

Abstract
There exist a limited but growing number of biological metal centers whose properties lie conspicuously outside the realm of known inorganic chemistry. The synthetic analogue approach, broadly directed, offers a powerful exploratory tool that can define intrinsic chemical possibilities for these sites while simultaneously expanding the frontiers of fundamental inorganic chemistry. This speculative application of analogue study is exemplified here in the evolution of synthetic efforts inspired by the cluster chemistry of biological nitrogen fixation.
The study of synthetic small- molecule analogues is a proven chemical approach for the elucidation of biological metal centers. The principal utility of the method was delineated in a definitive early (in 1980) account (1): Synthetic analogues permit (i) the “deduction of minimal site structure (where unknown) based on coincident model-biomolecule properties or (where known) required for execution of biological function,” while further allowing (ii) the “detection of the influence of the biomolecular environment on the intrinsic properties of the site as represented by the model.” In recent years, the impact of synthetic studies, as construed along these original, corroborative† goals, has diminished due to impressive improvements in biological and biophysical techniques that are allowing the isolation, manipulation, and characterization of biomolecules in ever-increasing speed and detail. Moreover, the success of synthetic analogue efforts has rendered most common biological metal motifs well understood at the level of basic inorganic chemistry; issues of deeper biological interest concerning these sites are probably unapproachable through simple synthetic studies.
Synthetic analogue chemistry nevertheless remains an indispensable component in the armamentarium of bioinorganic investigation. The same advances in biomolecular science that are mitigating the power of corroborative synthetic methods are also revealing the existence of unforeseen, indeed unknown, chemistry within biological systems. These new frontiers in inorganic and bioinorganic chemistry reside in “aberrant” biological metal centers, sites restricted (apparently) to only a few select organisms and distinguished by the presence of unusual metals, peculiar spectroscopic signatures, and/or singular chemical reactivity. Where atomic-level definition is available, these centers are marked by surprising complexity that raise but do not answer underlying chemical questions concerning structure and function. Therein lies the third, speculative† objective of the synthetic analogue approach: (iii) the broad exploration of fundamental chemistry as motivated by a biological system, with the immediate goal of establishing possibilities and methodologies that ultimately will lead to corroborative synthetic analogues and a chemical understanding of the inspirational system. This rationale is not new to synthetic chemistry, but it is growing in prominence because of the obvious gaps in chemical knowledge highlighted by such extraordinary examples as nitrogenase, the photosynthetic water-oxidation complex, the hydrogenases, and the carbon monoxide dehydrogenases. We present here a case study of this speculative approach as applied to the molecular description of nitrogenase.
The Synthetic Problem of Nitrogenase
Biological nitrogen fixation, the conversion of dinitrogen to ammonia by the nitrogenase enzymes (Eq. 1), is the predominant entry point for nitrogen in the global nitrogen cycle (2–7).
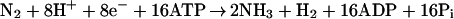 |
1 |
The foundational significance of the process and its intrinsic chemical formidability have prompted extensive, extended studies to attain a molecular description of this enzyme system. These efforts have met with limited success, and even simple molecular details such as the minimal requisite reaction stoichiometry (Eq. 1 is a limiting, but not necessarily obligate formulation), the location of substrate binding, and the oxidation states of the metals involved remain either unknown or in debate. Few problems in bioinorganic chemistry have proved as challenging and refractory.
Progress on this topic is constrained by the complexity of the enzyme and its recalcitrance to standard biochemical and biophysical probes. The system is inaccessible in a poised, “ready” state: Without dinitrogen substrate, the enzyme spontaneously reduces protons to dihydrogen. Substrate reduction requires a host of other obligatory events (protein docking/release, electron transfer/storage, ATP hydrolysis, proton transfer, and multisite substrate binding/release), leading to a multitude of concurrently populated enzyme states during turnover and to convoluted kinetics. Enzyme intermediates and inhibited forms are either unobserved or ill-defined. In short, there is no specific information on any active form of nitrogenase.
Molecular understanding of this enzyme therefore is confined to resting-state attributes. We note some salient features: In its best-studied form, the enzyme system consists of two separate soluble components, the Fe protein, the functionally obligate electron donor, and the MoFe protein, responsible for dinitrogen reduction; the MoFe protein contains two metallocluster types, the FeMo-cofactor and the P-cluster, both of which are biologically and chemically unprecedented; the site of nitrogen chemistry is inferred (but not absolutely proven!) to occur at the FeMo-cofactor, a complex heterometallic Mo-Fe-S cluster, whereas the P-cluster, a high-nuclearity Fe-S cluster, is thought to mediate electron transfer to the active site; the FeMo-cofactor can be isolated from the native protein as a stable, soluble, but inactive metallocluster that can be reincorporated into apoprotein with restoration of activity; and two related yet genetically separate nitrogenase variants are also known that are distinguished by the total absence of molybdenum in both types, the presence of a single vanadium in the cofactor of one, and the apparent absence of any metal save iron in the other. Finally, incisive structural data on the metalloclusters has become available, as presented in the following sections, and this information above all else has framed the problem for speculative synthetic analogue efforts.
X-Ray Absorption Spectroscopy (1978–1992)
The first structural insight into the FeMo cofactor originated from Mo-K edge x-ray absorption spectroscopy of the native protein and the isolated cofactor cluster. The extended x-ray absorption fine structure (EXAFS) analysis substantiated the existence of a Mo-Fe-S cluster, revealing the nearest neighbors of molybdenum as three to four sulfur atoms at 2.36(2)-Å distance and two to three iron atoms at 2.72(3) Å (8); the molybdenum environments of free and protein-bound cofactor were found to be essentially indistinguishable by this technique. Combined with the compositional data for the FeMo-cofactor (early analyses suggested [MoFe6–8S4–6]), these results offered the first reasonable foundation for synthetic work.
The initial EXAFS results suggested a [MoFe3S3] heterometallic cuboidal structure as one possible local geometry about the molybdenum center. Appropriate synthetic analogues were achieved quickly through self-assembly reactions employing [MoS4]2− as both an organizing component and a source of molybdenum and sulfide (9, 10); a recent, highly efficient variation of this synthetic method (11) is illustrated in Scheme S1. The heterocubane clusters thus obtained possessed molybdenum environments in excellent agreement with the original EXAFS analysis and with subsequent X-ray absorption near edge structure (XANES) studies that indicated a fac-octahedral Mo(IV) coordination sphere in the FeMo cofactor consisting of three light (2p) atom donors and three sulfurs. Later studies would reveal the existence of an alternate nitrogenase with a vanadium-containing cofactor; synthetic [VFe3S4] cubane clusters (12), prepared in a manner similar to the molybdenum-based syntheses, likewise were determined to have a heterometal environment congruent by EXAFS criterion to that of the biological cluster. To date, these systems are still the most accurate synthetic structural representations for the heterometallic portion of their respective cofactors. The synthetic development of [MFe3S4] clusters presaged a wider occurrence of the motif in biological systems, specifically as void-filled “inorganic mutants” of 3-Fe sites in ferredoxins (13) and, more recently, in distorted form within the C-cluster of Ni-containing carbon monoxide dehydrogenase (refs. 14 and 15; Fig. 1). Synthetic methodology has kept pace with these developments: In addition to the thiometallate routes used for the early transition metal [MFe3S4] clusters, pathways starting from trinuclear Fe-S clusters have allowed access to late metal derivatives as well (Scheme S2), and well defined, synthetic examples of the [MFe3S4] cubane core are now known for M = V, Nb, Mo, W, Re, Co, Ni, Cu, Ag, Tl (13, 16). The accessibility, stability, and recurrence of the heterometallic cubane motif suggest that additional biological examples may surface in the future.
Scheme 1.

Direct synthesis of a single [MoFe3S4] heterometallic cubane cluster.
Figure 1.

Other [MFe3S4] cubane motifs in biology: void-filled 3-Fe clusters (Left) and the C- cluster in a Ni-Fe-S carbon monoxide dehydrogenase (Right). A different structure is available (at lower resolution) for the C-cluster in an analogous enzyme from a different organism that contains the [NiFe3S4] cubane cluster in a less-distorted form (15). M = Mn, Co, Ni, Cu, Zn, Cd, Ga, Tl; N*, histidine; S*, cysteinate.
Scheme 2.

Synthesis of a [NiFe3S4] heterometallic cubane cluster.
The composition of the FeMo-cofactor also drew attention to the synthesis of higher-nuclearity Fe-S clusters of similar stoichiometry. This interest was predicated on the optimistic notion that the nitrogenase cofactor was representative of some general cluster type of high thermodynamic stability and ready kinetic accessibility. Both propositions seemed reasonable at the time, given the stability of isolated cofactor and the chemistry of biological Fe-S clusters as understood then.‡ Clusters of compositional note during this period spanned a range of core structures and terminal ligands (ref. 17; Fig. 2). Although most of these clusters were not directly useful as synthetic analogues (an unsurprising outcome in light of the imprecise analytical data and fragmentary structural information available for the cofactor), they nonetheless contributed tangibly to the understanding of synthetic Fe-S chemistry. Certain abiological phosphine-ligated species, however, would emerge serendipitously as the progenitors for the next generation of cofactor-related synthetic clusters.
Figure 2.

Selected higher-nuclearity Fe-S clusters. (Y, z) = (N2H4, 4), (OH/N3/CN, 5), (S, 6); X, monoanions (halide/thiolate/aryloxide).
The First Macromolecular Structures (1992–2002)
The macromolecular structure determination of the MoFe protein (18, 19) marked a watershed in nitrogenase chemistry and in Fe-S biochemistry generally. The diffraction data (2.7-Å resolution in 1992) allowed the first direct visualization of the enzyme active site, yielding a structural model for the FeMo-cofactor as a [MoFe7S9] cluster composed of two sulfur-voided [M4S3] cuboidal fragments linked by three μ2-sulfur bridges (Fig. 3). This structure corroborated the x-ray absorption spectroscopy predicted local environment about molybdenum but also revealed a number of startling features: six three-coordinate iron sites at the cluster center, each slightly pyramidalized inward; open, nonrhomb cluster faces about those anomalous iron sites, in contrast to the [Fe2S2] rhombs that typically constitute Fe-S cluster geometries; and a seemingly vacant cavity within the cluster core. Later, higher-resolution (2.0-Å) protein crystal structures (20) would show the P-cluster to be a symmetric [Fe8S7] structure (Fig. 3) comprising, in the native state (PN), two [Fe4S3] cuboidal halves, vertex-fused through a μ6-sulfide and further interconnected by two μ2-cysteinate bridges; after oxidation (the POX state), the cluster was found rearranged to a more open, asymmetric geometry, with additional protein ligation provided by a serine side-chain oxygen and a backbone amide nitrogen. All these cluster structures were remarkable from a synthetic perspective, and new chemistry was clearly needed to understand their properties.
Figure 3.

Metalloclusters in the nitrogenase MoFe protein as derived from protein crystallography (pre-2002). N*, histidine; O*, serine/serinate; S*, cysteinate.
As part of synthetic studies that preceded the macromolecular structures, phosphine-ligated [MFe4S6] (M = Mo, V) species were prepared in a specific attempt to access new (Mo,V)-Fe-S clusters for comparison with the FeMo-cofactor (Scheme S3; refs. 21 and 22). The biological correspondence would turn out not to involve molybdenum at all but rather the remaining Fe4S6 cluster fragment, which resembled the cofactor model in possessing a sulfur-voided cuboidal geometry, nonrhomb faces with μ2-sulfide bridges, and, for the phosphine-ligated sites, curious trigonal pyramidal metal geometries that are distributed and flattened (if the axial ligands are ignored) in a manner analogous to the three-coordinate sites in the biological cluster. If the [MoFe3S4] cubane core, less one sulfide, can be said to represent one end of the FeMo-cofactor, then the [MFe4S6] core, less the heterometal, would constitute the remainder. The idiosyncratic, cofactor-like features of the [MFe4S6] clusters (i.e., trigonal pyramidal metal sites and nonrhomb faces) are discernible elsewhere only in the [Fe6,7S6]-X-PR3 clusters shown in Fig. 2 (21); a phosphine ligand environment therefore surfaced as the common denominator for these structural features, perhaps due to the preferential stabilization of reduced oxidation states by this ligand class relative to anion donors. This perceived relationship, conjoined with speculation that the FeMo-cofactor might be a predominantly ferrous cluster in the inactive, structurally characterized state, encouraged deeper investigation into phosphine-ligated Fe-S species.
Scheme 3.

Synthesis of [MFe4S6] heterometallic clusters. (M, x) = (Mo, 4), (V, 7).
Reduction of phosphine-ligated [MFe3S4] cubanes (M = Fe, Mo, V) yielded edge-bridged double-cubane [M2Fe6S8] clusters that approach the core composition and overall extended geometry (as opposed to more compact, higher-symmetry structures) of symmetrized versions of the FeMo-cofactor (23–25). In an effort to realize clusters with cofactor-exact 8:9 metal/sulfide stoichiometries, reactions of these double-cubane products with sulfide reagents were explored; we note two relevant discoveries. First, treatment of a [Mo2Fe6S8] cluster with two equivalents of (Et4N)SH resulted not in insertion of sulfide ligands but rather the isolation of a reduced cluster of unaltered composition but rearranged core geometry (Scheme S4, a; ref. 26). This rearrangement was notable in the emergence of tight metal–metal interactions that give rise to ostensibly three-coordinate iron sites (if an Fe—Fe bond is excluded from consideration); this represents the closest approach thus far to a three-coordinate iron-sulfide environment in synthetic chemistry. Second, if related but more reduced [M2Fe6S8] clusters are reacted with three to four equivalents of the same hydrosulfide reagent, net incorporation of sulfur is achieved, with formation of rearranged cluster cores of the desired [M8S9] composition (Scheme S4, b; ref. 27). Despite the constitutional similarity, the resultant clusters do not adopt cofactor-like cores but instead bear a striking near-congruence to the native form of the nitrogenase P-cluster (if bridging μ2-sulfides are substituted for the μ2-cysteinates in the biological system). This structural motif also occurs in other, more complex clusters derived from sulfidation of reduced edge-bridged double-cubane species (27) and therefore seems to reflect intrinsically accessible and stable chemistry. If this sulfide-induced rearrangement is general for juxtaposed, reduced Fe-S cubane clusters, this transformation could represent the chemical basis by which the P-cluster is assembled. The existence of substantive redox- and sulfide-dependent rearrangements in extended, higher-nuclearity clusters in both synthetic and biological (i.e., POX vs. PN) contexts also introduces other ramifications. First, the prospect of cluster rearrangement suggests a possible role during the function of the FeMo- cofactor; this is discussed further in the next section. Second, the compositional and geometric similarities between the FeMo-cofactor and PN-cluster structures (both are octanuclear with cuboidal halves) and their occurrence within the same protein hint at a chemical and evolutionary relationship by core rearrangement. These are intriguing propositions for further study.
Scheme 4.

Selected reactions of edge-bridged double-cubane clusters. LnM = (Et3P)(Cl4cat)Mo for Cl4cat-ligated products or TpM for Tp-ligated products; Cl4cat, tetrachlorocatecholate(2−); Tp, hydrotris(pyrazolyl)borate(1−); (M, x = z) = (Mo, 3), (V, 4).
A different synthetic approach emphasizes the construction of the individual sulfur-voided cuboidal subunits within the FeMo-cofactor. The isolated [Fe4S3] core is a long-known but exceedingly rare Fe-S motif, and examples are limited to the venerable homoleptic nitrosyl cluster known as Roussin's black anion {[Fe4S3(NO)7]−} and to phosphine derivatives of more recent vintage {[Fe4S3(NO)4(PR3)3]0,+} (28). Coucouvanis et al. (29) have prepared analogous heterometallic [MoFe3S3] clusters that resemble the other subunit of the cofactor by carbonylation of an edge-bridged double-cubane cluster (Scheme S4, c); this reaction presumably proceeds by formation of a CO-ligated single-cubane species followed by phosphine-coupled desulfurization. From these few examples, it seems at present that strong-field, low-valent (subferrous) iron sites are necessary to stabilize [MFe3S3] cuboidal cores in discrete form.
The Interstitial Atom (2002)
Very recently, a high-resolution crystallographic analysis (1.16 Å) of the MoFe protein has revealed a μ6-interstitial atom§ at the center of the FeMo-cofactor (Fig. 4; ref. 30). This discovery, if correct, is arguably the most significant addition to nitrogenase knowledge since the advent of the first macromolecular structures. Although the new structure refinement did not allow identification of this ligand with certainty, the electron density and bond distances are consistent with a light (2p) element, perhaps carbon, nitrogen, or oxygen, and the resolution dependence of the electron density best matches the curve generated by a nitrogen atom. These considerations plus the universal acceptance of the FeMo-cofactor as the site of nitrogen reduction have led to the tentative assignment of nitride as the newfound core ligand.
Figure 4.

Revised FeMo-cofactor structure derived from the most recent, high-resolution macromolecular structure determination. N*, histidine; S*, cysteinate.
The most obvious consequence of this finding stems directly from its impact on cluster environment: The revelation of the internal ligand makes all iron centers in the cofactor four-coordinate and (distorted) tetrahedral. This revision eliminates the most chemically troubling aspect of the original structural model, the bizarre three-coordinate, unsaturated iron sites that have remained noticeably absent in synthetic systems, and it removes any need to invoke arcane explanations such as metal–metal bonding, very reduced iron states, or indiscernible hydride ligands to account for the resting-state cluster geometry. Although the FeMo-cofactor is still singular and remarkable from an inorganic perspective, it can now be viewed (reassuringly) as a structurally and chemically plausible, albeit unusual, weak-field Fe-S cluster.
The existence of this central atom raises related questions of provenance and function. Does this ligand originate from cluster biosynthesis or cluster reactivity? If the former, does this ligand have some function as an organizing or templating agent during cluster assembly? If the latter, does the internal ligand represent adventitious trapped material (nitride from substrate, oxide from water?) within a dead-end cluster form outside the catalytic cycle, or is it an essential participant in or mediator of cluster function? In keeping with the nature of this subject, the addition of a single atom has raised far more questions than answers.
The structural revision of the FeMo- cofactor nevertheless offers a cautionary mechanistic tale. In the original cofactor model, the presence of six putative three-coordinate iron sites, together with the apparent existence of an all-Fe nitrogenase, led to wide speculation (ref. 31 and references cited therein) concerning the participation of iron in dinitrogen reduction and, in some conjectures, the consequent incorporation of nitrogen ligation as part of the Fe-S cluster core during catalysis. The amended cofactor model now contains only coordinatively stable metal centers and no obvious substrate binding sites. There is an irony here: The addition of the interstitial donor removes much of the impetus behind Fe-mediated mechanistic hypotheses, yet if the ligand turns out to be nitride, it could also provide the first (and only) direct observational evidence¶ to support those original proposals! The seeming absence of a binding site in the resting-state cofactor structure suggests that structural changes are needed before the cluster becomes substrate-competent. If the interstitial ligand actually participates in the reaction chemistry, either directly during turnover or indirectly by activation/release before catalysis, dramatic rearrangements of the cluster core geometry will certainly occur. We have noted the observation of major redox- and chemically induced rearrangements in synthetic clusters of roughly comparable composition and geometry. The implications raised by the new interstitial ligand and the synthetic chemistry underscore the hazards in formulating mechanistic hypotheses based on resting-state perceptions.
New Synthetic Frontiers
The structural revision of the FeMo- cofactor necessitates a reappraisal of synthetic strategy. The focus of immediate attention is, of course, the new monatomic ligand. What is its identity? This is the overriding first concern, because the chemistries of carbide, nitride, and oxide (if these are indeed the only candidates) differ radically. Although this issue is currently unsettled, we will assume it to be nitride for the present discussion.
In molecular transition metal chemistry,‖ nitride is found most commonly as a terminal ligand (≈300 structurally characterized complexes); to sustain its 3− formal charge at a single metal site, a multiply bonded metal-nitride moiety and a high-valent metal are practical criteria, and known examples are restricted to groups 5–8. As bridging ligands, nitrides have been observed to span anywhere from two to six metal centers, with dinuclear bridges being most prevalent (≈150 complexes). For μ2-nitrides, multiple bonding remains important, although the lower per-metal bond order and greater charge delocalization expand the range of acceptable environments relative to the terminal binding mode. In addition, localization of the multiple bond and asymmetric bridging are widespread in these systems, particularly for mixed [M(μ2-N)M′] units; very disparate local metal environments therefore are possible within the dinuclear fragment as long as one site is an early-to-middle transition element in a moderate-to-high oxidation state.
Of greatest interest in the present context are cluster-bound nitrides, i.e., bridging nitrides of μ3 or higher, which exist within a smaller set of compounds (≈100 total, about evenly distributed for μ3–μ6 cases). These nitride complexes fall neatly into three general classes: (i) mid- to high-valent early-transition metal clusters (groups 4–6), which describe all complexes containing μ3- nitrides and can be viewed as a further extension of the environment described for terminal and μ2 species; (ii) low-valent middle- to late-transition metal carbonyl clusters (mainly groups 8–9 but also heterometallic cores containing these elements plus group 6–10 metals), in which almost all nitrides with the highest bridging modes (μ4–6) occur; and (iii) Au(I) phosphine clusters, which form a meager set of μ4- and μ5-N compounds distinguished by their unique aurophilic structures. To some extent, the preference of oxidized early metal clusters for lower nitride-bridging modes can be attributed to their electron deficiency, which makes π donation from the nitride bridge more favorable than additional σ-derived bridging.** For the reduced, electron-rich metal centers in the carbonyl and gold clusters, additional bridging seems advantageous in compensating for the basicity of the nitride ligand.
The various nitride-bridging modes manifest regular preferences for coordination geometry (Table 1). True interstitial nitrides, as defined by completely internal coordination within a cluster polyhedron, are encountered at μ4,5 for Au(I) and μ5,6 for transition metal clusters. From the existing examples, bridging modes in excess of μ6 seem unfavorable for nitride; in cases where the potential interstitial nuclearity can exceed six, the internal nitrides are invariably found coordinated to only a pentanuclear (μ5) subset.
Table 1.
Cluster-bridging nitride geometries and representative complexes
| Bridging geometry | Representative cluster | CSD refcode* |
|---|---|---|
| μ3 | ||
| Pyramidal | [{Cp*Ti(μ-NH)}3(μ3-N)] | SAXSEP |
| T-shaped | [{CpMo(CO)2}2{CpMo(O)}(μ3-N)] | BAXFEL |
| Trigonal planar† | [{CpMo(O)(SMe)(μ3-N)}{(CpMo)2(μ2-SMe)3}] | GOTZO |
| μ4 | ||
| Sawhorse | [Fe4(μ4-N)(CO)12]− | PRUFEB |
| Basal‡ trigonal pyramidal† | [PtRu3(μ-H)(μ4-N)(CO)10P(i-Pr)3] | QUDMOR |
| Interstitial tetrahedral§ | [Au4(μ4-N)(PPh3)4]+ | CUGPUP |
| μ5 | ||
| Basal‡ square pyramidal | [Fe5(μ5-N)(μ-CO)2(CO)12]− | CABROM10 |
| Interstitial distorted¶ | [PtRh10(μ5-N)(μ-CO)10(CO)11]3− | BENPOZ |
| Interstitial trigonal bipyramidal§ | [Au5(μ5-N)(PPh3)5]2+ | SEVJUY |
| μ6 | ||
| Interstitial octahedral | [Fe6(μ6-N)(μ-CO)3(CO)12]3− | ZUCWAV |
| Interstitial trigonal prismatic | [Co6(μ6-N)(μ-CO)9(CO)6]− | PIMNCO |
Cambridge Structural Database, version 5.23.
Rare.
Exposed nitrides centered in (or protruding slightly from) the basal face (triangular or square) of a pyramidal coordination polyhedron.
Au(I) only.
Usually found within high-nuclearity clusters.
It is clear from our brief survey that the environment in the FeMo-cofactor is without direct precedent in nitride chemistry. The dissimilarities arise not from simple differences in ligand set or cluster geometry but rather from fundamental divergences in essential metal properties. For example, iron nitrides are known in tetra-, penta-, and hexanuclear carbonyl clusters (Fig. 5) and in oxidized dinuclear species supported by multidentate ancillary ligands (32). Likewise, the trigonal prismatic μ6-N interstitial geometry occurs in metal carbonyl clusters (see Table 1). Weak-field, exchange-labile, redox-active environments typical of the iron sites in Fe-S clusters, however, do not exist in nitride chemistry as it currently stands. The development and adaptation of new and existing fundamental nitride chemistry to this end is therefore a clear and present synthetic challenge that must be addressed in conjunction with (and perhaps before) efforts aimed at the systematic preparation of (Mo-)Fe-S-N clusters. We note that the recent chemistry of iron-amide and -imide clusters (33) may have some bearing on this topic. These clusters (Fig. 6) are ligated by basic nitrogen anion donors that are chemical cousins to nitride, evince properties that broadly resemble those of Fe-S clusters, and, in terms of metal attributes, are perhaps more closely connected to hypothetical cofactor-relevant nitride environments than existing iron-nitride species. Their synthesis and reactivity characteristics therefore could provide guidance and possible starting points for further investigation directed toward weak-field iron-nitride chemistry.
Figure 5.

Representative nitride-containing iron-carbonyl clusters: [Fe4(μ4-N)(CO)12]− (Left), [Fe5(μ5-N)(μ-CO)2(CO)12]− (Center), and [Fe6(μ6-N)(μ-CO)3(CO)12]3− (Right). See Table 1 for corresponding Cambridge Structural Database refcodes.
Figure 6.

Representative weak-field iron-imide clusters. X, monoanions (halide/thiolate).
Prospects
Traditionally, synthetic inorganic chemistry has provided the molecular intuition needed to interpret metal behavior in biological systems. Today, in a reversal of circumstance, metallobiomolecules are posing fundamental inorganic questions whose answers lie outside our existing knowledge. It is a fitting symmetry that the relationship between inorganic chemistry and biology has come full circle.
Speculative synthetic analogues can play a critical role in bridging the two domains and advancing both. The continuing synthetic investigation of nitrogenase illustrates the synergies of this approach. As described in this account (and we have not discussed the extensive enzyme-inspired studies aimed at functional simulation), the speculative approaches motivated by this demanding problem have provided insight into the metalloclusters of nitrogenase. Equally important, these efforts have also contributed broadly to basic areas of inorganic chemistry that would probably not have been examined otherwise; the intrinsic value of such exploration should not be underestimated. The most recent revision of the FeMo-cofactor structure presents yet another opportunity to expand our understanding of the biological system in particular and of fundamental inorganic chemistry in general.
The synthetic problem of nitrogenase, nevertheless, remains unsolved. To this we give Captain Ahab's oath: “Aye, aye! and I'll chase him round Good Hope, and round the Horn, and round the Norway Maelstrom, and round perdition's flames before I give him up.”
Acknowledgments
We thank Dr. R. Panda for assistance. Research on the subject of this perspective has been supported by National Institutes of Health Grant GM-28856 (to R.H.H.), National Science Foundation Grant CHE- 9984645 (to S.C.L.), and an Arnold and Mabel Beckman Foundation Young Investigator Award (to S.C.L.).
Footnotes
The distinction between corroborative and speculative studies has been made previously (34). Our present classification subsumes the original conceptions for both categories as largely corroborative while reserving the speculative designation for biological situations that demand new chemical perceptions.
The prospect of easy synthetic access has waned in recent years with the discovery of an extensive biosynthetic apparatus, external to the nitrogenase enzyme, for cofactor assembly.
The original void in the cofactor core now seems to be an artifact of Fourier-series termination effects arising from the limited resolution of the earlier data.
There is also a strangely appropriate correspondence to industrial nitrogen fixation (the Haber–Bosch process), where iron-coordinated, surface-bound nitrides are the first chemical intermediates observed after dissociative chemisorption of dinitrogen (see ref. 35).
Our analysis is based on a survey of the Cambridge Structural Database, version 5.23 (April 2002) (Cambridge University, Cambridge, England).
This correlation is not absolute, as demonstrated by isolated instances of μ5- and μ6-nitride in penta- and hexanuclear amidoimidonitrido Zr(IV) clusters (Cambridge Structural Database refcodes VUHHIP and VUHHOV) (see ∥ footnote).
References
- 1.Ibers J A, Holm R H. Science. 1980;209:223–235. doi: 10.1126/science.7384796. [DOI] [PubMed] [Google Scholar]
- 2.Howard J B, Rees D C. Chem Rev (Washington, DC) 1996;96:2965–2982. doi: 10.1021/cr9500545. [DOI] [PubMed] [Google Scholar]
- 3.Burgess B K, Lowe D J. Chem Rev (Washington, DC) 1996;96:2983–3011. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 4.Eady R R. Chem Rev (Washington, DC) 1996;96:3013–3030. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]
- 5.Smith B E. Adv Inorg Chem. 1999;47:160–218. [Google Scholar]
- 6.Christiansen J, Dean D R, Seefeldt L C. Annu Rev Plant Physiol Plant Mol Biol. 2001;52:269–295. doi: 10.1146/annurev.arplant.52.1.269. [DOI] [PubMed] [Google Scholar]
- 7.Burgess B K. Chem Rev (Washington, DC) 1990;90:1377–1406. [Google Scholar]
- 8.Cramer S P, Hodgson K O, Gillum W O, Mortenson L E. J Am Chem Soc. 1978;100:3398–3407. [Google Scholar]
- 9.Wolff T E, Berg J M, Hodgson K O, Frankel R B, Holm R H. J Am Chem Soc. 1979;101:4140–4150. [Google Scholar]
- 10.Wolff T E, Berg J M, Power P P, Hodgson K O, Holm R H. Inorg Chem. 1980;19:430–437. [Google Scholar]
- 11.Fomitchev D V, McLauchlan C C, Holm R H. Inorg Chem. 2002;41:958–966. doi: 10.1021/ic011106d. [DOI] [PubMed] [Google Scholar]
- 12.Kovacs J A, Holm R H. Inorg Chem. 1987;26:702–711. [Google Scholar]
- 13.Johnson M K, Duderstadt R E, Duin E C. Adv Inorg Chem. 1999;47:1–82. [Google Scholar]
- 14.Dobbek H, Svetlitchnyi V, Gremer L, Huber R, Meyer O. Science. 2001;293:1281–1285. doi: 10.1126/science.1061500. [DOI] [PubMed] [Google Scholar]
- 15.Drennan C L, Heo J, Sintchak M D, Schreiter E, Ludden P W. Proc Natl Acad Sci USA. 2001;98:11973–11978. doi: 10.1073/pnas.211429998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou J, Raebiger J W, Crawford C A, Holm R H. J Am Chem Soc. 1997;119:6242–6250. [Google Scholar]
- 17.Dance I, Fisher K. Prog Inorg Chem. 1994;41:637–803. [Google Scholar]
- 18.Kim J, Rees D C. Nature. 1992;360:553–560. doi: 10.1038/360553a0. [DOI] [PubMed] [Google Scholar]
- 19.Mayer S M, Lawson D M, Gormal C A, Roe S M, Smith B E. J Mol Biol. 1999;292:871–891. doi: 10.1006/jmbi.1999.3107. [DOI] [PubMed] [Google Scholar]
- 20.Peters J W, Stowell M H B, Soltis S M, Finnegan M G, Johnson M K, Rees D C. Biochemistry. 1997;36:1181–1187. doi: 10.1021/bi9626665. [DOI] [PubMed] [Google Scholar]
- 21.Nordlander E, Lee S C, Cen W, Wu Z Y, Natoli C R, Di Cicco A, Filipponi A, Hedman B, Hodgson K O, Holm R H. J Am Chem Soc. 1993;115:5549–5558. [Google Scholar]
- 22.Cen W, MacDonnell F M, Scott M J, Holm R H. Inorg Chem. 1994;33:5809–5818. [Google Scholar]
- 23.Zhou H-C, Holm R H. Inorg Chem. 2003;42:11–21. doi: 10.1021/ic020464t. [DOI] [PubMed] [Google Scholar]
- 24.Demadis K D, Campana C F, Coucouvanis D. J Am Chem Soc. 1995;117:7832–7833. [Google Scholar]
- 25.Hauser C, Bill E, Holm R H. Inorg Chem. 2002;41:1615–1624. doi: 10.1021/ic011011b. [DOI] [PubMed] [Google Scholar]
- 26.Osterloh F, Achim C, Holm R H. Inorg Chem. 2001;40:224–232. doi: 10.1021/ic000617h. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Zuo J-L, Zhou H-C, Holm R H. J Am Chem Soc. 2002;124:14292–14293. doi: 10.1021/ja0279702. [DOI] [PubMed] [Google Scholar]
- 28.Goh C, Holm R H. Inorg Chim Acta. 1998;270:46–54. [Google Scholar]
- 29.Coucouvanis D, Han J, Moon N. J Am Chem Soc. 2002;124:216–224. doi: 10.1021/ja0110832. [DOI] [PubMed] [Google Scholar]
- 30.Einsle O, Tezcan F A, Andrade S, Schmid B, Yoshida M, Howard J B, Rees D C. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 31.Lovell T, Li J, Case D A, Noodleman L. J Am Chem Soc. 2002;124:4545–4547. doi: 10.1021/ja012311v. [DOI] [PubMed] [Google Scholar]
- 32.Dutta S K, Beckmann U, Bill E, Weyhermüller T, Wieghardt K. Inorg Chem. 2000;39:3355–3364. doi: 10.1021/ic0001107. [DOI] [PubMed] [Google Scholar]
- 33.Duncan J S, Nazif T M, Verma A K, Lee S C. Inorg Chem. 2003;42:1211–1224. doi: 10.1021/ic025825j. [DOI] [PubMed] [Google Scholar]
- 34.Hill H A O. Chem Br. 1976;12:119–123. [Google Scholar]
- 35.Schlögl R. In: Handbook of Heterogeneous Catalysis. Ertl G, Knözinger H, Weitkamp J, editors. Vol. 4. New York: VCH; 1997. pp. 1697–1748. [Google Scholar]