

Abstract
This perspective discusses the ways that advanced paramagnetic resonance techniques, namely electron-nuclear double resonance (ENDOR) and electron spin-echo envelope modulation (ESEEM) spectroscopies, can help us understand how metal ions function in biological systems.
The invitation to write this article explained, “Perspective articles are intended to survey, from the author's distinctive perspective, some frontier aspects of the featured field and to highlight important recent developments and challenges.” Within that definition, I shall discuss the ways that advanced paramagnetic resonance techniques, namely electron-nuclear double resonance (ENDOR) spectroscopy (1) and its junior partner electron spin-echo envelope modulation (ESEEM) spectroscopy (2), help us understand how metal ions function in biological systems and provide information in all of the critical categories listed in Table 1. In these early days of proteomics, where the primary sequence of all proteins in an organism are knowable in principle, and increasingly in fact, and where large-scale efforts are under way to determine the resting-state x-ray crystal structures of these proteins, this article is a representative response to the broader question: what roles do spectroscopies play? The answer, of course, is many, and important ones.
Table 1.
Spectroscopically retrievable information
| Enzyme mechanism | |
| 3D structure | |
| Metal-ion | |
| Coordination geometry | |
| Valence | |
| Identification | |
| Protein dynamics | |
| Electronic and magnetic properties |
Background
Key information about the composition, bonding, and structure of a paramagnetic metal center can be obtained by analysis of the electron-nuclear hyperfine and nuclear quadrupole couplings (3) of nuclei associated with endogenous and exogenous metal ligands, enzyme-bound substrates, inhibitors, and products, as well as the metal ions themselves. In principle, these couplings can be derived from splittings observable in a center's EPR spectrum. For most metallo-biomolecules, however, a variety of factors make these interactions unresolvable for most (or any) nuclei, and the chemical information is lost.
ENDOR and ESEEM spectroscopies (3–7) retrieve the missing information. They provide an NMR spectrum of those nuclei that interact with the electron spin of the paramagnetic center, and such spectra display orders-of-magnitude-better resolution than the EPR spectrum of the center. Resolved features in these NMR spectra are characterized by frequencies, called ENDOR frequencies, that give the hyperfine coupling (and quadrupole coupling for nuclear spins greater than one-half). To derive bonding and geometric information requires a set of measurements taken over a range of orientations of the molecular frame relative to the external field. Although the samples used in ENDOR/ESEEM studies of metalloenzymes almost always are frozen solutions (but see, for example, ref. 8), and thus contain a random distribution of all active-site orientations, we have shown that the desired information nonetheless can be obtained through analysis of a 2D dataset comprised of “orientation-selective” ENDOR spectra collected at fields (g values) across the EPR envelope of frozen-solution (5, 9).
ENDOR detects the actual NMR transitions as induced by an incident radio-frequency (rf) field. The field is applied, in conjunction with a microwave field, to a sample in the cavity of an EPR spectrometer; the NMR transitions are not detected directly, but rather through a change in the EPR signal intensity. In short, one has “EPR-detected” NMR, hence the term “double resonance.” In an ESEEM experiment the amplitude of an electron spin echo is modulated at the ENDOR frequencies without the application of a rf field. It can be argued that the bioinorganic community became fully aware of these techniques through the seminal ESEEM studies of Cu(II) centers by Peisach and Mims (10), even though they were preceded by bioinorganic applications of ENDOR to both iron–sulfur proteins (11) and hemes (12).
These methods are inherently broad-banded, and as a result it is possible to detect ENDOR or ESEEM signals from every type of nuclear spin. Thus, with proper isotopic labeling it is possible to characterize every atom associated with a paramagnetic center (5, 13); because the electron spin is the “detector,” the methods are more sensitive than conventional NMR and see “nonstandard” nuclei (e.g., 2H, 14N, 17O, 57Fe, and 95,97Mo) as easily as “standard” nuclei (1H, 13C, and 15N). With these benefits goes the additional virtue of selectivity. Only nuclei that have a hyperfine interaction with the electron-spin system being monitored give an ENDOR signal. For example, it is possible to examine 57Fe resonances from one metal cluster without interference from other clusters, as in nitrogenase (14) and from a cluster in the presence of excess inorganic Fe (see below). The obvious limitation to paramagnetic resonance methods, of course, is that they require a paramagnetic center, be it metal ion or radical. However, as illustrated below, even this can be overcome in certain circumstances.
There are reasons to emphasize ENDOR over ESEEM as the tool of “first resort,” and this article does, although not exclusively.
Information Recovered
These methods contribute detailed information of all of the types listed in Table 1, but most importantly do so not only for resting-state metal centers, but also for reactive intermediates (as we shall see), which seldom are amenable to x-ray diffraction methods (15). The information indeed includes the determination of such electronic properties as metal-ion valencies (in resting enzymes and their reactive intermediates): recent examples include the dinuclear centers in [Ni,Fe]hydrogenase (16) and ribonucleotide reductase (17), and perhaps most dramatically, the catalytic molybdenum–iron cofactor of nitrogenase (14). However, it includes the other types of information of Table 1, too, ranging from the location of a single electron (hole) and a single proton in a high-valent intermediate (18), a task for which ENDOR spectroscopy is perhaps uniquely suited, to information about dynamics (19), an area where ENDOR/ESEEM spectroscopies might have been thought unsuitable. As a result of all this, in favorable cases these methods can definitively characterize elusive enzymic mechanisms, the “highest” goal of Table 1.
The use of multinuclear continuous wave and pulsed ENDOR techniques in addressing bioinorganic problems can be illustrated by two, representative, recent examples: one involves determining the coordination sphere of catalytic metal ion and the 3D binding geometry of a key reactant; the other identifies the reactive intermediate in what is perhaps the most, and certainly one of the most, extensively studied enzymic reactions. These (and many of the citations above) are taken from our own work (a more extensive discussion of both the techniques and their applications can be found in ref. 20), not least because doing so ensured that the deadline set by the editors could in fact be met. A third example describes the first ENDOR and ESEEM study of metal ions associated with nucleic acids.
Dual Role of the Catalytically Active [4Fe-4S]+ Cluster of PFL-AE.
In recent years, the “radical S-adenosylmethionine” superfamily of enzymes has been found to use Fe-S clusters and S-adenosylmethionine (AdoMet) to generate catalytically essential radicals (23, 24). A key mechanistic question regarding this family is the role of the cluster in this process. In particular, the [4Fe-4S] cluster of enzymes in the Fe-S/AdoMet family has a “unique” iron site that is not coordinated to the enzyme by a cysteinyl sulfur; does this Fe have a catalytic function, as is the case for aconitase (25)?
In collaboration with Joan Broderick and coworkers (21, 22), we used EPR and 35-GHz pulsed ENDOR spectroscopy to explore the role of this cluster in the pyruvate formate-lyase (PFL) activating enzyme (PFL-AE), a representative member of this family that generates a stable glycyl radical on the enzyme PFL. In doing so we generated information about both the AdoMet binding geometry and the coordination sphere of the unique cluster Fe, thereby discovering that the cluster plays a dual role.
Three-dimensional structure information.
2H and 13C pulsed ENDOR spectroscopy was performed on [4Fe-4S]+-PFL-AE (S = 1/2) in the presence of AdoMet labeled at the methyl position with either 2H or 13C (denoted [1+/AdoMet]) (21). The observation of substantial 2H and 13C hyperfine couplings from the labels showed that AdoMet binds adjacent to the 4Fe cluster. The cofactor was found to bind in the same geometry to both the 1+ and 2+ states through ENDOR measurements on the [4Fe-4S]2+ state of PFL-AE, which had been made EPR-active through cryoreduction of the frozen [4Fe-4S]2+/AdoMet complex to form the reduced state (denoted [2+/AdoMet]red) trapped in the structure of the oxidized state.
Modeling of the through-space electron-nuclear dipolar interaction between the cluster electron spin and the methyl-13C and -2H of AdoMet, as derived from orientation-selective, 2D ENDOR datasets, showed that the shortest distance between an AdoMet methyl hydrogen and an iron of the cluster is ≈3.7(2) Å, with a distance of ≈4.9(6) Å from the methyl carbon to this iron. Most intriguingly, the analysis further disclosed a through-bond, isotropic contribution to the 13C interaction, which requires overlap between orbitals on the cluster and on AdoMet. The most plausible origin is a dative interaction between a negatively charged sulfide of the cluster and the positively charged sulfur of AdoMet, rather than with the unique Fe of the cluster (Fig. 1).
Figure 1.

Representation of the proposed interactions of AdoMet with the [4Fe-4S] cluster of PFL-AE, as derived from 2H, 13C, 14,15N, and 17O ENDOR spectroscopy (21, 22).
Coordination sphere.
This mechanism includes no role for the “unique” cluster iron. However, 35-GHz pulsed ENDOR spectroscopic studies of the [4Fe-4S]+/AdoMet complex labeled specifically with 17O and 13C in the carboxyl group and 15N in the amino group of the methionine fragment showed that both groups are coordinated to the unique iron of the [4Fe-4S] cluster in a classical five-membered-ring N/O chelate (Fig. 1; ref. 22).
Thus, the two roles of the cluster: chelation by the unique Fe “anchors” the nonreacting end of AdoMet, thereby positioning the reactive sulfonium linkage for electron transfer to AdoMet via the sulfide–sulfonium interaction (Fig. 1), which leads to cleavage of the sulfonium–adenosyl bond and subsequent radical chemistry.
Dioxygen Activation by Heme Enzymes: P450cam.
This case describes the determination of the catalytically active intermediate in the hydroxylation reaction carried out by cytochrome P450cam (26).
Dioxygen activation is one of the major functions of biological metal centers, and ENDOR spectroscopy has been used to study heme, nonheme iron (27), diiron (28), and noniron dioxygen-activating enzymes (29). Heme oxygenations in particular are perhaps the most widely studied bioinorganic reactions: during the 1990s, roughly one paper on cytochromes P450 alone was published every five hours (Martin Newcomb, personal communication). The chemically relevant portion of the enzymatic cycle for the monoxygenation of substrate, RH, begins with one-electron reduction of the oxy-ferroheme [O2Fe(P)] enzyme, and ends with the hydroxylated product, water, and the ferriheme state,
 |
1 |
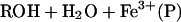 |
It had long been thought that heme hydroxylations included the intermediates sketched in Fig. 2 (30), with the rate-limiting step for hydroxylation being the reduction of the dioxygen-bound ferrous state. However, none of the three intermediates in Fig. 2 had been detected or characterized before our work, and each of the three is potentially capable of reacting with a substrate (31). Roman Davydov of my laboratory therefore initiated a program designed to supply the missing information. We use cryogenic γ-irradiation (32) of an oxy-ferrous enzyme to inject the electron that initiates the catalytic process (Eq. 1, Fig. 2). Subsequent stepwise annealing stages permit an enzyme to traverse its catalytic cycle, through to product formation, while the combination of EPR and ENDOR measurements provides an optimal means of characterizing the individual stages in catalysis.
Figure 2.

Potential reactive intermediates formed on one-electron reduction of and proton delivery to an oxy-ferroheme (31).
This approach has been applied to P450cam in collaboration with Steve Sligar (26), to heme oxygenase in collaboration with Masao Ikeda-Saito and coworkers (33), and to nitric oxide synthase in collaboration with Bettie Sue Siler Masters, John Dawson, and coworkers (34). The purposes of this article are best served by describing the ENDOR measurements that establish Compound I as the hydroxylating species in P450cam (26), despite the fact that neither we nor others have characterized Compound I of a P450.
The EPR and ENDOR results obtained during cryoreduction and annealing of oxy-P450cam included the following: (i) Cryoreduction generates the end-on, “peroxo” intermediate, which converts at cryogenic temperatures to the hydroperoxo-ferriheme species (Fig. 2). (ii) On brief annealing at ≈200 K, the hydroperoxo-ferriheme state disappears while the 5-exo-hydroxycamphor enzymatic product forms quantitatively and stereospecifically, with no spectroscopic evidence for the buildup of a Compound I intermediate during this process. Fig. 3 presents the two alternate hydroxylation pathways that begin with the hydroperoxo-ferriheme: direct reaction by this intermediate (left pathway) and intervening formation of Compound I as the (undetected) reactive species (right pathway).
Figure 3.

The two alternate hydroxylation pathways for P450cam that begin with the hydroperoxo-ferriheme: direct reaction by this intermediate on the left and intervening formation of Compound I as the (undetected) reactive species on the right. The 1H ENDOR signal from H(C5) of product showed that the reaction follows the right pathway (26).
The actual pathway was disclosed by 1,2H ENDOR data from the primary product state. The observation of two signals with large hyperfine couplings, one from the nonexchangeable C5 proton remaining on the hydroxycamphor product and the other from the exchangeable hydroxyl, showed that the product state contains hydroxycamphor trapped in a nonequilibrium geometry, with its hydroxyl group bound to the heme ferric ion. Fig. 3 (right pathway) shows that such a primary product arises when Compound I is the reactive, hydroxylating species. The alternate mechanism, direct hydroxylation by the remote oxygen atom of a hydroperoxo-ferriheme, instead would leave a water/hydroxide bound to Fe (Fig. 3, left pathway), contrary to observation. Thus, the cryoreduction/ENDOR measurements show that Compound I is the hydroxylating intermediate in P450cam, even though it is too reactive to accumulate for direct detection, even at ≈200 K.
Catalytic Mn2+ Site of the Hammerhead Ribozyme.
Metal ions also are critical to the structure and function of RNA, yet it is quite challenging to define a precise coordination environment in RNA under solution conditions. Thus it is noteworthy that DeRose, Britt, and coworkers (35–37) recently introduced the use of ENDOR and ESEEM methods to study the catalytic metal ions of ribozymes (RNA-based enzymes).
The hammerhead ribozyme is a chemically active RNA molecule, in which an “enzyme” strand performs a site-specific phosphodiester bond cleavage reaction on a hybridized “substrate” strand (Fig. 4). It is highly activated by either a Mg2+ or a Mn2+ ion, and under appropriate conditions it binds a single Mn2+ that is critical to activity (37). ENDOR and ESEEM were used to examine the coordination sphere of this metal ion, with Mn2+GMP serving as a model. The results indicated direct coordination by a guanine nitrogen (35, 36); more recent measurements on a hammerhead ribozyme specifically 15N-labeled at the G10.1 site prove that this base is the Mn ligand (M. Vogt, C. G. Hoogstraten, S. Lahiri, C. Aznar, R. D. Britt, and V. J. DeRose, unpublished work). 1,2H and 31P ENDOR and ESEEM further give information about water coordination and inner- and outer-sphere phosphate interactions in Mn2+-containing ribozyme and nucleotide complexes (35, 37).
Figure 4.

Hammerhead ribozyme model with “enzyme” strand in gray and “substrate” strand in blue. The Mn(II) site critical to activity, as deduced from 1H, 31P, and 14,15N ENDOR/ESEEM, is displayed as Inset (35–37).
The Future?
Always dangerous to predict. However, one can envisage a continuing growth in the application of advanced paramagnetic resonance techniques to bioinorganic problems. Certainly one aspect of growth will involve technical developments. Development of multifrequency and time-domain approaches (6, 8, 38) will continue. Although it is our view that the vicinity of 35 GHz in general provides an optimum compromise between competing demands of spectral dispersion and sample handling, numerous biological problems do demand a higher frequency, and certain spectroscopic constraints will sometimes demand a lower frequency. But expansion into new areas of bioinorganic chemistry is perhaps most important. This is illustrated for proteins and nucleic acids by the examples given above.
Acknowledgments
Our experiments were performed on spectrometers built by Clark Davoust and use approaches devised by Dr. Peter Doan. Above all, they depend on the bioinorganic and biochemical wizardry of our collaborators, some of whom are named above. This work is supported by National Institutes of Health Grant HL 13531, National Science Foundation Grant MCB-9904018, and U.S. Department of Agriculture Grant 99-35305-8643.
References
- 1.Feher G. Phys Rev. 1959;114:1219–1244. [Google Scholar]
- 2.Mims W B. Proc R Soc London. 1965;283:452–457. [Google Scholar]
- 3.Abragam A, Bleaney B. Electron Paramagnetic Resonance of Transition Ions. New York: Dover; 1986. [Google Scholar]
- 4.Lowe D J. ENDOR and EPR of Metalloproteins. Austin, TX: R. G. Landes; 1995. [DOI] [PubMed] [Google Scholar]
- 5.DeRose V J, Hoffman B M. Methods Enzymol. 1995;246:554–589. doi: 10.1016/0076-6879(95)46025-x. [DOI] [PubMed] [Google Scholar]
- 6.Schweiger A, Jeschke G. Principles of Pulse Electron Paramagnetic Resonance. Oxford: Oxford Univ. Press; 2001. [Google Scholar]
- 7.Prisner T, Rohrer M, MacMillan F. Annu Rev Phys Chem. 2001;52:279–313. doi: 10.1146/annurev.physchem.52.1.279. [DOI] [PubMed] [Google Scholar]
- 8.Coremans J W A, Poluektov O G, Groenen E J J, Canters G W, Nar H, Messerschmidt A. J Am Chem Soc. 1996;118:12141–12153. [Google Scholar]
- 9.Hoffman B M, Martinsen J, Venters R A. J Magn Res. 1984;59:110–123. [Google Scholar]
- 10.Mims W B, Peisach J. Biochemistry. 1976;15:3863–3869. doi: 10.1021/bi00662a033. [DOI] [PubMed] [Google Scholar]
- 11.Fritz J, Anderson R, Fee J, Palmer G, Sands R H, Tsibris J C M, Gunsalus I C, Orme-Johnson W H, Beinert H. Biochim Biophys Acta. 1971;253:110–133. doi: 10.1016/0005-2728(71)90239-8. [DOI] [PubMed] [Google Scholar]
- 12.Scholes C P, Isaacson R A, Feher G. Biochim Biophys Acta. 1972;263:448–452. doi: 10.1016/0005-2795(72)90098-0. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman B M, DeRose V J, Doan P E, Gurbiel R J, Houseman A L P, Telser J. Biol Magn Reson. 1993;13:151–218. [Google Scholar]
- 14.Lee H-I, Hales B J, Hoffman B M. J Am Chem Soc. 1997;119:11395–11400. [Google Scholar]
- 15.Moffat K. Chem Rev (Washington, DC) 2001;101:1569–1581. doi: 10.1021/cr990039q. [DOI] [PubMed] [Google Scholar]
- 16.Huyett J E, Carepo M, Pamplona A, Franco R, Moura I, Moura J J G, Hoffman B M. J Am Chem Soc. 1997;119:9291–9292. [Google Scholar]
- 17.Sturgeon B E, Burdi D, Chen S, Huynh B H, Edmondson D E, Stubbe J, Hoffman B M. J Am Chem Soc. 1996;118:7551–7557. [Google Scholar]
- 18.Huyett J E, Doan P E, Gurbiel R, Houseman A L P, Sivaraja M, Goodin D B, Hoffman B M. J Am Chem Soc. 1995;117:9033–9041. [Google Scholar]
- 19.Florens L, Schmidt B, McCracken J, Ferguson-Miller S. Biochemistry. 2001;40:7491–7497. doi: 10.1021/bi0101188. [DOI] [PubMed] [Google Scholar]
- 20. Hoffman, B. M. (2003) Accounts Chem. Res., in press. [DOI] [PubMed]
- 21.Walsby C, Hong W, Broderick W E, Creek J, Ortillo D, Broderick J B, Hoffman B. J Am Chem Soc. 2002;124:3143–3151. doi: 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- 22.Walsby C J, Ortillo D, Broderick W E, Broderick J B, Hoffman B M. J Am Chem Soc. 2002;124:11270–11271. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 23.Cheek J, Broderick J B. J Biol Inorg Chem. 2001;6:209–226. doi: 10.1007/s007750100210. [DOI] [PubMed] [Google Scholar]
- 24.Frey P A. Annu Rev Biochem. 2001;70:121–148. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
- 25.Werst M M, Kennedy M C, Beinert H, Hoffman B M. Biochemistry. 1990;29:10526–10532. doi: 10.1021/bi00498a015. [DOI] [PubMed] [Google Scholar]
- 26.Davydov R, Makris T M, Kofman V, Werst D W, Sligar S G, Hoffman B M. J Am Chem Soc. 2001;123:1403–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- 27.Rocklin A M, Tierney D L, Kofman V, Brunhuber N M W, Hoffman B M, Christoffersen R E, Reich N O, Lipscomb J D, Que J L. Proc Natl Acad Sci USA. 1999;96:7905–7909. doi: 10.1073/pnas.96.14.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeRose V J, Liu K E, Lippard S J, Hoffman B M. J Am Chem Soc. 1996;118:121–134. [Google Scholar]
- 29.Van Camp H L, Wei Y H, Scholes C P, King T E. Biochim Biophys Acta. 1978;537:238–246. doi: 10.1016/0005-2795(78)90507-x. [DOI] [PubMed] [Google Scholar]
- 30.Sono M, Roach M P, Coulter E D, Dawson J H. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 31.Newcomb M, Shen R, Choi S-Y, Toy P H, Hollenberg P F, Vaz A D N, Coon M J. J Am Chem Soc. 2000;122:2677–2686. [Google Scholar]
- 32.Davydov R, Kuprin S, Graslund A, Ehrenberg A. J Am Chem Soc. 1994;116:11120–1128. [Google Scholar]
- 33.Davydov R, Kofman V, Fujii H, Yoshida T, Ikeda-Saito M, Hoffman B. J Am Chem Soc. 2002;124:1798–1808. doi: 10.1021/ja0122391. [DOI] [PubMed] [Google Scholar]
- 34.Davydov R, Ledbetter-Rogers A, Martasek P, Larukhin M, Sono M, Dawson J H, Masters B S, Hoffman B M. Biochemistry. 2002;41:10375–10381. doi: 10.1021/bi0260637. [DOI] [PubMed] [Google Scholar]
- 35.Hoogstraten C G, Grant C V, Horton T E, DeRose V J, Britt R D. J Am Chem Soc. 2002;124:834–842. doi: 10.1021/ja0112238. [DOI] [PubMed] [Google Scholar]
- 36.Morrissey S R, Horton T E, Grant C V, Hoogstraten C G, Britt R D, DeRose V J. J Am Chem Soc. 1999;121:9215–9218. [Google Scholar]
- 37.Morrissey S R, Horton T E, DeRose V J. J Am Chem Soc. 2000;122:3473–3481. [Google Scholar]
- 38.Bar G, Bennati M, Nguyen H-H T, Ge J, Stubbe J, Griffin R G. J Am Chem Soc. 2001;123:3569–3576. doi: 10.1021/ja003108n. [DOI] [PubMed] [Google Scholar]