

Abstract
The structures and the O—O and M—O bonding characters of a series of reported side-on (η2) 1:1 metal complexes of O2 are analyzed by using density functional theory calculations. Comparison of the calculated and experimental systems with respect to O—O bond distance, O—O stretching frequency, and O—O and M—O bond orders provides new insights into subtle influences relevant to O2 activation processes in biology and catalysis. The degree of charge transfer from the generally electron-rich metals to the dioxygen fragment is found to be variable, such that there are species well described as superoxides, others well described as peroxides, and several cases having intermediate character. Increased charge transfer to dioxygen takes place via overlap of the metal dxy orbital with the in-plane π* orbital of O2 and results in increased M—O bond orders and decreased O—O bond orders. Comparison of theory and experiment over the full range of compounds studied suggests that reevaluation of the O—O bond lengths determined from certain x-ray crystal structures is warranted; in one instance, an x-ray crystal structure redetermination was performed at low temperature, confirming the theoretical prediction. Librational motion of the coordinated O2 is identified as a basis for significant underestimation of the O—O distance at high temperature.
With the longstanding goal of understanding in detail how dioxygen binds and is activated at metal centers in biological and catalytic systems, great effort has been expended to characterize the structures, physicochemical properties, and reactivity of metal–dioxygen complexes (1–8). As the initially formed species in most oxidation processes, 1:1 metal-O2 adducts are of special interest, particularly in view of their postulated involvement in enzymes that react with dioxygen at an isolated monometallic active site, such as Cu in dopamine β-monooxygenase or galactose oxidase (9). Two binding modes have been identified in 1:1 metal-O2 complexes, end-on (η1) and side-on (η2). These adducts have been further defined as superoxo or peroxo complexes, primarily on the basis of x-ray structural data (O—O bond distance) and vibrational spectroscopy (O—O stretching frequency, νOO) (1–4). Thus, compounds with an O—O bond length of ≈1.4–1.5 Å and νOO between ≈800 and 930 cm−1 are designated as peroxides, whereas those with O—O ≈1.2–1.3 Å and νOO ≈1,050–1,200 cm−1 fall into the superoxide category. On this basis, and keeping in mind the caveat that it is often difficult to assess experimentally the actual amount of charge transferred to dioxygen on complexation, the majority of the known η2 1:1 compounds are generally agreed to be best described as peroxo complexes (1, 5, 8).
Reports of η2-superoxo complexes are more rare, with structurally characterized examples limited to 1 (10), 2 (11), 3 (K. Qin, C. D. Incarvito, A.L.R., and K.H.T., unpublished results), 4 (12), and (TpMe2)2SmO2 (TpMe2, Tris(3,5-dimethylpyrazol-1-yl)hydroborate) (13) (Scheme S1). In the course of the investigation of 6, for which intermediate character of the dioxygen unit was found (14, 15), we noted puzzling incongruities among the data reported for these complexes (Table 1). For example, whereas 1 and 2 (as reported in ref. 11, denoted “high T”; see below) show similarly short O—O distances in their x-ray structures, the measured νOO for 1 is 151 cm−1 higher than that measured for 2 (high T). The O—O distance in 4 is 0.065 Å greater than in 2 (high T), yet the νOO for 4 is higher by 111 cm−1. Moreover, the experimentally determined νOO values for 2 and 6 are virtually identical, yet x-ray data and density functional level of theory calculations for 6 indicate that it has a >0.1 Å longer O—O distance. The apparent lack of a correlation between O—O distance and νOO value for these systems (all of which exhibit 18O-isotope shifts consistent with a simple harmonic oscillator) led us to investigate these and several other representative complexes (Table 1) using electronic structure calculations at the density functional level. These studies reveal variation of the degree of charge transfer from metal to coordinated dioxygen across the series of η2 complexes. In addition, comparison of theory and experiment brings to light several key correlations and suggests reevaluation of O—O distances determined by x-ray crystallography for several cases. The anomalous distances are rationalized and the theoretical results confirmed through consideration of new low temperature x-ray crystal structural data for 2.
Scheme 1.

Table 1.
Experimental and theoretical O—O bond lengths, vibrational frequencies, and Mayer bond orders (B.O.)
| Molecule | Ref.* | Electronic ground state | O—O bond length, Å
|
νoo [Δ18O, Δ18O2], cm−1
|
Mayer B.O.
|
|||
|---|---|---|---|---|---|---|---|---|
| Experiment | Calc (t) | Experiment | Calc (t) | O—O | M—O | |||
| 1 | 10 | 1A′ | 1.22(3) | 1.329 | 1112 [†, 52] | 1124 [31, 66] | 1.174 | 0.565 |
| 2 (low T) | 11‡ | 3A" | 1.355(3) | 1.380 | 961 [24, 53] | 987 [25, 52] | 0.995 | 0.812 |
| 2 (high T) | 11 | 1.262(8) | ||||||
| 3 | §, 12 | 3A′ | 1.325(3) | 1.339 | 1104 [†, 60] | 1086 [31, 61] | 1.167 | 0.640 |
| 4 | 12 | ¶ | 1.327(5) | ¶ | 1072 [†, 65] | ¶ | ¶ | ¶ |
| 5 | 12 | 3A′ | † | 1.361 | 1027 [†, 58] | 1034 [28, 59] | 1.092 | 0.718 |
| 6 | 14, 15 | 1A | 1.44(2) | 1.376 | 968 [25, 51] | 1013 [28, 57] | 1.049 | 0.594 |
| 7 | 26 | 1A | 1.411(18) | 1.400 | 891 [†, 52] | 941 [26, 53] | 1.001 | 0.681 |
| 8 | 27 | 4B2 | † | 1.377 | 983 [26, 50] | 1009 [34, 60] | 1.039 | 0.874 |
| 8 | 28 | 5B2 | 1.421(5) | 1.430 | † | 937 [25, 52] | 1.003 | 0.770 |
| 9 | 34 | ¶ | 1.43 | ¶ | 892 [†, †] | ¶ | ¶ | ¶ |
| (TpMe2)2SmO2 | 13 | ¶ | 1.319 | ¶ | 1124 [†, †] | ¶ | ¶ | ¶ |
| O2 | 29 | 3Σg | 1.208 | 1.220 | 1549 [43, 87] | 1550 [44, 89] | 1.772 | |
O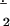
|
30 | 2Πg | 1.363 | (1090)‖ | 1097 [31, 63] | 1.256 | ||
| LiO2 | 29 | 2A2 | † | 1.364 | 1097 [30, 62] | 1097 [31, 63] | 1.271 | 0.437 |
| NaO2 | 31 | 2A2 | † | 1.369 | 1094 [30, 61] | 1087 [30, 65] | 1.301 | 0.328 |
| NiO2 | 32 | 3B1 | † | 1.382 | 966 [26, 52] | 982 [27, 55] | 1.091 | 0.860 |
| BrAlO2 | 33 | 1A1 | † | 1.691 | 524 [12, 23] | 543 [13, 25] | 0.877 | 1.007 |
Except where noted, reference cited is source of experimental data.
Not reported.
Value for O—O bond length is from this work.
K. Qin, C. D. Incarvito, A.L.R., and K.H.T., unpublished results.
Not computed.
Value is an estimate from experimental data, for anion well separated from any counterion.
Materials and Methods
Computational Methods.
All molecular geometries were fully optimized at the density functional level of theory by using the exchange and correlation functionals of Perdew and coworkers (16, 17) as modified by Adamo and Barone (mPWPW91, ref. 18). Atomic orbital basis functions were taken for metals and Cl from the compact relativistic effective core potential basis CEP-31G (19), for N and O from the 6–311G* basis, for B and C from the 6–31G basis, and for H from the minimal STO-3G basis (20). For superoxide radical anion, the oxygen basis set was 6–311+G*. Exploratory calculations indicated that, for the properties studied here, inclusion of diffuse basis functions on N and O had a significant effect only for anionic O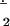 .
.
This combination of functionals and basis sets was chosen after a survey of several methods to assess their performance with respect to computation of structural parameters and vibrational frequencies for the relevant and well characterized compound [(TMPDA)2Cu2(μ-O)2]2+ (TMPDA, N,N,N′,N′-tetramethyl-1,3-propanediamine). Key experimental data for this molecule include Cu—O and Cu—Cu bond lengths of 1.81 and 2.85 Å (from near-edge x-ray absorption fine structure measurements) (21); predicted values at the mPWPW91 level are 1.837 and 2.869 Å. The key stretching frequency of the bis(μ-oxo)dicopper core has a measured Raman frequency of 608 cm−1 with a frequency shift of 26 cm−1 for doubly 18O-labeled material (21); the corresponding values at the mPWPW91 level are predicted to be 608 cm−1 (28 cm−1).
All presented computational data are for species in their electronic ground states as verified by the stability of the Kohn–Sham determinants to spin and symmetry changes (see Table 1 for electronic ground-state symmetries). For all structures having singlet electronic ground states, unrestricted Kohn–Sham determinants were compared with restricted analogs to verify stability of the latter. Restricted determinants were used for 7t, LiO2, NaO2, NiO2, and BrAlO2; unrestricted determinants were used for all other cases. Harmonic vibrational frequencies were computed analytically for all species to ensure their character as local minima and to evaluate isotope effects on O—O stretching frequencies.
Oxygen–oxygen and metal–oxygen bond orders were computed from the mPWPW91 density matrices by the method of Mayer (22). Partial atomic charges (23) were also computed by the methods of Löwdin (24) and Mulliken (25); however, because no significant correlations between charges and other properties were identified, we will not discuss these data. Oxygen–atom and metal orbital spin densities were determined from Mulliken population analysis.
X-Ray Crystallography.
A Bruker/Nonius (Billerica, MA) platform goniometer with an APEX charge-coupled device detector and a Kryo–Flex low-temperature unit was used to collect data. The structure was processed with the parameters from the earlier room-temperature determination (11). All nonhydrogen atoms were refined with anisotropic thermal parameters and hydrogen atoms were treated as idealized contributions. All crystallographic software is contained in the smart, saint, and shelxtl program libraries (Bruker/Nonius). Relevant data are listed in Table 2.
Table 2.
Crystal data and structure refinement information for 2
| Empirical formula | C24H40BCoN6O2 |
| Formula weight | 514.36 |
| Temperature | 150(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Monoclinic |
| Space group | P2(1)/n |
| Unit cell dimensions | a = 9.5549(6) Å |
| b = 29.8935(18) Å | |
| c = 9.6027(6) Å | |
| β = 101.8720(11)° | |
| Volume | 2,684.1(3) Å3 |
| Z, Z′ | 4, 1 |
| Density (calculated) | 1.273 Mg/m3 |
| Absorption coefficient (MoKα) | 0.671 mm−1 |
| Crystal color, size | deep red-brown, 0.34 × 0.13 × 0.08 mm3 |
| Reflections collected | 29,356 |
| Independent reflections | 5,703 [R(int) = 0.0342] |
| Completeness to θ = 26.75° | 99.9% |
| Absorption correction | Empirical (SADABS) |
| Maximum and minimum transmission | 0.9483 and 0.8041 |
| Data/restraints/parameters | 5703/0/307 |
| Goodness-of-fit on F2 | 1.456 |
| Final R indices [I > 2σ(I)] | R1 = 0.0440, wR2 = 0.1229 |
| R indices (all data) | R1 = 0.0523, wR2 = 0.1277 |
| Largest difference peak and hole | 0.674 and −0.489 e.Å−3 |
Results and Discussion
To better understand the nature of the interactions between the metal and the O2 fragments in various 1:1 η2 adducts, we here undertake electronic structure calculations at the density functional level for several examples. As summarized in Scheme S1 and Table 1, our theoretical models (labeled with a t to facilitate discussion distinguishing them from the experimentally characterized systems) include all substituents and thus are exact replicas for the cases 2 (11), 3 (K. Qin, C. D. Incarvito, A.L.R., and K.H.T., unpublished results; ref. 12), 5 (12), 6 (14, 15), and 7 (26). In the case of 1 (10), the theoretical model simplifies the 5-isopropyl groups on the pyrazole rings for computational efficiency, and for the same reason the phenyl rings pendant on the porphinato ligands in 8 (27) and 8− (28) were deleted in the theoretical models. In addition to these various transition–metal complexes, we also consider the following pertinent small molecules: molecular oxygen (O2, 29), the free superoxide radical anion (O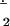 ) (30), sodium and lithium superoxide MO2 (M, Na or Li) (29, 31), NiO2 (32), and BrAlO2 (33).
) (30), sodium and lithium superoxide MO2 (M, Na or Li) (29, 31), NiO2 (32), and BrAlO2 (33).
For the sake of further comparison, we include in our analysis reported experimental data for 4 (12), 9 (34), and (TpMe2)2SmO2 (13), but theoretical models are not constructed. In the case of 4, we wish to avoid possible difficulties associated with the counterion of this cationic metal–dioxygen complex. With respect to 9, Kitajima et al. (34) reported two isomeric forms (“blue” and “brown”) having identical νOO values and O—O bond lengths but varying in whether a hydrogen bond was observed between the pyrazole and oxygen fragments in the crystal structures. We elect not to address this complication here. Finally, the complex (TpMe2)2SmO2 is sufficiently large to make any reasonable model impractical.
A comparison of the theoretical and experimental structures indicates excellent agreement in general for heavy-atom bond lengths and bond angles in the organic ligands and, for the most part, about the transition metals (agreement is typically within the reported experimental error in the x-ray crystal structures). One exception, however, is that significant discrepancies were observed in several instances for geometrical data associated with the coordinated oxygen fragment, particularly the O—O distance. These discrepancies prompted us to further investigation.
Table 1 lists measured and computed O—O bond lengths, measured and computed O—O vibrational frequencies, and computed O—O and M—O Mayer bond orders for the indicated species. A rapid quantitative measure of relationships between the various tabulated variables is provided by the Pearson R correlation matrix, which is published as Table 3 in supporting information on the PNAS web site, www.pnas.org.
The computed νOO values are of very high quality, as indicated by the excellent correlation with the experimental numbers (R = 0.997, 16O only). As further demonstration of the agreement between these values, the data approximately fit the equation y = mx + b, where m is unity and b is zero; linear regression of the theoretical frequencies on the experimental ones subject to the constraint of zero intercept delivers a regression line (slope = 0.988) with an F value of 1,372.7. As a final quantitative measure, the mean unsigned error in the computed frequencies is 18.0 cm−1, which is ≈2% of the average frequency. A separate examination of 18O isotope effects also shows very good agreement between theory and experiment. In 10 cases, there is agreement to within 1 cm−1, and the remaining cases are also very close with the exception of 1, where IR spectral interpretation was complicated both by compound impurities and overlapping IR bands (10).
Interestingly, correlation coefficients between the computed (t) O—O bond lengths and the νOO values, both measured and predicted, also are near unity in absolute value (R = 0.925 and 0.937, respectively). Such high correlations suggest a near linear relationship between these two properties. The quality of this relationship is illustrated in Fig. 1, which includes all experimental and theoretical points for which both data are available except for BrAlO2, which is discussed further below. Although there clearly appears to be, overall, a significant linear relationship, there are two significant outliers to the correlation, namely the experimental points for 1 and 2 (in the case of 2, the originally reported O—O bond length of 1.262 Å is plotted as “high T”). When these two points are included, the Pearson R for the best fit line to the data in Fig. 1 is 0.781, but if they are removed from the regression, then R jumps to 0.950.
Figure 1.

Plot of O—O stretching frequencies (νOO, cm−1) vs. O—O bond lengths (Å) for all cases where both data are available (BrAlO2 case excluded). Blue circles represent theoretical data points, and red squares represent experimental ones. Cases where experiment and theory are simultaneously available are labeled. The dashed line represents a least-squares linear fit to all data shown, excluding the experimental points for 1 and 2 (high T).
Such significant deviation in these points merits closer attention. Note that the O—O bond lengths reported for 1 and, originally, for 2 from x-ray crystal structural analysis are extraordinarily short (Table 1). In the case of 1, the length is essentially that of molecular oxygen, which, to maintain charge neutrality, would imply that the metal is Cu(I), a situation that seems highly unlikely. Moreover, the measured O—O stretching frequency for 1 is reasonably close to the values determined for 4 and (TpMe2)2SmO2, which have bond lengths of 1.327 and 1.319 Å, respectively. It would certainly be unusual for an O—O bond 0.1 Å shorter than the ones found in the latter two compounds to have so similar a stretching frequency. The originally reported bond length in 2 (1.2628 Å) is longer than that for molecular oxygen but still suggests little charge transfer from a formal Co(I) species. The two other molecules having O—O stretching frequencies closest to that reported for 2 are 6 and 8, both of which are reported to have O—O bond lengths close to 1.4 Å.
On the basis of this analysis, and with the assumption that systematic error in reported νOO values should be relatively small, it would seem that reevaluation of the crystal structural data for 1 and 2 is warranted with respect to the O—O bond lengths. Both structures were originally obtained at 22°C (high T). We have redetermined the structure of 2 at −123°C (low T) and find an O—O distance of 1.3553 Å, significantly longer than the distance reported previously (1.2628 Å) and in close agreement with that predicted by theory (1.380 Å). In all other respects, the low and high T structures are identical; the average Co—N distance is identical in both, and the average Co—O is 1.81 Å at 22°C and 1.83 Å at −123°C. Collection of data at high T on the same crystal used for the low T structure reproduced the structural parameters reported in 1990 (11). Thus, we can conclude that, whereas there are generally only minor and usually chemically insignificant differences in bond parameters as a function of the temperature of data collection, in this case, the difference in the O—O distance is extraordinarily large and reproducible. Moreover, from the corroborating evidence from the theoretical calculations, the high T O—O distance is too short by ≈0.1 Å.¶ We strongly suspect a similar problem with the room temperature structure of 1, although this has not been confirmed yet because of difficulties encountered in reproducing the synthesis of 1 according to the published report (10).
The basis for the significant difference between the O—O distances determined at low and high T may be understood by considering Fig. 2, in which low T and high T views of the N3CoO2 core of 2 are provided. Although the thermal ellipsoids are all larger in the high T structure, as expected, those of the oxygen atoms are especially enlarged and reveal that the O2 group is actively oscillating/rotating about an axis defined by the Co atom and the midpoint of the O—O bond. The effect of this motion (libration) on a bond distance is well documented (35–37) and always shortens the distance, although generally the difference is <0.003 Å and is seldom chemically important. The vibration/rotation activity of the O2 ligand in this case is especially large because its constraints are few, and its position, as determined crystallographically, is an averaged composite of all orientations in ≈1016 unit cells. Thermal motion is modeled as an ellipsoid as an approximation for motion that actually occurs in an arc. As the motion increases in amplitude, the approximation becomes progressively less satisfactory and shortens the distance as a function of the cosine of the librational angle. It is worth noting that this is an extreme example of the effects of libration, and in such cases only low-temperature data are reliable. When crystallographically determined distances obtained at higher temperatures differ greatly from well founded expectations, repeating the measurements at a much lower temperature would be advisable.
Figure 2.

Thermal ellipsoid (50%) figures for the N3CoO2 core of 2 determined at 22 and −123°C.
Given the strong correlation between O—O bond length and vibrational frequency, it is of significant interest to examine the extent to which variation may be associated with O—O bond order, and moreover the degree to which O—O bond order and M—O bond order are interrelated. The correlation between the computed Mayer O—O bond order and the experimental (or computed) O—O stretching frequency is very high (R = 0.916 and 0.903, respectively; the latter correlation is presented graphically in Fig. 3). The bond order is computed to be maximal for molecular oxygen, where it has a value of 1.772, and next highest for bare superoxide and its sodium and lithium adducts, where it takes on values of ≈1.3. This represents a reduction of ≈0.5, which is about the magnitude expected considering that reduction of molecular oxygen takes place by electron transfer into a π* antibonding orbital. In the limit of peroxide dianion, with two electrons transferred into the already half-occupied π* system of molecular oxygen, one would expect an O—O bond order of ≈1.0. This value is most closely approached for 7t (1.001) and 8t− (1.003), consistent with their formulations as Pd(II) and Mn(III) peroxide complexes, respectively (26, 28). Most of the remaining transition–metal–dioxygen complexes have O—O bond orders intermediate between these extremes (1.0–1.7), with the smallest bond order predicted for 2 and the largest for 1. These values are consistent with the comparatively low and high O—O stretching frequencies reported for 2 and 1, respectively.
Figure 3.

Predicted O—O stretching frequencies (cm−1) vs. Mayer O—O bond orders (unitless) using data in Table 1 (BrAlO2 case excluded). The dashed line represents a least-squares linear fit to the data shown.
The case of BrAlO2 exhibits a still smaller O—O bond order of 0.877. This molecule is computed to have a separation between the two oxygen atoms of 1.691 Å, a strikingly long distance consistent with a bond order prediction of less than one. Similarly long bond lengths have been predicted for Group 2 MO2 compounds by using levels of theory that provide good agreement with experimental matrix IR spectra (38, 39). Thus, it would appear that just as the transition–metal systems discussed above sample a continuum of O—O bond orders intermediate between peroxide and superoxide, other molecules can continue to extend the continuum into the region of bond orders below one. This reduced bond order derives from depletion of oxygen in-plane π bonding density through donation to the empty 3s orbital of Al3+. The combination of a fully occupied in-plane π* and a partially occupied in-plane π orbital leads to a net “negative” π bond order, thereby decreasing the normal bond order of unity expected for an isolated peroxide dianion. In the BrAlO2 case, sufficient O—O bonding remains so that a true O—O stretching frequency is still observed (it exhibits an 18O isotope shifts consistent with the mode being a pure O—O stretch), but the frequency itself is a very small 524 cm−1 (predicted 543 cm−1).‖
A separate point of interest is the degree to which O—O and M—O bond orders are found to be reasonably well (inversely) correlated ( R = −0.877). In the transition–metal complexes, population of the in-plane oxygen π* orbital takes place primarily via population of the metal dxy orbital, with which it overlaps (Scheme S2). Thus, decreased O—O bond orders should be associated with increased M—O bond orders, and this trend is indeed what is observed. Put differently, increasingly electron-rich transition–metal atoms reduce oxygen more effectively to the limit of peroxide dianion, and that reduction takes place in a fashion that increases metal–oxygen covalency. Again BrAlO2 represents a good example of the extreme in this relationship: its low O—O bond order is associated with the highest computed M—O bond order, 1.007. Related trends in the degree of electron transfer between metals and coordinated O2 have been noted for matrix-isolated alkali metal species as well (40–42).
Scheme 2.

In those cases having singlet ground states, there is an interesting technical aspect of the calculations that sheds some light on the issue of whether covalency or superexchange coupling is more responsible for that state's stability. It is well established empirically that singlets showing dominant superexchange coupling tend to be characterized by restricted→unrestricted instabilities in their density functional level of theory Kohn–Sham (KS) determinants, and that a qualitative measure of the degree to which superexchange dominates is the expectation value of the S2 operator applied to the KS determinant (43, 44). A perfect singlet biradical has an expectation value for S2 of 1.0, whereas a perfect covalent singlet shows no restricted instability and has an S2 expectation value of 0.0. For the transition–metal singlets studied here, 1t, 6t, and 7t, the expectation values of S2 were computed to be 0.701, 0.396, and 0.000. Thus, 7t is computed to be highly covalent, as expected for a Pd(II)–peroxide complex. Compound 1t, on the other hand, has substantial biradical character, although it is not complete, and is well described as a Cu(II)–superoxide species. The expectation value of S2 for the copper compound 6t indicates it has more covalent character than 1t, albeit that character is not complete. Such a situation might be expected given the considerably stronger donating character of the β-diketiminate ligand in 6t compared with the Tris(pyrazolyl)hydroborate ligand in 1t (45). One concludes, then, that 6t should show properties intermediate between a Cu(II)–superoxide and a Cu(III)–peroxide, as suggested in prior analysis (15).
Finally, a key issue concerns the degree to which there is a correlation between the range of O—O bond orders/lengths described here and the presence of unpaired spin density on the O2 fragment. In isolated superoxide radical anion, there is obviously a single unpaired spin that is entirely associated with the O2 π* system. Population analysis indicates that for LiO2 and NaO2 the same situation holds, which is consistent with these species being essentially ionic in character. The π* orbital containing the unpaired spin in these latter cases is the “out-of-plane” one, i.e., the plane containing the atoms is the nodal plane for this molecular orbital's constituent p atomic orbitals (the other π* orbital is formally doubly occupied).
When there is some possibility for covalent interaction between the O2 fragment and its associated metal, however, it is less obvious how spin will be distributed in a molecule having net superoxide-like O—O bond order. Chromium compound 3 seems reasonably well described as a “classical” superoxide: a spin density of 0.90 electrons is computed for the out-of-plane O2 π* orbital, and this density is antiferromagnetically coupled to a spin density of 2.98 electrons on the metal to deliver the observed overall triplet state. When the Cl ligand in 3 is replaced with Ph to form 5, however, the unpaired spin density on the O2 fragment is reduced to 0.74 electrons. This reduction in unpaired spin density not due not to a loss of density from O2, but rather to a gain of density of opposite spin, i.e., the out-of-plane π* orbital is more nearly doubly occupied in 5 than in 3 and there is a concomitant loss of spin density on the metal.
Widely varying fractional unpaired spin densities in the O2 out-of-plane π* orbital are computed for the other transition–metal complexes. In particular, the values are 0.48 for 1, 0.42 for 8, 0.28 for 2, 0.18 for 8−, and 0.00 for 6 and 7. Restricting ourselves to the simplest analysis for the sake of brevity, such fractional unpaired spin densities derive from mixing between the out-of-plane O2 π* orbital and the aligned metal dx2-y2 orbital (giving rise to δ-type bonding and antibonding orbitals; Fig. 4). Thus, for instance, if the lower-energy bonding hybrid MO formed from these two fragments contains exactly one electron and the antibonding orbital is empty, the unpaired spin density on the O2 fragment will be exactly the percentage of the O2 π* orbital that is contributed to form the bonding hybrid MO. An interesting variation arises when the bonding hybrid MO is doubly occupied, the antibonding hybrid is empty, and the contributions of the O2 π*, and metal dx2-y2 orbitals to both MOs are equal, i.e., 50% each. In that case, there is one electron's worth of net paired spin density in the out-of-plane π* orbital of the O2 fragment, but no unpaired spin density at all; this situation roughly corresponds to cases 6 and 7, although a complete analysis requires consideration of all MOs and not simply the two described here for illustrative purposes. It may be debated whether this last example should properly be referred to as a superoxide, based on electron count, or a (strongly donating) peroxide, based on closed-shell character. Given the quantitative values provided above, we leave it to the reader to decide this point, and simply note that it is the total amount of density in the O2 fragment orbitals, irrespective of spin polarization, that dictates the total bond order, and that this bond order takes on a continuous range of values, with these values being correlated with O—O bond lengths and stretching frequencies.
Figure 4.

Hybridization of out-of-plane O2 π* orbital with metal dx2-y2 orbital to create bonding and antibonding δ-type MOs. In this drawing, the contribution from each fragment to the final hybrids is about equal, but in any particular system these contributions will vary based on the relative starting energy levels of the different fragments (reflecting their intrinsic fragment electronegativities).
Conclusion
Density functional calculations accurately reproduce the structures of a variety of 1:1 η2-O2 complexes, many of which are relevant to biological and catalytic O2 activation processes, and provide new insights into the properties of the MO2 unit. Comparison of theory and experiment for the series of complexes reveals a statistically significant linear relationship between the O—O bond lengths and stretching frequencies. On the basis of this result, the reported O—O bond distances for 1 and 2 appear to be in error, which we confirm for the case of 2 by a redetermination of its x-ray crystal structure at low temperature. The reproducible difference in O—O bond distances between the low- and high-temperature data collections is attributed to librational motion of the coordinated O2, which results in an underestimation of the O—O distance at high temperature.
The O—O distances and stretching frequencies further correlate with O—O bond order, such that increased distances correspond to decreased νOO values and bond orders. In addition, the O—O and M—O bond orders are inversely correlated, consistent with variable charge transfer from the metal to the O2 fragment through covalent interaction of the metal dxy orbital with the in-plane π* orbital of O2. The degree of charge transfer depends closely on the nature of the metal and its supporting ligands, pointing toward the significance of subtle tuning of metal–dioxygen adduct properties and reactivity in catalytic systems.
Supporting Information
Cartesian coordinates and electronic energies for all computed structures, Pearson R values, and x-ray crystallographic data for 2 (Crystollographic Information File) can be found in Supporting Dataset, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
We thank Prof. E. I. Solomon for the personal communication of unpublished data on 1 and the National Institutes of Health (Grant GM47365 to W.B.T.), the National Science Foundation (Grant CHE-0203346 to C.J.C.), and the U.S. Department of Energy (Grant ER14273 to K.H.T.) for financial support.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Note that the Pearson R between calculated and experimental O—O distances increases to 0.940 from 0.781 when the low T value for 2 is used and the experimental value for 1 is omitted.
We note that the relationship between the O—O bond length and the O—O stretching frequency for BrAlO2 is not particularly well fit by the correlation line of Fig. 1. However, there is certainly no rigorous basis for a simple linear relationship between these two quantities in any case, so it is perhaps not too surprising that this should be the case. Thus, although we have emphasized the near linear relationship between O—O bond length and stretching frequency over the reduced range spanned by the transition–metal complexes, and the extent to which the deviations from this linearity exhibited by 1 and 2, as originally reported, prompted us to examine certain structural details in these molecules more closely, we do not intend to suggest that such linear behavior should necessarily extend outside the range studied here.
References
- 1.Vaska L. Acc Chem Res. 1976;9:175–183. [Google Scholar]
- 2.Valentine J S. Chem Rev. 1973;73:235–245. [Google Scholar]
- 3.Gubelmann M H, Williams A F. Struct Bonding (Berlin) 1983;55:1–65. [Google Scholar]
- 4.Hill H A O, Tew D G. In: Comprehensive Coordination Chemistry. Wilkinson G, Gillard R D, McCleverty J A, editors. Vol. 2. Oxford: Pergamon; 1987. pp. 315–333. [Google Scholar]
- 5.Dickman M H, Pope M T. Chem Rev. 1994;94:569–584. [Google Scholar]
- 6.Momenteau M, Reed C A. Chem Rev. 1994;94:659–698. [Google Scholar]
- 7.Karlin K D, Tolman W B, Kaderli S, Zuberbühler A D. J Mol Catal A. 1997;117:215–222. [Google Scholar]
- 8.Girerd J-J, Banse F, Simaan A J. In: Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations. Meunier B, editor. Vol. 97. Berlin: Springer; 2000. pp. 145–177. [Google Scholar]
- 9.Klinman J P. Chem Rev. 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 10.Fujisawa K, Tanaka M, Moro-oka Y, Kitajima N. J Am Chem Soc. 1994;116:12079–12080. [Google Scholar]
- 11.Egan J, J W, Haggerty B S, Rheingold A L, Sendlinger S C, Theopold K H. J Am Chem Soc. 1990;112:2445–2446. [Google Scholar]
- 12.Qin K, Incarvito C D, Rheingold A L, Theopold K H. Angew Chem Int Ed. 2002;41:2333–2335. doi: 10.1002/1521-3773(20020703)41:13<2333::AID-ANIE2333>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Loppnow G R, McDonald R, Takats J. J Am Chem Soc. 1995;117:7828–7829. [Google Scholar]
- 14.Spencer D J E, Aboelella N W, Reynolds A M, Holland P L, Tolman W B. J Am Chem Soc. 2002;124:2108–2809. doi: 10.1021/ja017820b. [DOI] [PubMed] [Google Scholar]
- 15.Aboelella N W, Lewis E A, Reynolds A M, Brennessel W W, Cramer C J, Tolman W B. J Am Chem Soc. 2002;124:10660–10661. doi: 10.1021/ja027164v. [DOI] [PubMed] [Google Scholar]
- 16.Perdew J, Wang Y. Phys Rev B. 1992;45:13244–13249. doi: 10.1103/physrevb.45.13244. [DOI] [PubMed] [Google Scholar]
- 17.Burke K, Perdew J P, Wang Y. In: Electronic Density Functional Theory: Recent Progress and New Directions. Dobson J F, Vignale G, Das M P, editors. New York: Plenum; 1998. pp. 81–121. [Google Scholar]
- 18.Adamo C, Barone V. J Chem Phys. 1998;108:664–675. [Google Scholar]
- 19.Stevens W J, Krauss M, Basch H, Jasien P G. Can J Chem. 1992;70:612–629. [Google Scholar]
- 20.Hehre W J, Radom L, von Schleyer P R, Pople J A. Ab Initio Molecular Orbital Theory. New York: Wiley; 1986. [Google Scholar]
- 21.Mahadevan V, DuBois J L, Hedman B, Hodgson K O, Stack T D P. J Am Chem Soc. 1999;121:5583–5584. [Google Scholar]
- 22.Mayer I. Chem Phys Lett. 1983;97:270–277. [Google Scholar]
- 23.Cramer C J. Essentials of Computational Chemistry: Theories and Models. Chichester, U.K.: Wiley; 2002. [Google Scholar]
- 24.Löwdin P-O. J Chem Phys. 1950;18:365–375. [Google Scholar]
- 25.Mulliken R S. J Chem Phys. 1955;23:1833–1840. [Google Scholar]
- 26.Stahl S S, Thorman J L, Nelson R C, Kozee M A. J Am Chem Soc. 2001;123:7188–7189. doi: 10.1021/ja015683c. [DOI] [PubMed] [Google Scholar]
- 27.Urban M W, Nakamoto K, Basolo F. Inorg Chem. 1982;21:3406–3408. [Google Scholar]
- 28.VanAtta R B, Strouse C E, Hanson L K, Valentine J S. J Am Chem Soc. 1987;109:1425–1434. [Google Scholar]
- 29.Andrews L, Smardzewski R R. J Chem Phys. 1973;58:2258–2261. [Google Scholar]
- 30.Rolfe J, Holzer W, Murphy W F, Bernestein J H. J Chem Phys. 1968;49:963–975. [Google Scholar]
- 31.Smardzewski R R, Andrews L. J Chem Phys. 1972;57:1327–1333. [Google Scholar]
- 32.Huber H, Ozin G A. Can J Chem. 1972;50:3746–3747. [Google Scholar]
- 33.Bahlo J, Himmel J-J, Schnöckel H. Inorg Chem. 2002;41:2678–2689. doi: 10.1021/ic011313d. [DOI] [PubMed] [Google Scholar]
- 34.Kitajima N, Komatsuzaki H, Hikichi S, Osawa M, Moro-oka Y. J Am Chem Soc. 1994;116:11596–11597. [Google Scholar]
- 35.Cruickshank D W J. Acta Crystallogr. 1956;9:757–758. [Google Scholar]
- 36.Busing W R, Levy H A. Acta Crystallogr. 1964;17:142–146. [Google Scholar]
- 37.Dunitz J D. X-Ray Analysis and the Structure of Organic Molecules. Ithaca, NY: Cornell Univ. Press; 1979. [Google Scholar]
- 38.Bauschlicher C W, Jr, Partridge H, Sodupe M, Langhoff S R. J Phys Chem. 1992;96:9259–9264. [Google Scholar]
- 39.Andrews L, Chertihin G V, Thompson C A, Dillon J, Byrne S, Bauschlicher C W., Jr J Phys Chem. 1996;100:10088–10099. [Google Scholar]
- 40.Bakac A, Scott S L, Espenson J H, Rodgers K R. J Am Chem Soc. 1995;117:6483–6488. [Google Scholar]
- 41.Andrews L. In: Cryochemistry. Moskovits M, Ozin G, editors. New York: Wiley; 1976. pp. 195–230. [Google Scholar]
- 42.Lever A B P, Ozin G A, Gray H B. Inorg Chem. 1980;19:1823–1824. [Google Scholar]
- 43.Lovell T, McGrady J E, Stranger R, Macgregor S. Inorg Chem. 1996;35:3079–3080. doi: 10.1021/ic951564k. [DOI] [PubMed] [Google Scholar]
- 44.Ruiz E, Cano J, Alvarez S, Alemany P. J Comput Chem. 1999;20:1391–1400. [Google Scholar]
- 45.Randall D W, DeBeer S, Holland P L, Hedman B, Hodgson K O, Tolman W B, Solomon E I. J Am Chem Soc. 2000;122:11632–11648. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.