterized virulence factor of S.marcescens involved in cytotoxicity (e.g. Poole and Braun, 1988a,b; Hertle, 2002).

Fig. 6. Addition of iron increases the virulence of the mutant 10H1. Kinetics of killing at 20°C of fer-15 hermaphrodites transferred as L4 worms from OP50 to 10H1 on standard NGM medium (closed squares) or on NGM medium supplemented with 0.1 mM iron (open squares).
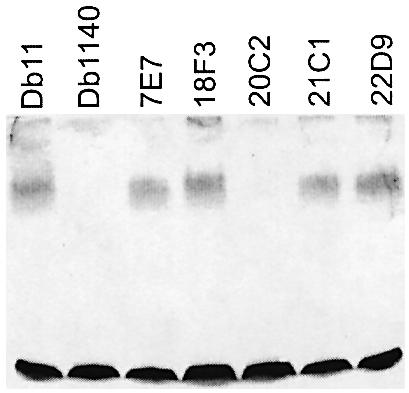
Fig. 7. The mutant 20C2 is deficient in O-antigen. Silver-stained Tricine–SDS–PAGE gel of LPS prepared from different S.marcescens strains. Db1140 and 20C2 had no visible O-antigen ladder (upper broad band). The lower band corresponds to the LPS core.
Regarding the other mutants, for 8E2, 10H4 and 7D1/22D9 absolutely no information is available regarding the function of the corresponding homologous proteins. For 23E6, the gene downstream of the insertion site is a homologue of the E.coli yjcE gene which encodes a presumed Na+–H+ exchanger (but see Verkhovskaya et al., 2001). For 7A8, the disrupted gene potentially encodes a DJ-1/PfpI domain-containing protein. This domain is common to a variety of protein families, including proteases and transcriptional regulators. For 7F1, the corresponding protein contains an amino oxidase domain. In the case of 21C1, the homologous protein contains an ATPase domain. For the remainder, a putative function could be assigned based on sequence comparisons (Table II; Supplementary data).
In the clone 8G1, a gene that encodes a homologue of the two-component system sensor BaeS of Xanthomonas axonopodis is disrupted. Two-component signal transduction systems regulate the expression of multiple genes in response to the environment and are known virulence factors in many pathogenic bacteria (Hoch, 2000). 8H1 contains an insertion upstream of a homologue of mgtB that encodes a highly conserved putative Mg2+ transport ATPase (see below). In 10E5, the transposon insertion is in a gene coding for a homologue of a TonB-dependent ferrisiderophore receptor and would be predicted to have a defect in iron uptake, as for 10H1. However, the difference in survival of worms on Db11 and 10E5 was small and precluded statistically significant tests of the rescuing capacity of exogenous iron.
Clone 22D4 contains a transposon inserted within a homologue of the chaperone gene ibpB. In E.coli, IbpB has been shown to protect enzymes from inactivation by heat and oxidants (Kitagawa et al., 2002). In Db11, it could serve to protect the bacteria during their growth in the intestine. 18F3 has an insertion in a homologue of the gene wbeiT of Edwardsiella ictaluri that encodes a UDP- glucose-4-epimerase and is within an O-antigen biosynthesis gene cluster (see DDBJ/EMBL/GenBank accession No. 16648662). No difference in LPS structure was observed in Tricine–SDS–PAGE analysis between 18F3 and Db11 (Figure 7), but, as this method can only detect major changes in LPS structure, we cannot exclude the possibility that 18F3 has a defect in its LPS. Lastly, 7E7 has an insertion in a clear homologue of galR of Y.pestis encoding the repressor of the gal operon, but there is no obvious link between this insertion and the observed reduced virulence of the mutant.
Interspecies conservation of virulence genes
Seven of the 12 S.marcescens disrupted loci conserved in other bacterial species have homologues in P.aeruginosa. The infection of C.elegans by P.aeruginosa is currently the best characterized nematode infection model (Tan and Ausubel, 2000; Aballay and Ausubel, 2002). We inactivated two of these homologous genes in the P.aeruginosa strain PA14 by targeted gene disruption to determine whether they encoded virulence factors. We chose the gene PA3713 of unknown function, corresponding to 7F1, and PA0151, corresponding to 10E5 and thus encoding a ferrisiderophore receptor, as mentioned above. During infection of worms, a significant reduction in virulence was observed at 20 and 25°C for both mutants compared with PA14 (Figure 8; data not shown). Neither gene had previously been identified as being required for full virulence in PA14.

Fig. 8. Mutants of P.aeruginosa for the homologues of the genes disrupted in the Db11-derived mutants 10E5 and 7F1 (PA14-M151 and PA14-M3713, respectively) are attenuated in C.elegans. Kinetics of killing of C.elegans infected by P.aeruginosa strain PA14 (closed squares), PA14 mutant M151 (open circles) and PA14 mutant M3713 (open triangles). Worms were grown on NGM plates at 20°C, and 40–50 N2 hermaphrodites were used in each test. The curves are representative of at least two independent trials. The differences between PA14 and M151 or M3713 are statistically significant (p < 0.0004 and p < 0.023, respectively).
Discussion
By performing a large-scale screen for mutants with a reduced virulence during the infection of C.elegans, we have identified factors that contribute to the pathogenicity of the S.marcescens strain Db11 in a range of different infection models. Given the nature of the transposon used to generate the mutants (de Lorenzo et al., 1990), the observed attenuation could be linked to the disruption of a single gene or, in the case of an insertion within an operon, could be due to a polar effect on the expression of downstream genes. Because this study involved a relatively large number of mutants, and since the genome sequence of S.marcescens was not available, we were not able to demonstrate directly for each mutant that the single mini-Tn5 Cm insertion provokes the attenuation. Never theless, in a number of cases there is a clear link between the molecular lesion and the inferred function of the disrupted gene. In the case of 21C4, the transposon is inserted within the shlBA operon. ShlB is a specific outer-membrane transporter and activator of the hemolysin ShlA (Ondraczek et al., 1992). ShlA specifically interacts with eukaryotic membranes to create pores that lead to cell lysis (Schiebel and Braun, 1989). As would be predicted, the mutant 21C4 has a reduced cytotoxicity (Figure 5B). The mutant 20C2 contains an insertion in a homologue of wzm that codes for an LPS transporter. It is a rough mutant, with a detectable difference in its LPS structure (Figure 7). In many Gram-negative bacteria, including S.marcescens, LPS O-antigen plays an important role in both resistance to host defences and adherence (Palomar et al., 1993, 1995; Lerouge and Vanderleyden, 2002). Additionally, the in vitro cytotoxic activity of S.marcescens smooth strains is higher than that of rough strains, perhaps due to the direct stabilization of hemolysin by interaction with the LPS O-chain (Poole and Braun, 1988a), consistent with the observed difference in cytotoxicity between Db11 and 20C2 (Figure 5B). 10H1 contains a mini-Tn5 Cm insertion within a gene cluster homologous to the V.cholerae vibriobactin or E.coli enterobactin siderophore biosynthesis operon (Wyckoff et al., 2001). Siderophores are an integral part of the iron-uptake mechanisms of diverse bacteria. Since this metal is present at only 10–9 M in the environment, they are a necessary complement to the capacity of bacteria to take up free iron by passive transport (Braun and Braun, 2002). As predicted, the clone produced a reduced level of siderophores and its reduced virulence was rescued by the addition of iron to the medium (Figure 6). Although iron is often a limiting factor for the growth and virulence of bacterial pathogens in vivo (Braun, 2001), this has not previously been demonstrated to be the case for S.marcescens. The locus disrupted in 10H1 (and in 10E5) is distinct from the molecularly characterized S.marcescens iron-uptake mechanisms (Angerer et al., 1992; Debarbieux and Wandersman, 2001). For both 7F1 and 10E5, the targeted disruption of the homologous genes in P.aeruginosa PA14 resulted in a marked reduction of virulence (Figure 8). Taken together, these observations strongly support the notion that it is the identified insertion that is causal for the attenuation in the virulence in the different S.marcescens mutants.
The identification of S.marcescens virulence genes will contribute to a fuller understanding of the mechanisms that underlie the infection of the nematode by Db11 and by other bacterial pathogens, including PA14 as illustrated above. It may have a broader relevance as, for example, the locus disrupted in 8H1 is homologous to the S.typhimurium mgtCB locus which lies within a Salmonella pathogenicity island (SPI-3) and is necessary for full virulence during the infection of mice (Blanc-Potard et al., 1999). A homologue for the gene disrupted in 7D1/22D9 has only been described for S.typhimurium and Y.pestis. Interestingly, in Db11 this gene appears to have undergone a local duplication, resulting in tandem copies (see Supplementary data). In many bacterial pathogens chromosomally encoded virulence factors are clustered. Comparisons with the genome sequences of other species suggest that the genes disrupted in 8E2, 8G1 and 20C2 may lie close together on the Db11 chromosome (see Supplementary data), conceivably within a pathogenicity island. The confirmation of such a possibility must await the determination of the complete Db11 genome sequence (see http://www.sanger.ac.uk/Projects/S_marcescens/).
A previous screen using C.elegans led to the identification of eight P.aeruginosa PA14 genes necessary for full virulence (Tan et al., 1999). Of these, five encode regulators involved in quorum sensing and/or two-component signal transduction (e.g. GacA). In the current screen, only two of the 21 characterized mutants, 8G1 and 7E7, would appear to have a defect in a regulatory system, containing insertions within homologues of the two-component sensor baeS and of galR, the repressor of the gal operon, respectively. While neither screen is saturated, this may reflect the difference between the screening protocols. It could also reflect an intrinsic difference between the two bacterial species. For example, Db11 does not produce any detectable N-acyl homoserine lactone (G.Salmond, personal communication), the most frequent meditator of quorum sensing. The P.aeruginosa gene gacA has recently been shown to be essential for biofilm formation (Parkins et al., 2001). The formation of biofilms by pathogens is often linked to their virulence during infection. Db11 is capable of forming biofilms but, as no biofilm-deficient mutants were present in the bank screened (J.M.Ghigo, personal communication), we were unable to address the role of biofilm formation in the infection and killing of C.elegans by S.marcescens.
Biochemical assays of lipase and protease activities, factors known to contribute to virulence (Hejazi and Falkiner, 1997), revealed that the mutants were indistinguishable from the wild-type strain (data not shown). Thus, as previously noted for PA14 (Tan et al., 1999), screening uniquely on the basis of a defect in such known pathogenicity-related factors would not have identified the mutants described here, emphasizing the utility of in vivo screening. Finally, we showed that the hemolysin operon previously found to be necessary for the colonization of the rat urinary tract (Marre et al., 1989) is also required for the full virulence of S.marcescens during a clinically relevant murine lung infection model. This clearly demonstrates the utility of the C.elegans–Db11 model for the identification of S.marcescens virulence factors important for the infection of a broad range of hosts.
Materials and methods
Strains and media
The S.marcescens strains Db11 and Db1140 were originally isolated by H.Boman (Flyg et al., 1980; Flyg and Xanthopoulos, 1983). Strain 2170 (Suzuki et al., 2001) was obtained from T.Watanabe (Niigata, Japan), ATCC 274 (Rius et al., 1994), Sma 3, Sma 12 and Sma 13 (1047, 3888 and 0028, respectively, in the collection of H.Auken) were obtained from G.Salmond (Cambridge, UK) and SM365 [also known as W225 (V.Braun, personal communication)] was a gift from L.Debarbieux (Paris, France). The strain Db11-GFP was obtained by conjugation with XL-1 Blue (Stratagene) carrying the plasmid pUFR-GFP (a gift from Dr W.Deng, University of British Columbia). Pseudomonas aeruginosa PA14 (Rahme et al., 1995) was a gift from M.W.Tan (Stanford, CA), E.coli OP50 was obtained from the Caenorhabditis Genetic Center (CGC); its fluorescent derivative OP50-GFP is described elsewhere (Labrousse et al., 2000). The complete medium for bacterial culture and maintenance was LB with appropriate antibiotics when required.
The wild-type N2 (Bristol) strain and the conditional sterile fer-15(b26) II were obtained from the CGC. They were cultured as described previously (Sulston and Hodgkin, 1988).
Construction of insertional S.marcescens Db11 mutants by conjugation
Both donor (E.coli S17–1 λpir/pUT-Cm) and recipient (S.marcescens Db11; TetR, KanR, SmR) were grown overnight at 37°C in LB (in the presence of chloramphenicol for the donor strain). One millilitre of each culture was centrifuged and pellets were washed twice in LB and resuspended in 1 ml of LB. One hundred microlitres of each resuspended culture was mixed together, spread onto a nitrocellulose filter on an LB agar plate and incubated for 6 h at 37°C. Bacteria were scraped off the filter in 1 ml phosphate-buffered saline (PBS). Dilutions of 10–2 and 10–3 were plated onto LB agar supplemented with 10 µg/ml Tet (to kill the donor strain) and 30 µg/ml Cm (to select the transconjugants) and were incubated for 48 h at 30°C. Two thousand CmR clones were retrieved, pooled and frozen down in 10% glycerol–LB. The library was stored at –80°C.
Caenorhabditis elegans screen
The pooled bank of 2000 CmR clones was replated and 2300 clones were picked. Individual Db11 mini-Tn5 Cm mutants were inoculated into 200 µl of LB medium in 96-well plates (supplemented with 30 µg/ml Cm) and incubated for 8 h at 37°C. Fifty microlitres of each culture was spread on NGM agar previously deposited into 24-well plates and incubated for 12 h at 37°C. After 1 h at room temperature, each well was seeded with ∼100 L1 stage hermaphrodite fer-15 worms. Plates were incubated at 25°C and examined for live worms each day. In the control plates inoculated with the parental strain Db11, all the worms were dead after 8–10 days. Each clone was tested four times in a preliminary screen. Among the 2300 clones tested in this way, 11 were clearly attenuated in their virulence as they supported the growth and survival of worms for at least 15 days. A further 119 clones were judged as potentially attenuated, as they supported the growth of at least one individual worm for a minimum of 13 days in at least one test. These 119 clones were subjected to a second round of screening during which each was again tested four times. Those that supported the growth and survival of worms for at least 13 days (in at least one test) or for 12 days (in at least two experiments), or those for which from the four independent trials the sum of [maximal survival (days)—10 days] was greater than 3 days were selected (a total of a further 12 clones; see Supplementary data). The survival of worms on each of the 23 chosen clones was then measured to determine the precise time course of infection.
Caenorhabditis elegans killing assay
Assays of S.marcescens killing were performed as previously described (Pujol et al., 2001). Briefly, five 60 mm NGM plates were inoculated with a drop of an overnight culture of Db11, or a specific clone, and incubated at 37°C for 8–10 h. Plates were allowed to cool to room temperature and seeded with L4 stage hermaphrodite N2 worms (10 per plate). Plates were then incubated at 25°C and scored each day for live worms. At least two replicates were performed for each selected clone. Worms were transferred to new plates each day. A worm was considered dead when it no longer responded to touch. Worms that died as a result of becoming stuck to the wall of the plate were excluded from the analysis. To study the influence of worm age on the infection by Db11, worms were cultivated on OP50 for different lengths of time before transfer to Db11.
Assaying the pathogenicity associated with dead bacteria
Bacteria were grown overnight in LB at 37°C. The culture was centrifuged for 15 min at 6000 r.p.m. and the supernatant was passed through 0.22 µm filters (Millipore). When required, the bacteria in the cell pellet were killed by heating at 70°C for 30 min. NGM plates were inoculated with a drop of the cell pellet resuspended either at the original cell density in the case of the live bacteria or at 10 times the original density for the heat-killed bacteria.
Shifting experiments
L4 hermaphrodite N2 worms grown on OP50 were transferred to Db11 plates. At selected times, worms were washed once with Thimerosal (Sigma; 0.1% w/v in M9) to kill the bacteria adhering to the surface and then three times in M9 buffer. Worms were then transferred to plates containing either OP50 or Db11. Worm mortality was scored as described above. To determine the effect of Db11 upon the grinder of the nematode, L4 hermaphrodite N2 worms were deposited on NGM plates containing Db11 and allowed to feed at 25°C for 1–4 h. Then they were transferred to NGM plates without bacteria for 15 min and deposited on plates containing OP50-GFP for 5 min. Finally, they were harvested and deposited in a drop of M9 on a 2% agar pad and observed using a fluorescent videomicroscope (DMIRBE, Leica).
Drosophila strain and infections
Cinnabar brown flies were reared on a standard cornmeal medium. For infections, overnight S.marcescens cultures were diluted and grown in LB medium to an OD (595 nm) of 0.8. The culture was then diluted 1:100 in LB and kept on ice. A pulled glass needle was filled with this bacterial suspension. Next, 4.6 nl of the diluted suspension, corresponding to 50–100 bacteria, were injected into the thorax of each fly using a Nanoject II injector (Drummond). As a control, 4.6 nl of the bacterial suspension was injected into a drop of LB, plated on LB agar plates and counted after overnight incubation at 37°C. In each experiment, batches of at least 20 females were injected. Flies that did not recover from the injection procedure were discarded. The viability of the flies was assessed at 20°C over a period of at least 24 h. In all experiments, Db11 was injected as a positive control; LB was injected in one batch of flies as a negative control. Each clone was tested at least twice.
Cellular cytotoxicity assay
Immortalized human bronchial epithelial cells (16HBE14o–) (Cozens et al., 1994) were seeded at 104 cells/well in 10 µl of MEM supplemented with 5% fetal calf serum and antibiotics in 96-well plates and incubated for 16 h at 37°C. The MEM was then replaced with RPMI 1640 medium without antibiotics, fetal calf serum or phenol red but with 25 mM HEPES, and incubated at 37°C for 4 h before infection. Bacteria were cultivated overnight with appropriate antibiotics and then seeded in fresh LB. When they were in exponential phase, the bacteria were pelleted, washed, resuspended in RPMI 1640 and added to the cultured 16HBE14o– cells at a multiplicity of infection of 20. After 2 h, the cytotoxicity was determined by measuring the release of the cytosolic enzyme lactate dehydrogenase (LDH) into the medium using the Cytotoxicity Detection Kit (Roche). The cytotoxicity index was calculated as the ratio of the average experimental values obtained for each strain (less the values obtained without bacteria) to the value obtained for cells treated with 1% Triton X-100. Each strain was tested in triplicate in each experiment and experiments were repeated four times.
Murine respiratory tract infection
For intranasal infections, bacteria were grown for 16 h in LB medium, harvested and diluted in 0.01 M PBS pH 7.5. BALB/c mice (8–10 weeks old; Charles River, Germany) were anaesthetized with a mixture of ketamine hydrochloride and xylazine hydrochloride and subsequently inoculated with ∼2 × 107 c.f.u. in 30 µl into both nostrils. Mortality of animals was monitored daily. All experiments were approved by the local authorities.
Molecular analysis of mini-Tn5 Cm mutants
Genomic DNA from each clone was prepared using the Nucleospin kit (Macherey–Nagel) or with the standard chloroform–isoamyl alcohol extraction technique. This DNA was digested with PstI (which does not cut the transposon) and the fragments were cloned into pSP72 (Promega). XL-1 Blue cells were thermotransformed with the product of the ligation and selected at 37°C on LB agar Cm (30 µg/ml), and the recombinant plasmids were purified by WizardPrep (Promega). The genomic fragments were partially sequenced using primers JEP 131 (5′-CGA ACT TGT GTA TAA GAG TCA G-3′) and JEP 132 (5′-CTA GGC GGC CAG ATC TGA TCA A-3′). Sequences were analysed using the BLAST suite of programs (Altschul et al., 1990) at GenBank (http://www.ncbi.nlm.nih.gov/BLAST/).
LPS analysis
Extracts of bacteria were prepared as described (Coyne et al., 1994). The samples were analysed by Tricine–SDS–PAGE gel (Schägger and von Jagow, 1987) and the LPS was silver stained (Tsai and Frasch, 1982).
Construction of the P.aeruginosa PA14 mutants M151 and M3713
Internal DNA fragments from PA0151 and PA3713 genes were PCR amplified using the oligonucleotide pairs 151O1/151O2 (5′-GCTA CCG CTT CAG CCA GGC TGC CGC-3′/5′-GTG GTC GGC GGT GGC GGT GAA ATT G-3′) and 3713O1/3713O2 (5′-CAG ATG GGC TGG GAG AAG AAG ACC T-3′/5′-AGC AGG TAG TCG CGG TAG CTG GTC T-3′), respectively. The fragments were cloned into pCR2.1 (Invitrogen) yielding pCRM151 and pCRM3713, respectively. These plasmids, which do not replicate in P.aeruginosa, were introduced into PA14 by electroporation. The insertional mutants M151 and M3713 were selected on Pseudomonas isolation agar plates containing carbenicillin (500 µg/ml). The location of the plasmid insertion within the chromosome of the mutants was checked by PCR with appropriate oligonucleotide pairs [the universal or reverse M13 primers with V151 (5′-AAA AGC GAC CGA CAC GAG CAA CCC-3′) and V3713 (5′-AGC AAT CTG TGA ACA TGG CTC CCC-3′) for M151 and M3713, respectively].
Statistical analysis
One-sided rank log tests [within the PRISM (Graphpad) software package] were used to assess the similarity between two groups. Values of p < 0.05 were considered statistically significant. The mean lifespan was calculated using Microsoft Excel.
DDBJ/EMBL/GenBank accession numbers
The sequences derived from the mutants, with the exception of 21C4, have been deposited in the DDBJ/EMBL/GenBank database with the accession Nos AJ549760–AJ549799.
Supplementary data
Supplementary data are available at http://www.ciml.univ-mrs.fr/EWBANK_jonathan/SuppMat/Screen/Kurz.html.
Acknowledgments
Acknowledgements
We thank S.Garvis, E.Pradel and N.Pujol for critical comments and discussions, L.Carta, L.Debarbieux, J.M.Ghigo, G.Salmond and T.Watanabe for gifts of strains and/or communicating unpublished results, E.Termine for DNA sequencing and Ulrich Voegl for putting J.J.E. and I.S. in contact. Nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources. This work was supported by the Université de la Méditerranée, institutional grants from the CNRS and INSERM, a MENRT PRFMMIP grant and a CNRS ATIPE to J.J.E. C.L.K. received an MENRT predoctoral fellowship.
References
- Aballay A. and Ausubel,F.M. (2002) Caenorhabditis elegans as a host for the study of host–pathogen interactions. Curr. Opin. Microbiol., 5, 97–101. [DOI] [PubMed] [Google Scholar]
- Alexandrakis G., Alfonso,E.C. and Miller,D. (2000) Shifting trends in bacterial keratitis in south Florida and emerging resistance to fluoroquinolones. Ophthalmology, 107, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Angerer A., Klupp,B. and Braun,V. (1992) Iron transport systems of Serratia marcescens. J. Bacteriol., 174, 1378–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc-Potard A.B., Solomon,F., Kayser,J. and Groisman,E.A. (1999) The SPI-3 pathogenicity island of Salmonella enterica. J. Bacteriol., 181, 998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun V. (2001) Iron uptake mechanisms and their regulation in pathogenic bacteria. Int. J. Med. Microbiol., 291, 67–79. [DOI] [PubMed] [Google Scholar]
- Braun V. and Braun,M. (2002) Active transport of iron and siderophore antibiotics. Curr. Opin. Microbiol., 5, 194–201. [DOI] [PubMed] [Google Scholar]
- Carbonell G.V., Alfieri,A.F., Alfieri,A.A., Vidotto,M.C., Levy,C.E., Darini,A.L. and Yanaguita,R.M. (1997) Detection of cytotoxic activity on Vero cells in clinical isolates of Serratia marcescens. Braz. J. Med. Biol. Res., 30, 1291–1298. [DOI] [PubMed] [Google Scholar]
- Carbonell G.V., Della Colleta,H.H., Yano,T., Darini,A.L., Levy,C.E. and Fonseca,B.A. (2000) Clinical relevance and virulence factors of pigmented Serratia marcescens. FEMS Immunol. Med. Microbiol., 28, 143–149. [DOI] [PubMed] [Google Scholar]
- Couillault C. and Ewbank,J.J. (2002) Diverse bacteria are pathogens of Caenorhabditis elegans. Infect. Immun., 70, 4705–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne M.J. Jr, Russell,K.S., Coyle,C.L. and Goldberg,J.B. (1994) The Pseudomonas aeruginosa algC gene encodes phosphoglucomutase, required for the synthesis of a complete lipopolysaccharide core. J. Bacteriol., 176, 3500–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozens A.L., Yezzi,M.J., Kunzelmann,K., Ohrui,T., Chin,L., Eng,K., Finkbeiner,W.E., Widdicombe,J.H. and Gruenert,D.C. (1994) CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol., 10, 38–47. [DOI] [PubMed] [Google Scholar]
- Darby C., Hsu,J.W., Ghori,N. and Falkow,S. (2002) Caenorhabditis elegans: plague bacteria biofilm blocks food intake. Nature, 417, 243–244. [DOI] [PubMed] [Google Scholar]
- Debarbieux L. and Wandersman,C. (2001) Folded HasA inhibits its own secretion through its ABC exporter. EMBO J., 20, 4657–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lorenzo V., Herrero,M., Jakubzik,U. and Timmis,K.N. (1990) Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing and chromosomal insertion of cloned DNA in Gram-negative eubacteria. J. Bacteriol., 172, 6568–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewbank J.J. (2002) Tackling both sides of the host–pathogen equation with Caenorhabditis elegans. Microbes Infect., 4, 247–256. [DOI] [PubMed] [Google Scholar]
- Flyg C. and Xanthopoulos,K.G. (1983) Insect pathogenic properties of Serratia marcescens. Passive and active resistance to insect immunity studied with protease-deficient and phage-resistant mutants. J. Gen. Microbiol., 129, 453–464. [DOI] [PubMed] [Google Scholar]
- Flyg C., Kenne,K. and Boman,H.G. (1980) Insect pathogenic properties of Serratia marcescens: phage-resistant mutants with a decreased resistance to Cecropia immunity and a decreased virulence to Drosophila. J. Gen. Microbiol., 120, 173–181. [DOI] [PubMed] [Google Scholar]
- Gan Y.H., Chua,K.L., Chua,H.H., Liu,B., Hii,C.S., Chong,H.L. and Tan,P. (2002) Characterization of Burkholderia pseudomallei infection and identification of novel virulence factors using a Caenorhabditis elegans host system. Mol. Microbiol., 44, 1185–1197. [DOI] [PubMed] [Google Scholar]
- Grimont P.A. and Grimont,F. (1978) The genus Serratia. Annu. Rev. Microbiol., 32, 221–248. [DOI] [PubMed] [Google Scholar]
- Haddy R.I., Mann,B.L., Nadkarni,D.D., Cruz,R.F., Elshoff,D.J., Buendia,F.C., Domers,T.A. and Oberheu,A.M. (1996) Nosocomial infection in the community hospital: severe infection due to Serratia species. J. Fam. Pract., 42, 273–277. [PubMed] [Google Scholar]
- Hejazi A. and Falkiner,F.R. (1997) Serratia marcescens. J. Med. Microbiol., 46, 903–1012. [DOI] [PubMed] [Google Scholar]
- Hertle R. (2002) Serratia marcescens hemolysin (ShlA) binds artificial membranes and forms pores in a receptor-independent manner. J. Membr. Biol., 189, 1–14. [DOI] [PubMed] [Google Scholar]
- Hertle R., Hilger,M., Weingardt-Kocher,S. and Walev,I. (1999) Cytotoxic action of Serratia marcescens hemolysin on human epithelial cells. Infect. Immun., 67, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch J.A. (2000) Two-component and phosphorelay signal transduction. Curr. Opin. Microbiol., 3, 165–170. [DOI] [PubMed] [Google Scholar]
- Kitagawa M., Miyakawa,M., Matsumura,Y. and Tsuchido,T. (2002) Escherichia coli small heat shock proteins, IbpA and IbpB, protect enzymes from inactivation by heat and oxidants. Eur. J. Biochem., 269, 2907–2917. [DOI] [PubMed] [Google Scholar]
- Labrousse A., Chauvet,S., Couillault,C., Kurz,C.L. and Ewbank,J.J. (2000) Caenorhabditis elegans is a model host for Salmonella typhimurium. Curr. Biol., 10, 1543–1545. [DOI] [PubMed] [Google Scholar]
- Lerouge I. and Vanderleyden,J. (2002) O-antigen structural variation: mechanisms and possible roles in animal/plant–microbe interactions. FEMS Microbiol. Rev., 26, 17–47. [DOI] [PubMed] [Google Scholar]
- Mallo G.V., Kurz,C.L., Couillault,C., Pujol,N., Granjeaud,S., Kohara,Y. and Ewbank,J.J. (2002) Inducible antibacterial defence system in C.elegans. Curr. Biol., 12, 1209–1214. [DOI] [PubMed] [Google Scholar]
- Marre R., Hacker,J. and Braun,V. (1989) The cell-bound hemolysin of Serratia marcescens contributes to uropathogenicity. Microb. Pathog., 7, 153–156. [DOI] [PubMed] [Google Scholar]
- Nahlik M.S., Fleming,T.P. and McIntosh,M.A. (1987) Cluster of genes controlling synthesis and activation of 2,3-dihydroxybenzoic acid in production of enterobactin in Escherichia coli. J. Bacteriol., 169, 4163–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondraczek R., Hobbie,S. and Braun,V. (1992) In vitro activation of the Serratia marcescens hemolysin through modification and complementation. J. Bacteriol., 174, 5086–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Quinn A.L., Wiegand,E.M. and Jeddeloh,J.A. (2001) Burkholderia pseudomallei kills the nematode Caenorhabditis elegans using an endotoxin-mediated paralysis. Cell. Microbiol., 3, 381–394. [DOI] [PubMed] [Google Scholar]
- Palomar J., Montilla,R., Fuste,M.C. and Vinas,M. (1993) The role of O-antigen in susceptibility of Serratia marcescens to non-immune serum. Microbios, 76, 189–196. [PubMed] [Google Scholar]
- Palomar J., Leranoz,A.M. and Vinas,M. (1995) Serratia marcescens adherence: the effect of O-antigen presence. Microbios, 81, 107–113. [PubMed] [Google Scholar]
- Parkins M.D., Ceri,H. and Storey,D.G. (2001) Pseudomonas aeruginosa GacA, a factor in multihost virulence, is also essential for biofilm formation. Mol. Microbiol., 40, 1215–1226. [DOI] [PubMed] [Google Scholar]
- Poole K. and Braun,V. (1988a) Influence of growth temperature and lipopolysaccharide on hemolytic activity of Serratia marcescens. J. Bacteriol., 170, 5146–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole K. and Braun,V. (1988b) Iron regulation of Serratia marcescens hemolysin gene expression. Infect. Immun., 56, 2967–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole K., Schiebel,E. and Braun,V. (1988) Molecular characterization of the hemolysin determinant of Serratia marcescens. J. Bacteriol., 170, 3177–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol N. et al. (2001) A reverse genetic analysis of components of the Toll signalling pathway in Caenorhabditis elegans. Curr. Biol., 11, 809–821. [DOI] [PubMed] [Google Scholar]
- Rahme L.G., Stevens,E.J., Wolfort,S.F., Shao,J., Tompkins,R.G. and Ausubel,F.M. (1995) Common virulence factors for bacterial pathogenicity in plants and animals. Science, 268, 1899–1902. [DOI] [PubMed] [Google Scholar]
- Rius N., Sole,M., Francia,A. and Loren,J. (1994) Buffering capacity of pigmented and nonpigmented strains of Serratia marcescens. Appl. Environ. Microbiol., 60, 2152–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger H. and von Jagow,G. (1987) Tricine–sodium dodecyl sulfate–polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem., 166, 368–379. [DOI] [PubMed] [Google Scholar]
- Schiebel E. and Braun,V. (1989) Integration of the Serratia marcescens haemolysin into human erythrocyte membranes. Mol. Microbiol., 3, 445–453. [DOI] [PubMed] [Google Scholar]
- Schwyn B. and Neilands,J.B. (1987) Universal chemical assay for the detection and determination of siderophores. Anal. Biochem., 160, 47–56. [DOI] [PubMed] [Google Scholar]
- Sugiyama T., Kido,N., Kato,Y., Koide,N., Yoshida,T. and Yokochi,T. (1998) Generation of Escherichia coli O9a serotype, a subtype of E.coli O9, by transfer of the wb* gene cluster of Klebsiella O3 into E.coli via recombination. J. Bacteriol., 180, 2775–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston J. and Hodgkin,J. (1988) Methods. In Wood,W.B. (ed.), The Nematode Caenorhabditis elegans. Cold Spring Harbor Laboratory Press, Plainview, NY, pp. 587–606.
- Suzuki K., Uchiyama,T., Suzuki,M., Nikaidou,N., Regue,M. and Watanabe,T. (2001) LysR-type transcriptional regulator ChiR is essential for production of all chitinases and a chitin-binding protein, CBP21, in Serratia marcescens 2170. Biosci. Biotechnol. Biochem., 65, 338–347. [DOI] [PubMed] [Google Scholar]
- Tan M.W. and Ausubel,F.M. (2000) Caenorhabditis elegans: a model genetic host to study Pseudomonas aeruginosa pathogenesis. Curr. Opin. Microbiol., 3, 29–34. [DOI] [PubMed] [Google Scholar]
- Tan M.W., Rahme,L.G., Sternberg,J.A., Tompkins,R.G. and Ausubel,F.M. (1999) Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P.aeruginosa virulence factors. Proc. Natl Acad. Sci. USA, 96, 2408–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson N.R., Crow,M.A., McGowan,S.J., Cox,A. and Salmond,G.P. (2000) Biosynthesis of carbapenem antibiotic and prodigiosin pigment in Serratia is under quorum sensing control. Mol. Microbiol., 36, 539–556. [DOI] [PubMed] [Google Scholar]
- Tsai C.M. and Frasch,C.E. (1982) A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal. Biochem., 119, 115–119. [DOI] [PubMed] [Google Scholar]
- Verkhovskaya M.L., Barquera,B. and Wikstrom,M. (2001) Deletion of one of two Escherichia coli genes encoding putative Na+/H+ exchangers (ycgO) perturbs cytoplasmic alkali cation balance at low osmolarity. Microbiology, 147, 3005–3013. [DOI] [PubMed] [Google Scholar]
- Wyckoff E.E., Smith,S.L. and Payne,S.M. (2001) VibD and VibH are required for late steps in vibriobactin biosynthesis in Vibrio cholerae. J. Bacteriol., 183, 1830–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]