

Abstract
A series of mononuclear zinc thiolate complexes have been prepared and fully characterized. The reactions of the complexes with alkyl halides, leading to zinc halides and the corresponding thioethers, have been examined by kinetic methods. In toluene, the reactions obey a second-order rate law displaying activation parameters consistent with a SN2 attack of the zinc-bound thiolate on the carbon electrophile. Intramolecular hydrogen bonding of an amide N—H to the thiolate sulfur reduces the nucleophilicity and consequently, the rate of alkylation more than 30-fold at 25°C. The H-bonding shows an inverse H/D isotope effect of 0.33 (60°C) ascribed to differential H-bonding for the two isotopomers due to zero point energy differences. These model studies provide quantitative evaluation of H-bonding on reaction rates relevant to zinc thiol-activating proteins.
The biological functions of zinc are varied, with seminal roles including the structure and function of a wide range of metalloproteins (1). Structural zinc sites are typically characterized by tetrahedral coordination, in which the four ligands are protein residues. Prominent examples of structural zinc sites are the transcriptional activator zinc finger proteins (2), and the structural zinc in liver alcohol dehydrogenase (1), the latter defined by four Cys ligands coordinated to zinc. The lack of catalytic activity has been ascribed to the ligation by four protein-derived ligands yielding a coordinatively saturated ion (1). Nonetheless, higher coordinate complexes are well established for the d10 metal ion. Alternatively, functional, i.e., catalytic, zinc sites possess a single labile coordination site, generally occupied by water or hydroxide in the resting state of enzymes. The Lewis acidity of the metal reduces the pKa of coordinated water molecules, rendering zinc enzymes particularly potent in effecting hydrolysis reactions. Hydrolases (and lyases) are the most thoroughly understood class of zinc proteins, with leading examples including the mononuclear, carbonic anhydrase, carboxypeptidase A, thermolysin, matrix metalloproteinases, and the polynuclear zinc proteins, aminopeptidases, and alkaline phosphatase (1, 3). Despite the diverse array of substrates consumed by these proteins, the consensus mechanistic feature for all is nucleophilic attack of a zinc hydroxide on the electrophilic position of the substrate.
An emerging group of mononuclear zinc enzymes fashions carbon–sulfur bonds via the transalkylation of carbon electrophiles and thiol substrates (4–6). Members of this class include methyl reductase (7), the Ada DNA repair protein (8), farnesyl transferase (5), betaine homocysteine methyl transferase (9), epoxyalkane coenzyme M transferase (10), and the methionine synthases (11, 12). Whereas the physiological activity and active site ligation vary, aspects of their catalytic mechanisms seem common. Specifically, thiol ligation to zinc rendering an active site bound thiolate poised for nucleophilic attack of the electrophilic substrate has been proposed. In the methionine synthases and the Ada protein, spectroscopic evidence for zinc-thiolate coordination is available (8, 12). In both the cobalamin-dependent (Met H) and -independent (Met E) methionine synthases, incubation with selenohomocysteine led to x-ray absorption changes remarkably similar to those observed for proteins exposed to homocysteine, the natural substrate (12). Furthermore, selenium and zinc extended x-ray absorption fine structure data clearly support direct coordination in the case of selenohomocysteine. The fundamental steps depicted in Scheme S1 serve to highlight the similarities between zinc hydrolases and these thiol-activating proteins. The key elementary process in each is attack of a metal-ligated nucleophile (Zn–SR or Zn–OH) on an electrophilic substrate. However, important distinctions between the enzyme classes remain to be elucidated. Most relevant to the present study are the following two questions pertaining to the thiol activating enzymes. Is the active nucleophile a metal-coordinated or “free” thiolate? How does a single thiolate become alkylated at a metal site that contains multiple Cys ligands? The former issue was raised initially by Wilker and Lippard (13, 14) in their synthetic and mechanistic studies undertaken to model the Ada DNA repair system. The reaction of [Zn(SPh)4]2− with phosphotriesters was kinetically consistent with a dissociative pathway entailing deligation of thiolate from zinc before alkylation. In contrast, a number of laboratories have demonstrated that zinc thiolate complexes are cleanly alkylated by carbon electrophiles via a SN2 pathway, with the “bound” thiolate serving as nucleophile (15–18). Clearly, the nature of the zinc complexes and the medium used profoundly effect the course of these reactions. Perhaps more subtle is the observation that, in a thiolate-rich coordination sphere, e.g., the Zn(Cys)4 of Ada, only one zinc thiolate is selected for reaction. The factors that control such exquisite regiochemistry are not established. Access of the carbon electrophile to the thiolate sulfur may be a key in controlling the course of the enzymatic reactions (19). Another structural attribute that could serve to determine which thiolate is alkylated is hydrogen-bonding to zinc thiolates. For example, in cases where several of the zinc thiolates are participating in hydrogen-bonding interactions with H-bond donors, the residues could be less nucleophilic than thiolates not involved in such secondary interactions and consequently, react slower with incoming electrophiles. However, this hypothesis has yet to be tested (15).
Scheme 1.

This contribution details our efforts to design and apply small molecule zinc thiolate complexes to address these specific questions of mechanism (20). The borato ligands developed in our laboratories are ideally suited for this pursuit (Fig. 1; refs. 21 and 22). Synthesis of the appropriate monomeric zinc thiolate complexes permits evaluation of both the mechanism of alkylation by carbon electrophiles and the quantitative impact H-bonding plays on rates of reaction for the metal-bound thiolates. The application of hydrogen bonds to stabilize unusual structures (23, 24) and control spectroscopic and electronic properties of small molecules (25–27) has been an exciting and active area of investigation. The current results extend this approach to include a quantitative assessment of H-bonding in metal complexes to reaction rates (28).
Figure 1.

[S3] and [S2N] borato ligands used in this study and the numbering scheme used for the zinc thiolate complexes.
Experimental Methods
Manipulations involving organometallic reagents were performed under a nitrogen atmosphere either on a Schlenk line or in a Vacuum Atmospheres (Hawthorne, CA) glovebox. Elemental analyses were performed by Desert Analytics (Tucson, AZ). NMR spectra were recorded on either a 250-MHz or 400-MHz Bruker (Billerica, MA) spectrometer. [Ph(pztBu)BttBu]H, [Ph(pztBu)BttBu]Zn(CH3), and 1 were prepared as described (20) [[Ph(pztBu)BttBu] = phenyl(3-tert-butyl-pyrazolyl)bis((tert-butylthio)methyl)borate]. 2-Pivaloylaminophenyl disulfide and 4-pivaloylaminophenyl disulfide were prepared from the corresponding aminophenyl disulfides (29). N–deutero–2-pivaloylaminophenyl disulfide was prepared by refluxing the disulfide in C6H6/D2O for 72 h.
We first consider [Ph(pztBu)BttBu]Zn(SC6H4o-NHC(O)But), 2. 2-Pivaloylaminophenyl disulfide (460 mg, 1.1 mmol) was added to [Ph(pztBu)BttBu]Zn(CH3) (500 mg, 1.0 mmol) in 40 ml of tetrahydrofuran (THF) at 25°C. The solution was refluxed for 3 h. After filtration, the solvent was removed under reduced pressure, yielding a light yellow solid. The solid was washed with pentanes (2 × 10 ml) yielding a pale yellow solid. Yield: 450 mg (65%). [1H]NMR (CDCl3): δ 9.40 (br, NH, 1 H), 8.39 (d, (3′-C6H5)S, 1 H), 7.73 (d, (6′-C6H4)S, 1 H), 7.45 (d, (o-C6H5)B, 2 H), 7.32 (m, (m-C6H5)B, and (p-C6H5)B, 3 H), 7.17 (t, (5′-C6H4)S, 1 H), 6.99 (s, 5-pz, 1 H), 6.92 (t, (4′-C6H4)S, 1 H), 6.00 (s, 4-pz, 1 H), 2.32 (d, BCH2, 2 H), 2.12 (d, BCH2, 2 H), 1.47 (s, 3-C(CH3)3,9 H), 1.37 (s, SC(CH3)3,18 H), 1.15 (s, NC(CH3)3, 9 H). Fourier transform-IR (FT-IR) (KBr): νNH, 3,333 cm−1; νCO, 1,680 cm−1; (CH2Cl2) νNH, 3,329 cm−1; νCO, 1,674 cm−1. Anal. Calcd. (found) for C34H52BN3S3OZn: C, 59.1 (58.8); H, 7.58 (7.65); N, 6.08 (5.80).
Next we consider [Ph(pztBu)BttBu]Zn(SC6H4o-NDC(O)But), 2D. N–deutero–2-pivaloylaminophenyl disulfide (462 mg, 1.1 mmol) was added to [Ph(pztBu)BttBu]Zn(CH3) (500 mg, 1.0 mmol) in 40 ml of THF at 25°C. The solution was refluxed for 2 h. After filtration, the solvent was removed under reduced pressure, yielding a light yellow solid. The solid was washed with pentanes (2 × 10 ml) yielding a pale yellow solid. Yield: 420 mg (60%). 1H NMR (CDCl3): δ 8.35 (d, (3′-C6H5)S, 1 H), 7.71 (d, (6′-C6H4)S, 1 H), 7.43 (d, (o-C6H5)B, 2 H), 7.31 (m, (m-C6H5)B and (p-C6H5)B, 3 H), 7.15 (t, (5′-C6H4)S, 1H), 6.94 (s, 5-pz, 1 H), 6.89 (t, (4′-C6H4)S, 1 H), 5.95 (s, 4-pz, 1 H), 2.31 (d, BCH2, 2 H), 2.09 (d, BCH2, 2 H), 1.41 (s, 3-C(CH3)3,9 H), 1.30 (s, SC(CH3)3,18 H), 1.18 (s, NC(CH3)3, 9 H). FT-IR (KBr): νNH, 2,481 cm−1; νCO, 1,678 cm−1; (CH2Cl2) νNH, 2,482 cm−1; νCO, 1,671 cm−1.
Next we consider [Ph(pztBu)BttBu]Zn(SC6H4p-NHC(O)But), 3. 4-Pivaloylaminophenyl disulfide (580 mg, 1.1 mmol) was added to [Ph(pztBu)BttBu]Zn(CH3) (640 mg, 1.3 mmol) in 50 ml of CH3CN at 25°C. The solution was refluxed for 8 h. After filtration, the solvent was removed under reduced pressure, yielding a white solid. The product was extracted with CH2Cl2; then, the volatiles were removed under reduced pressure. The solid was washed with pentanes (2 × 10 ml) yielding a white solid. Yield: 420 mg (47%). [1H]NMR (CDCl3): δ 7.55 (d, (o-C6H5)B, 2 H), 7.51 (d, (o-C6H4)S, 2 H), 7.37 (m, (m-C6H5)B and (p-C6H5)B, 3 H), 7.32 (d, (m-(C6H4)S, 2 H), 7.24 (d, 5-pz, 1 H), 7.17 (br, HN, 1 H), 6.08 (d, 4-pz, 1 H), 2.02 (d, BCH2, 2 H), 1.50 (d, BCH2, 2 H), 1.39 (s, 3-C(CH3)3, 9 H), 1.28 (s, SC(CH3)3, 18 H), 1.12 (s, COC(CH3)3, 9 H). FT-IR (KBr): νNH, 3,312 cm−1; νCO, 1,659 cm−1; (CH2Cl2) νNH, 3,377 cm−1; νCO, 1,679 cm−1.
Next we consider [Ph(pztBu)BttBu]CdCl. [Ph(pztBu)BttBu]H (836 mg, 2 mmol) was added to a suspension of KH (80 mg, 2 mmol) in THF to form [Ph(pztBu)BttBu]K, which was isolated by reducing the solvent volume and precipitating with pentanes. [Ph(pztBu)BttBu]K (456 mg, 1 mmol) in 5 ml of CH2Cl2 was added to CdCl2 (183 mg, 1 mmol) in 20 ml of CH2Cl2/Et2O yielding a clear solution and white solid. After 3 h, the solution was filtered, and the volatiles were removed under reduced pressure. The resulting white solid was purified by recrystallization from CH2Cl2/Et2O. Yield: 450 mg (79%). [1H]NMR (CDCl3): δ 7.43 (d, (o-C6H5)B, 2 H), 7.29 (m, (m-C6H5)B and (p-C6H5)B, 3 H), 6.99 (s, 5-pz, 1 H), 6.01 (s, 4-pz, 1 H), 2.49 (d, BCH2, 2 H), 2.36 (d, BCH2, 1 H), 1.45 (s, 3-C(CH3)3, 9 H), 1.43 (s, SC(CH3)3, 18 H). Anal. Calcd. (found) for C23H38BCdClN2S2: C, 48.9 (48.9); H, 6.63 (6.77); N, 4.92 (4.96).
Next we consider [Ph(pztBu)BttBu]Cd(SPh), 4. KSPh (220 mg, 1.5 mmol) was added to [Ph(pztBu)BttBu]CdCl (800 mg, 1.4 mmol) dissolved in 30 ml of THF. After 6 h, the volatiles were removed under reduced pressure. The product was extracted with CH2Cl2. The CH2Cl2 solution was filtered, and the solvent was removed under reduced pressure. The resulting white solid was purified by recrystallization from CH2Cl2/pentanes. Yield: 733 mg (81%). [1H]NMR (CDCl3): δ 7.52 (d, (o-C6H5)S, 2 H), 7.45 (d, (o-C6H5)B, 2 H), 7.32 (m, (m-C6H5)B and (p-C6H5)B, 3 H), 7.13 (t, (m-C6H5)S, 2 H), 7.02 (m, (p-C6H5)S) and 5-pz, 2 H), 6.00 (s, 4-pz, 1 H), 2.37 (br, BCH2, 4 H), 1.38 (s, 3-C(CH3)3, 9 H), 1.24 (s, SC(CH3)3, 18 H). Anal. Calcd. (found) for C29H43BCdN2S3: C, 54.2 (54.5); H, 6.97 (6.78); N, 4.34 (4.38).
Reaction time courses were monitored by [1H]NMR spectroscopy. Concentrations of reactants and products were determined by referencing peak integrals to an internal standard, tetramethylsilane. Typical [1H]NMR parameters for kinetic studies included 16 scans per spectrum and 10–50 spectra per reaction. Reactions were conducted under pseudo first-order conditions, with [RX] > 20 [Zn(SAr)]. Reactions of 1 with alkyl halides were followed for three half-lives. The reaction between 2 (or 2D) and α-bromotoluene was analyzed by using the method of initial rates (30). In a typical experiment, 0.008 g of 1 was dissolved in 0.5 ml toluene-d8, the solution was transferred to a septum-sealed Wilmad (Buena, NY) NMR tube. Alkyl halide was added via a Hamilton syringe. Reported rate constants reflect the average of at least three replicate runs, with the errors reflecting one standard deviation. Activation parameters were derived from Eyring analysis of the temperature dependence of the rate constants over a temperature range of 35°C.
Crystallographic data were collected on a Bruker-AXS SMART APEX-CCD system with graphite-monochromatized Mo Kα radiation. Suitable crystals were mounted on a glass fiber under perfluoropolyether. Crystal, data collection, and refinement parameters are tabulated in Tables 3–13 (which are published as supporting information on the PNAS web site, www.pnas.org.). The systematic absences in the diffraction data are uniquely consistent with the space group P2 (1)/n for 2 and Pbca for 4. The structures were solved by using direct methods, completed by subsequent difference Fourier syntheses, and refined by full-matrix least-squares procedures. Absorption corrections were applied to the data by using sadabs. All non-hydrogen atoms were refined with anisotropic displacement coefficients. For 2, hydrogen atom H (1) was allowed to refine independently whereas all other hydrogen atoms were treated as idealized contributions. A complete list of bond distances and bond angles is contained in the Tables 3–13.
Results and Discussion
Structures.
Previously, we reported the synthesis and characterization of zinc phenylthiolate complexes of the tridentate borato ligands [PhTttBu] ([PhTttBu] = phenyltris((tert-butylthio)methyl)borate)and [Ph(pztBu)BttBu] that provide S3 and S2N donor environments, respectively, Fig. 1 (20). The monomeric complexes have the advantage of sulfur-rich coordination spheres, while possessing single thiolate ligands. These structures allow for alkylation at the unique thiolate thus simplifying product and kinetic analysis. Herein, we focus on a series of zinc thiolate complexes supported by the [S2N] tripodal ligand [Ph(pztBu)BttBu], in which the aryl thiolate is varied (Fig. 1). [Ph(pztBu)BttBu]Zn(SPh), 1, has been fully characterized, including by x-ray structural analysis. To assess the effect of hydrogen bonding, the related complex, 2, was prepared, which included a 2-pivalyl amide substituent on the phenyl thiolate. The ortho substituent allows for formation of a N—H–S bond contained in a five-membered ring. The solid state structure of 2 is quite similar to that of 1 (Fig. 2). The most relevant feature is the conformation of the amide group of the aryl thiolate that ensures a short N–S distance of 2.936(2) Å consistent with the N—H–S bond in the solid state (31). The amide hydrogen position was refined anisotropically permitting for establishment of the N—H, 0.79(2) Å and S–H, 2.49(2) Å distances. The Zn—S(thiolate) distance in 2 is 2.260(1) Å compared with 2.265(1) Å in 1. The very slight shortening of the Zn—S bond is due to the amide H-bond relieving the destabilization that results from the Zndπ—Spπ antibonding in 1 (32, 33). The H-bond is maintained in solution as indicated by FT-IR, (CH2Cl2) νN–H, 3,329 cm−1, and proton NMR, δ NH 9.34 (br), spectroscopic features (34, 35). The corresponding N–D complex, 2D, was prepared via deuterium incorporation into the amide before complexation to zinc. Complete deuterium exchange was indicated by the appropriate spectroscopic features, FT-IR, νN–D, 2,483 cm−1, [νN–H/νN–D = 1.34; calcd 1.37] and [2H]NMR, δ ND 9.23. Lastly, the zinc aryl thiolate complex possessing a pivalylamide substituent in the para position, 3, was synthesized. This complex serves as an important control for the kinetic reactions because its electronic structure is similar to that of 2. However, there is no possibility for intramolecular H-bond formation.†
Figure 2.

Thermal ellipsoid plot of 2. Thermal ellipsoids are drawn at the 30% probability level, and hydrogen atoms, except H(1), are omitted for clarity. Selected bond distances (in Å): Zn—N(1), 2.015(1); Zn—S(1), 2.4096(4); Zn—S(2), 2.3843(4); Zn—S(3), 2.2598(4).
Reaction of Zinc Thiolates with Alkyl Halides.
The zinc thiolate derivatives, 1–3, reacted with iodomethane or α-bromotoluene, generating the corresponding aryl thioether and [Ph(pztBu)BttBu]ZnX (X = I or Br) in quantitative yields (Eq. 1). Reaction rates were monitored by [1H]NMR spectroscopy in d8-toluene under pseudo first-order conditions, [RX] > 20[Zn(SAr)]. Representative time courses for the reaction of 1 with iodomethane are contained in Fig. 3. Reactions obeyed a second order rate expression (Eq. 2), first order in each reagent (Table 1). Activation parameters, ΔH‡ and ΔS‡, were derived from rate measurements over 35°C temperature ranges (Fig. 4, which is published as supporting information on the PNAS web site). The results are collected in Table 2. The clean second order behavior observed for all reactions is reflective of alkylation at the metal-bound thiolate. Were the alkylation reaction to proceed via an “unbound” thiolate§ larger rate constants would be expected because the latter is a more potent nucleophile (13). Given the low polarity medium, such a dissociative route is disfavored. The activation parameters are consistent also with an SN2 transition state; specifically, the small enthalpy term, ≈16 kcal/mol, and the ΔS‡ values, ≈−20 cal/mol-K, point to bond-making and bond-breaking and restricted entropy in the activated complex. However, these results are insufficient to distinguish between a SN2 transition state and a four-centered transition state, the latter proposed by Vahrenkamp and colleagues (16). In principle, the stereochemical outcome of alkylation with a chiral electrophile would discriminate between these alternate paths.
 |
 |
1 |
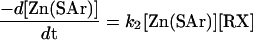 |
2 |
Introduction of an amido substituent in the ortho position of the aryl thiolate that hydrogen bonds to the sulfur, 2, reduced the rate of alkylation by α-bromotoluene by a factor of twenty relative to the parent complex, 1. Whereas the rate diminution is in accord with the decreased donor aptitude of the thiolate caused by the dipole interaction, this is not the only effect introduced by the amide group. To ferret out the electronic and steric contributions beyond those due to hydrogen bonding, complex 3, containing the amide in the para position, was evaluated. The thiolate sulfur of this complex will be more nucleophilic than in 1 due to the electron-releasing property of the amide, yet it will be similarly accessible sterically. Alkylation of 3 did occur more rapidly; however, the magnitude increase compared with 1 was modest (1.6 times), suggesting that the amide substituent does not significantly alter the electronic structure of the zinc thiolate. To establish the effect of steric encumbrance resulting from the ortho substituent in 2, the analogous N—CH3 amido complex was sought. However, this species has yet to be generated cleanly.¶ Alkylation of the N—D complex, 2D, provided an attractive opportunity to further probe the role of H-bonding. The measured kinetic isotope effect, kH/kD, is inverse with a value of 0.33 at 60°C. Given that the kinetic isotope effect (kie) is neither primary nor secondary, its magnitude is significant. Whereas the kie is expected to be inverse due to zero point energies differences that favor N—H in the hydrogen bonding position, the magnitude of the effect is beyond that anticipated when considering only N—H and N—D vibrational energy differences (36, 37). The origin of the kie is under continued investigation in this laboratory.
Figure 3.

Representative time courses for the reaction of 1 with iodomethane at 60°C under pseudo first-order conditions. For each reaction, the initial [1] was 27 mM and [CH3I] of 0.54 (♦), 0.81 (■), 1.08 (▴), and 1.35 (●) M. The solid lines represent nonlinear (exponential) least squares fits to the data.
Table 1.
Second-order rate constants for BrCH2C6H5 alkylation of [Ph(pztBu)BttBu]Zn(SAr) complexes in toluene at 60°C
| [Ph(pztBu)BttBu]Zn(SAr) | 104k2, M−1⋅s−1 | k2 (relative rates) |
|---|---|---|
| 1 | 28 | 21.5 |
| 2 | 1.3 | 1 |
| 2D | 3.9 | 3.0 |
| 3 | 44 | 33.8 |
Table 2.
Second-order rate constants and activation parameters for BrCH2C6H5 alkylation of [Ph(pztBu)BttBu]Zn(SAr) complexes in toluene
| [Ph(pztBu)BttBu]Zn(SAr) | k2, M−1⋅s−1, 25°C | ΔH‡, kcal/mol | ΔS‡, cal/mol-K |
|---|---|---|---|
| 1 | 2.1 × 10−4 | 16.1 ± 1.4 | −21.7 ± 3.8 |
| 2 | 6.2 × 10−6 | 18.5 ± 1.0 | −20.7 ± 2.6 |
| 2D | 1.5 × 10−5 | 16.9 ± 1.0 | −23.9 ± 2.9 |
| 1* | 5.8 × 10−5 | 16.7 ± 1.0 | −21.5 ± 2.7 |
Alkylation by CH3I.
Solvent Effects.
SN2 reactions show a predictable solvent dependence in that reaction rates increase as the solvent dielectric constant becomes larger. Therefore, to define the influence of solvent polarity of the reaction rate and to draw relevant comparisons between the present studies and rate constants published previously (15, 16), the reaction between 1 and iodomethane was monitored in CD2Cl2. Under pseudo first-order conditions, the time course of the reaction did not obey first order kinetics. Rather, plots of [1] vs. time were roughly linear through more than three half-lives. Such behavior is a hallmark of either a zero-order kinetic process or autocatalysis (30). Given the extensive studies in toluene demonstrating that the reactions were kinetically well behaved, the latter effect seemed more probable. Therefore, the reaction of 1 and iodomethane in CD2Cl2 was repeated in the presence of [Ph(pztBu)BttBu]ZnI (1 equiv. relative to the initial [1]) to ascertain the role of this product in the autocatalytic process. Under the condition of added product, the reaction displayed exponential decay profiles, Fig. 5 (which is published as supporting information on the PNAS web site), as expected for a reaction catalyzed by [Ph(pztBu)BttBu]ZnI. Conversely, added thioanisole did not alter the kinetic course of the reaction in CD2Cl2, thus ruling out its role as the source of catalysis. The second-order rate constant, k2 = 3.52 × 10−4 M−1⋅s−1 at 25°C, is six times larger than k2 measured in toluene. The rate constant measured in CD2Cl2 is ≈100 times smaller than for the methylation of other neutral zinc thiolate complexes. For example, Carrano and colleagues (15) reported rates for iodomethane addition to [L1O]Zn(SPh) of 2.5 × 10−2 M−1⋅s−1 and to [L1O]Zn(SBn) of 0.12 M−1⋅s−1. Tris(pyrazolyl)borate zinc thiolate complexes are also alkylated faster, [TpPh2]Zn(SPh), 7.2 × 10−2 M−1⋅s−1 ([TpPh2] = hydrido(3,5-diphenyl-1-pyrazolyl)borate); and [TpPh,Me]-Zn(SBn), 1.75 × 10−2 M−1⋅s−1 (27°C) ([TpPh,Me] = hydrido-(3-phenyl,5-methyl-1-pyrazolyl)borate) (16). The relative sluggishness of 1 may be reflective of either a steric impediment to attack or the role of the polarizable thioether ligands in modulating the nucleophilicity of the zinc-bound thiolate.
The proposed role of [Ph(pztBu)BttBu]ZnI in the reaction between 1 and iodomethane is depicted in Scheme S2. The transition state structure is that of a class SN2 path, with the thiolate of 1 serving as the entering nucleophile. For reactions in the low polarity toluene, the transition state collapses, yielding thioanisole coordinated to cationic [Ph(pztBu)BttBu]-Zn+ and an outer sphere iodide. Exchange of these entities results in the observed products. However, in the higher polarity CD2Cl2, [Ph(pztBu)BttBu]ZnI interacts with iodomethane via some measure of Zn–I ionization, to form a Zn–ICH3 adduct. This complex activates the iodomethane sufficiently to render the alkyl halide more reactive for subsequent methylations of 1. Whereas haloalkane adducts of zinc seem unprecedented, examples abound for silver and a number of transition metals (38). Furthermore, zinc iodoalkane interactions have been proposed at the active site of carboxypeptidase A as a means to inhibit the enzyme.
Scheme 2.

Metal Dependence.
Metal ion substitution at metalloprotein active sites represents an attractive and powerful approach to elucidate structure-function correlations. This approach is particularly important for zinc proteins because the native metal provides little spectroscopic information. The factors that drive activity are clearly subtle and not well-understood. For example, cadmium substituted Met E is completely inactive (4). In contrast, when the zinc in the Ada DNA repair protein is replaced with cadmium, activity is maintained (39). To develop a fundamental understanding of the differences in nucleophilicity between zinc and cadmium thiolate complexes (40), the reaction of isostructural complexes with iodomethane was evaluated. Alkylation of [Ph(pztBu)BttBu]Cd(SPh) at 323 K proceeded with k2 of 1.28 × 10−2 M−1⋅s−1. This rate constant is 17 times larger than for the reaction of the zinc complex, 1, under identical conditions, thus indicating that the cadmium thiolate is inherently more nucleophilic than the zinc thiolate (41). The result is in accord with the relative Lewis acidities of the metal ions, Zn > Cd. That is, cadmium as a poorer Lewis acid, should yield a more basic, and presumably more nucleophilic, thiolate complex. Wilker and Lippard (13, 14) demonstrated that, for the parent [M(SPh)4]2− complexes reacting with trimethylphosphate, the Zn analog was methylated 2.5 times faster than the Cd species. However, because the mechanism was determined to be dissociative, the relative rates are reflective of thiolate dissociative equilibria rather than a measure of the inherent metal thiolate nucleophilicity.
Summary and Prospectus.
These results provide quantitative assessment of the role of H-bonding in altering the reactivity of a metal thiolate. Specifically, the nucleophilicity of the zinc thiolate, 2, stabilized by an intramolecular amide N—H–S is more than an order of magnitude diminished relative to the zinc thiolate in 1. The inverse kie observed for the alkylation reactions of the H(D)-bonded complexes highlights both the sensitivity of the reaction to the H-bonding and provides evidence for a weaker N—D–S stabilization. Similar H-bonding interactions may serve to differentiate zinc thiolates in metalloprotein active sites where a single residue is selected for reaction.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Science Foundation (CHE-9974628 and CHE-0213260 to C.G.R.).
Abbreviations
- THF
tetrahydrofuran
- [Ph(pztBu)BttBu]
phenyl(3-tert-butyl-pyrazolyl)bis((tert-butylthio)methyl)borate
- kie
kinetic isotope effect
- FT-IR
Fourier transform-IR
Note
After submission of this paper a report (28) appeared describing the methylation of a zinc thiolate possessing an intramolecular amide hydrogen bond. Although a full kinetic analysis was not included, reaction rates for a single [MeI] at one temperature were detailed and are in agreement with the observations contained herein.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Spectroscopic data (FT-IR, NMR) are consistent with no H-bonding of the amide N—H.
This term is used to highlight the dissociative pathway; clearly, charge compensations require a thiolate to be matched with a cationic partner (ion pairing).
The o-N-methylpivalylamido disulfide has been prepared. Attempted conversions to the corresponding Zn(SAr) complex have led to decomposition, presumably due to the high temperatures required to initiate product conversion.
References
- 1.Lipscomb W N, Sträter N. Chem Rev. 1996;96:2375–2434. doi: 10.1021/cr950042j. [DOI] [PubMed] [Google Scholar]
- 2.Berg J M, Godwin H A. Annu Rev Biophys Biomol Struct. 1997;26:357–371. doi: 10.1146/annurev.biophys.26.1.357. [DOI] [PubMed] [Google Scholar]
- 3.Wilcox D E. Chem Rev. 1996;96:2435–2458. doi: 10.1021/cr950043b. [DOI] [PubMed] [Google Scholar]
- 4.Matthews R G, Goulding C W. Curr Opin Chem Biol. 1997;1:332–339. doi: 10.1016/s1367-5931(97)80070-1. [DOI] [PubMed] [Google Scholar]
- 5.Hightower K E, Fierke C A. Curr Opin Chem Biol. 1999;3:176–181. doi: 10.1016/s1367-5931(99)80030-1. [DOI] [PubMed] [Google Scholar]
- 6.Penner-Hahn J E. Indian J Chem A. 2002;41:13–21. [Google Scholar]
- 7.Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer R K. Science. 1997;278:1457–1462. doi: 10.1126/science.278.5342.1457. [DOI] [PubMed] [Google Scholar]
- 8.Lin Y X, Dotsch V, Wintner T, Peariso K, Myers L C, Penner-Hahn J E, Verdine G L, Wagner G. Biochemistry. 2001;40:4261–4271. doi: 10.1021/bi002109p. [DOI] [PubMed] [Google Scholar]
- 9.Evans J C, Huddler D P, Jiracek J, Castro C, Millian N S, Garrow T A, Ludwig M L. Structure. 2002;10:1159–1171. doi: 10.1016/s0969-2126(02)00796-7. [DOI] [PubMed] [Google Scholar]
- 10.Krum J G, Ellsworth H, Sargeant R R, Rich G, Ensign S A. Biochemistry. 2002;41:5005–5014. doi: 10.1021/bi0255221. [DOI] [PubMed] [Google Scholar]
- 11.Matthews R G. Acc Chem Res. 2001;34:681–689. doi: 10.1021/ar0000051. [DOI] [PubMed] [Google Scholar]
- 12.Peariso K, Zhou Z H S, Smith A E, Matthews R G, Penner-Hahn J E. Biochemistry. 2001;40:987–993. doi: 10.1021/bi001711c. [DOI] [PubMed] [Google Scholar]
- 13.Wilker J J, Lippard S J. Inorg Chem. 1997;36:969–978. doi: 10.1021/ic961082o. [DOI] [PubMed] [Google Scholar]
- 14.Wilker J J, Lippard S J. J Am Chem Soc. 1995;117:8682–8683. [Google Scholar]
- 15.Warthen C R, Hammes B S, Carrano C J, Crans D C. J Biol Inorg Chem. 2001;6:82–90. doi: 10.1007/s007750000171. [DOI] [PubMed] [Google Scholar]
- 16.Brand U, Rombach M, Seebacher J, Vahrenkamp H. Inorg Chem. 2001;40:6151–6157. doi: 10.1021/ic0105112. [DOI] [PubMed] [Google Scholar]
- 17.Grapperhaus C A, Tuntulani T, Reibenspies J H, Darensbourg M Y. Inorg Chem. 1998;37:4052–4058. doi: 10.1021/ic971599f. [DOI] [PubMed] [Google Scholar]
- 18. Bridgewater, B. M., Fillebeen, T., Friesner, R. A. & Parkin, G. (2000) J. Chem. Soc. Dalton Trans., 4494–4496.
- 19.Roehm P C, Berg J M. J Am Chem Soc. 1998;120:13083–13087. [Google Scholar]
- 20.Chiou S-J, Innocent J, Lam K-C, Riordan C G, Liable-Sands L, Rheingold A L. Inorg Chem. 2000;39:4347–4353. doi: 10.1021/ic000505q. [DOI] [PubMed] [Google Scholar]
- 21.Ohrenberg C, Ge P, Schebler P, Riordan C G, Yap G P A, Rheingold A L. Inorg Chem. 1996;35:749–754. [Google Scholar]
- 22. Chiou, S.-J., Ge, P., Riordan, C. G., Liable-Sands, L. & Rheingold, A. L. (1999) Chem. Commun., 159–160.
- 23.MacBeth C E, Golombek A P, Young V G, Yang C, Kuczera K, Hendrich M P, Borovik A S. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- 24.Momenteau M, Reed C A. Chem Rev. 1994;94:659–698. [Google Scholar]
- 25.Huang J, Ostrander R L, Rheingold A L, Leung Y C, Walters M A. J Am Chem Soc. 1994;116:6769–6776. [Google Scholar]
- 26.Okamura T, Takamizawa S, Ueyama N, Nakamura A. Inorg Chem. 1998;37:18–28. doi: 10.1021/ic970640b. [DOI] [PubMed] [Google Scholar]
- 27.Walters M A, Dewan J C, Min C, Pinto S. Inorg Chem. 1991;30:2656–2662. [Google Scholar]
- 28.Smith J N, Shirin Z, Carrano C J. J Am Chem Soc. 2003;125:868–869. doi: 10.1021/ja029418i. [DOI] [PubMed] [Google Scholar]
- 29.Ueno T, Inohara M, Ueyama N, Nakamura A. Bull Chem Soc Jpn. 1997;70:1077–1083. [Google Scholar]
- 30.Espenson J H. Chemical Kinetics and Reaction Mechanisms. New York: McGraw–Hill; 1995. [Google Scholar]
- 31.Hamilton W C, Ibers J A. Hydrogen Bonding in Solids. New York: Benjamin; 1968. [Google Scholar]
- 32.Darensbourg M Y, Longridge E M, Payne V, Reibenspies J, Riordan C G, Springs J J, Calabrese J C. Inorg Chem. 1990;29:2721–2726. [Google Scholar]
- 33.McGuire D G, Khan M A, Ashby M T. Inorg Chem. 2002;41:2202–2208. doi: 10.1021/ic011096g. [DOI] [PubMed] [Google Scholar]
- 34.Nesloney C L, Kelly J W. J Am Chem Soc. 1996;118:5836–5845. [Google Scholar]
- 35.Gallo E A, Gellman S H. J Am Chem Soc. 1993;115:9774–9788. [Google Scholar]
- 36.Bender B R. J Am Chem Soc. 1995;117:11239–11246. [Google Scholar]
- 37.Bender B R, Kubas G J, Jones L H, Swanson B I, Eckert J, Capps K B, Hoff C D. J Am Chem Soc. 1997;119:9179–9190. [Google Scholar]
- 38.Crabtree R H. Angew Chem Int Ed Engl. 1993;32:789–805. [Google Scholar]
- 39.Myers L C, Terranova M P, Ferentz A E, Wagner G, Verdine G L. Science. 1993;261:1164–1167. doi: 10.1126/science.8395079. [DOI] [PubMed] [Google Scholar]
- 40.Bakbak S, Incarvito C D, Rheingold A L, Rabinovich D. Inorg Chem. 2002;41:998–1001. doi: 10.1021/ic0109243. [DOI] [PubMed] [Google Scholar]
- 41.Kimblin C, Murphy V J, Hascall T, Bridgewater B M, Bonanno J B, Parkin G. Inorg Chem. 2000;39:967–974. doi: 10.1021/ic990682v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.