

Abstract
Iron is essential to life, but poses severe problems because of its toxicity and the insolubility of hydrated ferric ions at neutral pH. In animals, a family of proteins called transferrins are responsible for the sequestration, transport, and distribution of free iron. Comparison of the structure and function of transferrins with a completely unrelated protein hemopexin, which carries out the same function for heme, identifies molecular features that contribute to a successful protein system for iron acquisition, transport, and release. These include a two-domain protein structure with flexible hinges that allow these domains to enclose the bound ligand and provide suitable chemistry for stable binding and an appropriate trigger for release.
Metalloproteins offer a fascinating window into our evolutionary history. Metal ions provide chemical properties, such as redox activity or coordination ability, that expand the functional repertoire of proteins, which depend otherwise on the main group elements of C, H, N, O, P, S, and Se. On the other hand, biological systems have also had to compete with the intrinsic aqueous inorganic chemistry of metal ions and find ways to assimilate and incorporate them. Thus, the relationships between metal ions and proteins give insights not only into the ways in which protein function has evolved, in the context of complex three-dimensional protein structures, but also into the mechanisms that have evolved in living systems, to deal with and use elements from the earth's environment that have potentially hazardous properties.
Iron is the fourth most abundant element in the earth's crust and, after aluminium, the second most abundant metal. As a transition metal, it also has immensely useful ligand-binding and redox properties. It is therefore not surprising that it is used as an essential element by virtually all living species, in which it is a required cofactor in a multitude of proteins of diverse biological functions. At the same time, iron poses enormous problems. The very property that makes it attractive for biological redox processes, the easy one-electron interconversion of Fe(II) and Fe(III), also makes iron toxic, by generation of oxygen-derived radicals and other damaging species (1). Aerobic life also makes Fe3+ the thermodynamically favored species over Fe2+, leading to problems of solubilization; the solubility products of ferric hydroxides are such that the equilibrium concentration of hydrated Fe3+ cannot exceed 10−17 M at pH 7.
For these reasons, living organisms have evolved sophisticated molecular systems for the solubilization, sequestration, transport, and release of iron. Microorganisms, for example, use complex chelating molecules called siderophores to scavenge iron (2, 3) and devote considerable metabolic energy to their biosynthesis. Here, however, we focus on the proteins that have arisen later in evolution to deal with the requirements of more complex multicellular organisms, notably vertebrates. We consider two protein systems, the transferrins, which transport iron, and hemopexin, which transports heme, to address questions of protein design. How can the requirements of high-affinity binding for transport and protection be married with the needs of iron delivery to cells? What features make a successful protein for the uptake, transport, and distribution of iron and iron compounds?
Transferrins: Scavengers and Distributors of Ferric Iron
The name transferrin was originally given to the serum protein that binds and transports iron for delivery to cells. It is now also given to a wider family of homologous proteins that includes serum transferrin (sTf), lactoferrin (Lf), ovotransferrin (oTf), and melanotransferrin (mTf); for more comprehensive reviews, see refs. 4 and 5. Only sTf has a proven transport function, but all transferrins function to control the levels of free iron in the locations where they are found. Lf, for example, is present in milk, many other exocrine secretions, and white blood cells, and binds iron even more tightly than does sTf (6) and retains it to lower pH (7); its function appears to be primarily protective (8). The role of mTf, which is found in both membrane-bound and soluble forms (9), is uncertain but clearly relates in some way to the control of iron. All of these proteins are single-chain glycoproteins, with 670–690 amino acid residues and a molecular weight of ≈80 kDa. A proposed ancestral gene duplication is reflected in the twofold internal sequence repeat in these proteins, with ≈40% sequence identity between their N- and C-terminal halves. sTf, Lf, and oTf all bind two Fe3+ ions, with high affinity (Kd ≈ 10−20 M), together with two synergistically bound CO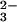 ions (4, 5), and this unique cation–anion relationship is one of the keys to understanding transferrin function (see below). mTf, in contrast, binds only one Fe3+ ion, as a result of amino acid substitutions in one binding site (10).
ions (4, 5), and this unique cation–anion relationship is one of the keys to understanding transferrin function (see below). mTf, in contrast, binds only one Fe3+ ion, as a result of amino acid substitutions in one binding site (10).
Polypeptide Fold.
The twofold sequence repeat of transferrins is reflected in their structures; sTf, Lf, and oTf (and by inference mTf) have all been shown by x-ray crystallography (11–15) to fold into two globular lobes, representing their N- and C-terminal halves (Fig. 1A). Both lobes have the same fold, comprising two domains that enclose a large cleft in which the iron binding site is located. There are subtle differences in the two lobes, in terms of structure, stability, and the ease of iron release (5, 16), and there is some evidence that the iron status of one lobe can influence binding or release from the other (16, 17). Nevertheless, the essential elements of transferrin function can be understood by consideration of the structure of either half-molecule. Two features of the protein structure are of particular importance. First, the binding cleft is hydrophilic, with many polar side chains and some 10–20 water molecules, as appropriate for binding an ionic species (Fe3+). Second, at the back of the binding cleft are two antiparallel β-strands that connect the two domains, and these contain a hinge that enables one domain to move relative to the other, opening or closing the cleft (Fig. 1B).
Fig 1.

(A) Ribbon diagram showing the characteristic bilobal structure of transferrins. Shown here is the iron-bound form of human Lf, with the N lobe on the left and the C lobe on the right. In each lobe, domain 1 is gold and domain 2 is green, with a single Fe3+ ion (red sphere) and CO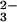 (orange) bound in the interdomain cleft. An α-helix (magenta, top) joins the two lobes; in Tf this is nonhelical. The C-terminal α-helix (pink) may play a role in communication between the lobes (57). (B) The conformational change that accompanies iron binding, shown here for the N lobe of human Tf (21). A hinge in the two β-strands that run behind the iron site allows one domain to move relative to the other. Two helices (blue) act as a fulcrum; one pivots on the other. (C) The canonical iron binding site of transferrins, shown here for the N lobe of human Lf, involves two tyrosine ligands, one aspartate, one histidine, and a bidentate CO
(orange) bound in the interdomain cleft. An α-helix (magenta, top) joins the two lobes; in Tf this is nonhelical. The C-terminal α-helix (pink) may play a role in communication between the lobes (57). (B) The conformational change that accompanies iron binding, shown here for the N lobe of human Tf (21). A hinge in the two β-strands that run behind the iron site allows one domain to move relative to the other. Two helices (blue) act as a fulcrum; one pivots on the other. (C) The canonical iron binding site of transferrins, shown here for the N lobe of human Lf, involves two tyrosine ligands, one aspartate, one histidine, and a bidentate CO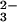 ion in a pocket formed by an arginine side chain and the N terminus of an α-helix. In the N lobe of sTfs, a pair of lysine residues forms a pH-sensitive interaction that assists iron release; these replace Arg-210 and Lys-301 shown here for Lf. (D) Comparison of the iron binding site found in transferrins (Left) with that in the bacterial periplasmic ferric binding protein (Right). In both cases a coordinating anion (carbonate and phosphate, respectively) is at the N terminus of a structurally homologous α-helix and a carboxylate ligand is contributed from a homologous loop. The histidine and two tyrosine ligands come from quite different parts of the structure, yet generate a binding site that is spatially and chemically almost identical. These and other figures were drawn with molscript (58) and rendered with raster3d (59).
ion in a pocket formed by an arginine side chain and the N terminus of an α-helix. In the N lobe of sTfs, a pair of lysine residues forms a pH-sensitive interaction that assists iron release; these replace Arg-210 and Lys-301 shown here for Lf. (D) Comparison of the iron binding site found in transferrins (Left) with that in the bacterial periplasmic ferric binding protein (Right). In both cases a coordinating anion (carbonate and phosphate, respectively) is at the N terminus of a structurally homologous α-helix and a carboxylate ligand is contributed from a homologous loop. The histidine and two tyrosine ligands come from quite different parts of the structure, yet generate a binding site that is spatially and chemically almost identical. These and other figures were drawn with molscript (58) and rendered with raster3d (59).
Iron Recognition and Binding.
The binding site is exquisitely tailored for binding iron as Fe3+; this is emphasized by the fact that although the binding constant for Fe3+ is ≈1020, that for Fe2+ is only ≈103 (18). With only one exception [in certain insect transferrins where a glutamine is substituted for histidine (19)], the iron ligands are the identical in every transferrin, in both the N and C lobe (5). Four protein ligands are provided, comprising one carboxylate oxygen, two phenolate oxygens, and one imidazole nitrogen, from the side chains of one Asp, two Tyr, and one His residue (Fig. 1C). These ligands are ideal for binding Fe3+, with three anionic oxygens (hard bases in the Pearson classification) and one neutral nitrogen. The 3− charge of the ligands also matches the 3+ charge of the metal ion. A second critical feature of the iron binding site, however, is that a bidentate carbonate ion fills the two remaining coordination sites. The charge on the anion is compensated for by a positively charged arginine side chain and the positive charge at the N terminus of an α-helix, which, with the arginine, creates a highly favorable anion binding pocket. In a classic example of molecular recognition the full hydrogen bonding potential of the CO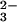 ion is used (5), and neither Fe3+ nor CO
ion is used (5), and neither Fe3+ nor CO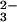 is bound significantly in the absence of the other.
is bound significantly in the absence of the other.
Iron Release.
Mechanically, the key feature in iron release is that the hinge in the two “backbone” strands at the back of the cleft enables one domain to rotate 50–60° relative to the other, opening the cleft and releasing iron (20–22). This conformational transition, from closed to open structure, is shown in Fig. 1B. What, however, triggers this transition? Iron is released in the context of receptor-mediated endocytosis, in which the iron-loaded sTf binds to cell surface receptors, is internalized, releases its iron within the endosome, and is then returned to the surface for more rounds of binding and transport (23, 24). Two factors appear to operate in facilitating iron release. First, there is evidence that the receptor itself may act to selectively “prize open” the C-lobe binding site (25). Second, the pH in the endosome is ≈5.5, substantially lower than the extracellular pH of 7.4, and at this lowered pH sTf spontaneously begins to release iron (16).
The precise nature of transferrin–receptor interactions is not known, but the effect of pH has been the subject of intensive studies. Inspection of the protein structure shows that in the closed, iron-bound form there are surprisingly few interactions between the two domains apart from those provided by the covalent bonds to iron. Networks of water molecules within the cleft between the two domains allow for easy access of protons to ligands. Thus, protonation of any of the iron ligands will weaken binding and favor domain opening. The first step appears to be protonation of the carbonate ion (26, 27), which initiates dissociation of the anion (27), and later steps implicate protonation of the histidine and/or tyrosine ligands (26). In the N lobe of sTfs, a pair of hydrogen-bonded lysines close to the iron center provides another pH-sensitive site; protonation of this “dilysine pair” would promote domain opening because the two residues are from opposing domains and should repel one another when both are charged (28–30). The full picture is probably that a series of protonations, beginning with the anion, generate a smooth transition toward the open state as iron binding is progressively weakened. Three protons are taken up on iron release (4).
Importance of Dynamics.
Conformational change is central to transferrin function. Whereas the importance of domain opening in iron release is obvious, dynamics also appears to play a vital role in iron binding. Kinetic and spectroscopic studies suggest that the CO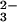 ion binds first (31, 32), and structural arguments support this (5). Once the anion has bound to the apo-protein, four of the six iron ligands (two carbonate oxygens and two tyrosines) will be associated together on one domain, and it is reasonable to suppose that this is the site of initial iron binding; indeed such intermediates have been defined crystallographically (33, 34). How, then, do domain closure and coordination to the other two ligands (Asp and His) occur, given that the latter are 9–10 Å away, on the other domain? Here, several crystal structures give a key insight. One crystal form of human apo-Lf has the N lobe open and the C lobe closed (20), whereas another has both open (35), and horse apo-Lf has been found with both lobes closed (36). Likewise, bacterial periplasmic binding proteins (PBPs), which resemble transferrins in fold and function, have also been observed in ligand-free but closed conformations (37). The strong suggestion is that in their iron-free form, transferrins can sample both open and closed states; if iron is bound to one domain, the other domain will then “lock on” as the protein samples the closed state, whereas if iron is not present it simply opens again (5, 38). The key to this is that there is a fine balance between open and closed states (39) in the absence of bound iron, with very little energy difference between them. Where the equilibrium lies may vary from one transferrin to another, but in broad terms it matters little, so long as the protein can sample both open and closed states, with small activation barriers between them.
ion binds first (31, 32), and structural arguments support this (5). Once the anion has bound to the apo-protein, four of the six iron ligands (two carbonate oxygens and two tyrosines) will be associated together on one domain, and it is reasonable to suppose that this is the site of initial iron binding; indeed such intermediates have been defined crystallographically (33, 34). How, then, do domain closure and coordination to the other two ligands (Asp and His) occur, given that the latter are 9–10 Å away, on the other domain? Here, several crystal structures give a key insight. One crystal form of human apo-Lf has the N lobe open and the C lobe closed (20), whereas another has both open (35), and horse apo-Lf has been found with both lobes closed (36). Likewise, bacterial periplasmic binding proteins (PBPs), which resemble transferrins in fold and function, have also been observed in ligand-free but closed conformations (37). The strong suggestion is that in their iron-free form, transferrins can sample both open and closed states; if iron is bound to one domain, the other domain will then “lock on” as the protein samples the closed state, whereas if iron is not present it simply opens again (5, 38). The key to this is that there is a fine balance between open and closed states (39) in the absence of bound iron, with very little energy difference between them. Where the equilibrium lies may vary from one transferrin to another, but in broad terms it matters little, so long as the protein can sample both open and closed states, with small activation barriers between them.
What Has Evolution to Tell Us?
The advantages of a bilobal structure (and of the gene duplication) has always been a puzzle. One suggestion has been that the larger size (relative to a single-lobe precursor) prolonged the lifetime of transferrin circulation by lessening its loss through the kidneys (40). However, bilobal transferrins are also found in insects, which lack the sophisticated circulatory systems of vertebrates. An alternative possibility is that the bilobal structure allows additional levels of control, for example through communication between the lobes (16, 17), or through variations in the orientations of the two lobes, which could give receptor discrimination.
A second question concerns the evolutionary precursor to the transferrins before the gene duplication. Can we find a single-sited, single-lobe transferrin relative and what would this tell us? Close scrutiny of the polypeptide folding in Lf (41) identified a potential evolutionary link with bacterial PBPs, which transport ions and small molecules through the periplasm of Gram-negative bacteria (42). These are half the size of transferrins, fold into two domains, and release their ligands by domain opening (43). Comparisons showed that not only does each transferrin lobe have the same fold as that in several anion-binding PBPs, but that the anion also binds in an analogous site (5, 41). Sequence identity between transferrins and PBPs is low (≈12–15% identity between the N lobe of Lf and the sulfate-binding PBP), but equally low sequence identity is also found between members of the PBP family. The similar fold and similar anion binding site and function suggested the possibility of divergent evolution from a common anion-binding precursor molecule, with the iron binding site in transferrins as a later evolutionary addition (5, 41).
Recently the structure of a PBP that transports Fe3+ ions (ferric binding protein, FBP) was determined (44). FBP also has the same fold as each half-transferrin, and a very similar iron binding site (Fig. 1D) that includes four protein ligands, one carboxylate (Glu), two phenolate (Tyr), and one imidazole (His). A phosphate ion and a water molecule complete the iron coordination. Remarkably, although the phosphate ion and the Glu ligand in FBP belong to structurally equivalent regions to those that contribute the carbonate ion and the Asp ligand in transferrins, the other three ligands come from quite different parts of the protein structure than their counterparts in transferrins.
The inferences are that (i) the transferrins are likely to have arisen by evolutionary divergence from an anion-binding precursor, shared with FBP and the anion-binding PBPs, (ii) in each case the metal-binding residues have been added later, in different places on the polypeptide but generating the same site spatially and chemically, and (iii) evolution has discovered the same iron binding apparatus twice, by convergence (44, 45). We conclude that this is a highly favorable set of ligands for binding Fe3+ ions and that the inclusion of an exogenous anion, with suitable pKa, in the coordination sphere may be likened to an Achilles heel that allows iron release to be initiated.
Hemopexin: Recycler and Transporter of Heme
Heme is used in a wide variety of biological processes, including respiration and energy transfer. Turnover of heme proteins, notably hemoglobin, leads to the release of heme into extracellular fluids, with potentially severe consequences for health. Like free iron, heme is a source of essential iron for invading bacterial pathogens (46) and is highly toxic because of its ability to catalyze free radical formation. Levels of free heme are usually low but can become dangerously high in conditions of hemolytic disease. Protection is given by hemopexin, a 60-kDa serum glycoprotein that sequesters heme with very high affinity (Kd < 10−12 M) from the bloodstream (47), transports it to specific receptors on liver cells, where it undergoes receptor-mediated endocytosis, and releases its bound heme into cells (48). It thus serves both to protect against heme toxicity and to conserve and recycle iron.
Polypeptide Fold.
Hemopexin is folded into two homologous domains of about 200 amino acid residues each, joined by a 20-residue linker (49, 50). There is ≈25% sequence identity between the two domains (50), consistent with an ancestral gene duplication. Both domains have the same fold (Fig. 2A), which comprises a striking and unusual arrangement of four β-sheet modules arranged in tandem as the blades of a four-bladed β-propeller (51). This forms a flat, disk-like structure with a narrow tunnel through the center and is a variant of the larger six-, seven-, and eight-bladed β-propeller domains (52) that are found in many cellular and cell-surface proteins that mediate protein–protein interactions (53). The crystal structure of the heme-bound form of hemopexin (54) shows that the two domains associate such that one edge of the C-terminal domain packs against the face of the N-terminal domain, in the vicinity of the central tunnel opening (Fig. 2B). Both domains appear to be rather rigid, stable structures. In contrast, the linker peptide is flexible. Even in the heme-bound form, part of the linker is disordered, and in apo-hemopexin, in the absence of heme, it is highly protease-sensitive (47, 49) and may simply act as a flexible tether between the domains.
Fig 2.

(A) Ribbon diagram showing the characteristic four-bladed β-propeller fold found for each domain of hemopexin. Each module (blade) comprises a four-stranded twisted β-sheet, with the four modules (shown in green, cyan, blue, and magenta from N to C terminus) arranged in tandem around a central tunnel, in which are bound several anions and cations. (B) Structure of the heme–hemopexin complex. The heme (green) is bound between the N- (blue) and C-terminal (red) β-propeller domains in a pocket that is bounded by the interdomain linker peptide. Two histidine residues (cyan) coordinate the heme iron. A disordered part of the linker that may be a “hot spot” where unwrapping of the heme begins (see text) is black. Ions bound in the central tunnel of each domain are purple (two Na+ and one Cl− in each domain and a phosphate in the C domain only). (C) Surface representation of the heme environment, showing how the porphyrin ring slots into a cleft between aromatic and other hydrophobic side chains (green) and basic side chains (blue) that pack around it. The heme propionates extend into the lower part of the cleft, below the aromatic residues, and interact with arginine and histidine residues. (D) Putative mode of heme binding to the isolated N-terminal domain of rabbit hemopexin. In this proposed model the heme binds to His-127 [as shown chemically (60)] and His-82, with one propionate extending into the central tunnel. Two cations (magenta) and one anion (yellow) also occupy this tunnel.
Heme Recognition and Binding.
Hemopexin binds a single heme molecule, between the two β-propeller domains, in a pocket bounded by the interdomain linker peptide (ref. 54; Fig. 2B). This linker plays a critical part in the creation of the binding site, forming its outer boundary and interacting with the heme to help stabilize the complex. The heme is bound by two histidine residues, His-213 from the linker and His-266 from the C-terminal domain, which coordinate the Fe(III) of the heme group. It is not completely buried, and the high heme affinity is undoubtedly generated by the host of noncovalent interactions that contribute to binding. The porphyrin ring is, apart from its two propionate substituents, hydrophobic, with highly delocalized π bonding. Accordingly, a large number of aromatic residues, all of them invariant in hemopexin sequences, pack around the heme (Fig. 2C), and with other hydrophobic residues provide a highly favorable binding environment. Unusually, the two heme propionates are also largely buried, but they are stabilized by basic residues that cluster around them, two arginines around one propionate and three histidines around the other (Fig. 2C). These may act as electrostatic anchors that guide the heme into place (54). The overall environment seems at first sight to be unusual, with its combination of hydrophobic and basic residues, but it is highly structured, with striking stacking of side chains that involves not only aromatic groups but also arginine side chains, histidine side chains, and the heme in π–π and cation–π interactions.
Heme Release.
As in the case of iron release from transferrin, heme is released from hemopexin in the low pH environment of the endosome, following receptor-mediated endocytosis (48). Little is known about the influence of the hemopexin receptor. However, it is likely that heme release begins with disruption of the way in which the linker peptide wraps around the heme, perhaps triggered by the binding of one or both of the rigid, disk-like domains to the receptor (54) or by protonation of some of the basic residues that cluster round the heme. Even in the heme–hemopexin complex the linker has a disordered region, which may act as a “hot spot” that initiates unwrapping. Protonation of His-213, the heme iron ligand that is contributed by the linker, could then lead to breakup of the complex. No structure is available for apo-hemopexin, but it is known that heme binding induces conformational changes that do not change the secondary structure, presumably through association of the two rigid domains (55). Heme binding also markedly increases resistance to heat (56) and proteolysis (49). These changes are consistent with a transition from a flexible, open form to the tight association of the domains seen in the crystal structure, in which association the heme plays a central part (54).
Evolution.
A curiosity in the hemopexin story is that when the isolated N-terminal domain is separated by proteolysis, it is found to bind heme on its own (49). This led to initial expectations that this domain would provide the heme binding site in intact hemopexin. As shown crystallographically (54), this is not the case, and the structure shown in Fig. 2A clearly represents the physiologically relevant mode of heme binding. Nevertheless, a plausible model can be built for binding to the N-terminal domain, in which heme slots into a crevice leading into the central tunnel (Fig. 2D). Perhaps this property is an evolutionary relic of an ancestral single-domain heme binding protein, predating the gene duplication that gave the present two-domain protein. The interdomain binding site in today's hemopexin evidently gives greater control of binding and release.
Common Principles: What Makes an Effective Protein for Binding, Transport, and Release?
Although these two protein families are seemingly very different in structure, a number of common principles emerge. In each case:
The protein provides a binding site (for Fe3+ or heme) between two protein domains. Each uses domains of completely different construction, but the principle is the same. This fold gives the possibility of complete enclosure of the ligand, but also provides a means of release.
There are only weak interactions (many of them water-mediated) between the domains, apart from those directly involving the bound ligand (Fe3+ or heme). Once ligand binding is weakened, therefore, the domains can readily move apart.
The appropriate chemistry is used to generate tight binding, a polar environment and an appropriate set of ligating residues for Fe3+, and aromatic and basic residues for the heme porphyrin ring with two His residues to coordinate its iron atom.
A trigger initiates release. In the case of transferrin, this is provided by receptor binding and by protonation of the carbonate ion and other residues in and around the iron site; in the case of hemopexin, this is probably provided by receptor binding, destabilization of the linker, and protonation of the His ligands.
Once binding is weakened the domains can move apart to complete the process of release.
The dynamics of multidomain assemblies play a role in both cases, well understood in the case of transferrins, but not yet in the case of hemopexin.
Acknowledgments
We thank our many collaborators and coworkers over the years who have contributed to this work, including Geoff Jameson, Gillian Norris, Sylvia Rumball, Clyde Smith, John Tweedie, Catherine Day, Richard Kidd, Rick Faber, Ross MacGillivray, Anne Mason, Massimo Paoli, Ann Smith, and Bill Morgan; Geoff Jameson and Chris Squire for careful reading of the manuscript; and the Health Research Council of New Zealand, the Marsden Fund, and the U.S. National Institutes of Health for funding support.
Abbreviations
sTf, serum transferrin
Lf, lactoferrin
PBP, periplasmic binding protein
References
- 1.Pierre J. L. & Fontecave, M. (1999) Biometals 12, 195-199. [DOI] [PubMed] [Google Scholar]
- 2.Braun V., Hantke, K. & Koster, W. (1998) Metal Ions Biol. Syst. 35, 67-145. [PubMed] [Google Scholar]
- 3.Stintzi A. & Raymond, K. N. (2002) in Molecular and Cellular Iron Transport, ed. Templeton, D. M. (Dekker, New York), pp. 273–320.
- 4.Aisen P. & Harris, D. C. (1989) in Iron Carriers and Iron Proteins, ed. Loehr, T. M. (VCH, New York), pp. 241–351.
- 5.Baker E. N. (1994) Adv. Inorg. Chem. 41, 389-463. [Google Scholar]
- 6.Aisen P. & Liebman, A. (1972) Biochim. Biophys. Acta 257, 314-323. [DOI] [PubMed] [Google Scholar]
- 7.Baker E. N., Baker, H. M. & Kidd, R. D. (2002) Biochem. Cell Biol. 80, 27-34. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez L., Calvo, M. & Brock, J. H. (1992) Arch. Dis. Child. 67, 657-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sekyere E., Food, M. R. & Richardson, D. R. (2002) FEBS Lett. 512, 350-352. [DOI] [PubMed] [Google Scholar]
- 10.Baker E. N., Baker, H. M., Smith, C. A., Stebbins, M. R., Kahn, M., Hellstrom, K. E. & Hellstrom, I. (1992) FEBS Lett. 298, 215-218. [DOI] [PubMed] [Google Scholar]
- 11.Anderson B. F., Baker, H. M., Dodson, E. J., Norris, G. E., Rumball, S. V., Waters, J. M. & Baker, E. N. (1987) Proc. Natl. Acad. Sci. USA 84, 1769-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson B. F., Baker, H. M., Norris, G. E., Rice, D. W. & Baker, E. N. (1989) J. Mol. Biol. 209, 711-734. [DOI] [PubMed] [Google Scholar]
- 13.Bailey S., Evans, R. W., Garratt, R. C., Gorinsky, B., Hasnain, S., Horsburgh, C., Jhoti, H., Lindley, P. F., Mydin, A., Sarra, R. & Watson, J. L. (1988) Biochemistry 27, 5804-5812. [DOI] [PubMed] [Google Scholar]
- 14.Hall D. R., Hadden, J. M., Leonard, G. A., Bailey, S., Neu, M., Winn, M. & Lindley, P. F. (2002) Acta Crystallogr. D 58, 70-80. [DOI] [PubMed] [Google Scholar]
- 15.Kurokawa H., Mikami, B. & Hirose, M. (1995) J. Mol. Biol. 254, 196-207. [DOI] [PubMed] [Google Scholar]
- 16.He Q.-Y. & Mason, A. B. (2002) in Molecular and Cellular Iron Transport, ed. Templeton, D. M. (Dekker, New York), pp. 95–123.
- 17.Ward P. P., Zhou, X. & Conneely, O. M. (1996) J. Biol. Chem. 271, 12790-12794. [DOI] [PubMed] [Google Scholar]
- 18.Harris W. R. (1986) J. Inorg. Biochem. 27, 41-52. [DOI] [PubMed] [Google Scholar]
- 19.Baker H. M., Mason, A. B., He, Q.-Y., MacGillivray, R. T. & Baker, E. N. (2001) Biochemistry 40, 11670-11675. [DOI] [PubMed] [Google Scholar]
- 20.Anderson B. F., Baker, H. M., Norris, G. E., Rumball, S. V. & Baker, E. N. (1990) Nature 344, 784-787. [DOI] [PubMed] [Google Scholar]
- 21.Jeffrey P. D., Bewley, M. C., MacGillivray, R. T. A., Mason, A. B., Woodworth, R. C. & Baker, E. N. (1998) Biochemistry 37, 13978-13986. [DOI] [PubMed] [Google Scholar]
- 22.Kurokawa H., Dewan, J. C., Mikami, B., Sacchettini, J. C. & Hirose, M. (1999) J. Biol. Chem. 274, 28445-28452. [DOI] [PubMed] [Google Scholar]
- 23.Klausner R. D., Ashwell, J. V., Van Renswoude, J. B., Harford, J. & Bridges, K. (1983) Proc. Natl. Acad. Sci. USA 80, 2263-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Octave J.-N., Schneider, Y.-J., Trouet, A. & Crichton, R. R. (1983) Trends Biochem. Sci. 8, 217-220. [Google Scholar]
- 25.Bali P. K. & Aisen, P. (1991) Biochemistry 30, 9947-9952. [DOI] [PubMed] [Google Scholar]
- 26.el Hage Chahine J. M. & Pakdaman, R. (1995) Eur. J. Biochem. 230, 1102-1110. [DOI] [PubMed] [Google Scholar]
- 27.MacGillivray R. T. A., Moore, S. A., Chen, J., Anderson, B. F., Baker, H., Luo, Y., Bewley, M., Smith, C. A., Murphy, M. E., Wang, Y., et al. (1998) Biochemistry 37, 7919-7928. [DOI] [PubMed] [Google Scholar]
- 28.Baker E. N. & Lindley, P. F. (1992) J. Inorg. Biochem. 47, 147-160. [DOI] [PubMed] [Google Scholar]
- 29.Dewan J. C., Mikami, B., Hirose, M. & Sacchettini, J. C. (1993) Biochemistry 32, 11963-11968. [DOI] [PubMed] [Google Scholar]
- 30.He Q.-Y., Mason, A. B., Tam, B. M., MacGillivray, R. T. A. & Woodworth, R. C. (1999) Biochemistry 38, 9704-9711. [DOI] [PubMed] [Google Scholar]
- 31.Kojima N. & Bates, G. W. (1981) J. Biol. Chem. 256, 12034-12039. [PubMed] [Google Scholar]
- 32.Zweier J. L., Wooten, J. B. & Cohen, J. S. (1981) Biochemistry 20, 3505-3510. [DOI] [PubMed] [Google Scholar]
- 33.Lindley P. F., Bajaj, M., Evans, R. W., Garratt, R. C., Hasnain, S. S., Jhoti, H., Kuser, P., Neu, M., Patel, K., Sarra, R., et al. (1993) Acta Crystallogr. D 49, 292-304. [DOI] [PubMed] [Google Scholar]
- 34.Mizutani K., Yamashita, H., Kurokawa, H., Mikami, B. & Hirose, M. (1999) J. Biol. Chem. 274, 10190-10194. [DOI] [PubMed] [Google Scholar]
- 35.Baker E. N., Anderson, B. F., Baker, H. M., Faber, H. R., Smith, C. A. & Sutherland-Smith, A. J. (1997) in Lactoferrin: Interactions and Biological Functions, eds. Hutchens, T. W. & Lonnerdal, B. (Humana, Totowa, NJ), pp. 177–191.
- 36.Sharma A. K., Rajashankar, K. R., Yadav, M. P. & Singh, T. P. (1999) Acta Crystallogr. D 55, 1152-1157. [DOI] [PubMed] [Google Scholar]
- 37.Flocco M. M. & Mowbray, S. L. (1994) J. Biol. Chem. 269, 8931-8936. [PubMed] [Google Scholar]
- 38.Faber H. R., Bland, T., Day, C. L., Norris, G. E., Tweedie, J. W. & Baker, E. N. (1996) J. Mol. Biol. 256, 352-363. [DOI] [PubMed] [Google Scholar]
- 39.Gerstein M., Anderson, B. F., Norris, G. E., Baker, E. N., Lesk, A. M. & Chothia, C. (1993) J. Mol. Biol. 234, 357-372. [DOI] [PubMed] [Google Scholar]
- 40.Williams J. (1982) Trends Biochem. Sci. 7, 394-397. [Google Scholar]
- 41.Baker E. N., Rumball, S. V. & Anderson, B. F. (1987) Trends Biochem. Sci. 11, 350-353. [Google Scholar]
- 42.Tam R. & Saier, M. H. J. (1993) Microbiol. Rev. 57, 320-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quiocho F. A. & Ledvina, P. S. (1996) Mol. Microbiol. 20, 17-25. [DOI] [PubMed] [Google Scholar]
- 44.Bruns C. M., Nowalk, A. J., Arvai, A. S., McTigue, M. A., Vaughan, K. G., Mietzner, T. A. & McRee, D. E. (1997) Nat. Struct. Biol. 4, 919-924. [DOI] [PubMed] [Google Scholar]
- 45.Baker E. N. (1997) Nat. Struct. Biol. 4, 869-871. [DOI] [PubMed] [Google Scholar]
- 46.Lee B. C. (1995) Mol. Microbiol. 18, 383-390. [DOI] [PubMed] [Google Scholar]
- 47.Hrkal Z., Vodrazka, Z. & Kalousek, I. (1974) Eur. J. Biochem. 43, 73-78. [DOI] [PubMed] [Google Scholar]
- 48.Smith A. & Hunt, R. T. (1990) Eur. J. Cell Biol. 53, 234-245. [PubMed] [Google Scholar]
- 49.Morgan W. T. & Smith, A. (1984) J. Biol. Chem. 259, 12001-12006. [PubMed] [Google Scholar]
- 50.Takahashi N., Takahashi, Y. & Putnam, F. W. (1985) Proc. Natl. Acad. Sci. USA 82, 73-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faber H. R., Groom, C. R., Baker, H. M., Morgan, W. T., Smith, A. & Baker, E. N. (1995) Structure (London) 3, 551-559. [DOI] [PubMed] [Google Scholar]
- 52.Fulop V. & Jones, D. T. (1999) Curr. Opin. Struct. Biol. 9, 715-721. [DOI] [PubMed] [Google Scholar]
- 53.Smith T. F., Gaitatzes, C., Saxena, K. & Neer, E. J. (1999) Trends Biochem. Sci. 24, 181-185. [DOI] [PubMed] [Google Scholar]
- 54.Paoli M., Anderson, B. F., Baker, H. M., Morgan, W. T., Smith, A. & Baker, E. N. (1999) Nat. Struct. Biol. 6, 926-931. [DOI] [PubMed] [Google Scholar]
- 55.Wu M.-L. & Morgan, W. T. (1994) Proteins Struct. Funct. Genet. 20, 185-190. [DOI] [PubMed] [Google Scholar]
- 56.Wu M.-L. & Morgan, W. T. (1993) Biochemistry 32, 7216-7222. [DOI] [PubMed] [Google Scholar]
- 57.Jameson G. B., Anderson, B. F., Norris, G. E., Thomas, D. H. & Baker, E. N. (1998) Acta Crystallogr. D 54, 1319-1335. [DOI] [PubMed] [Google Scholar]
- 58.Kraulis P. J. (1991) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 59.Merritt E. A. & Bacon, D. J. (1997) Methods Enzymol. 277, 505-524. [DOI] [PubMed] [Google Scholar]
- 60.Morgan W. T., Muster, P., Tatum, F., Kao, S., Alam, J. & Smith, A. (1993) J. Biol. Chem. 268, 6256-6262. [PubMed] [Google Scholar]