

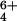
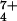
Abstract
The kinetics of proton-coupled electron-transfer (pcet) reactions are reported for Mn4O4(O2PPh2)6, 1, and [Mn4O4(O2PPh2)6]+, 1+, with phenothiazine (pzH). Both pcet reactions form 1H, by H transfer to 1 and by hydride transfer to 1+. Surprisingly, the rate constants differ by only 25% despite large differences in the formal charges and driving force. The driving force is proportional to the difference in the bond-dissociation energies (BDE >94 kcal/mol for homolytic, 1H → H + 1, vs. ≈127 kcal/mol for heterolytic, 1H → H− + 1+, dissociation of the O—H bond in 1H). The enthalpy and entropy of activation for the homolytic reaction (ΔH‡ = −1.2 kcal/mol and ΔS‡ = −32 cal/mol⋅K; 25–6.7°C) reveal a low activation barrier and an appreciable entropic penalty in the transition state. The rate-limiting step exhibits no H/D kinetic isotope effect (kH/kD = 0.96) for the first H atom-transfer step and a small kinetic isotope effect (1.4) for the second step (1H + pzH → 1H2 + pz•). These lines of evidence indicate that formation of a reactive precursor complex before atom transfer is rate-limiting (conformational gating), and that little or no N—H bond cleavage occurs in the transition state. H-atom transfer from pzH to alkyl, alkoxyl, and peroxyl radicals reveals that BDEs are not a good predictor of the rates of this reaction. Hydride transfer to 1+ provides a concrete example of two-electron pcet that is hypothesized for the O—H bond cleavage step during catalysis of photosynthetic water oxidation.
Keywords: bond-dissociation energy‖manganese‖hydrogen atom transfer‖proton transfer‖kinetic isotope effect
Atom- and ion-transfer reactions comprise a major class of chemical reactions found in biological systems, notably metabolic pathways (1–5). Reactions involving the net transfer of a hydrogen atom may be viewed in terms of two coupling limits with intermediate cases implicit (6): movement of a H atom between a donor/acceptor pair vs. independent proton/electron-transfer steps involving different electron and proton sites. As a class these are referred to as proton-coupled electron-transfer (pcet) processes (7).
An example arises in the case of the water-oxidizing/O2-evolving complex of photosynthesis [photosystem-II water-oxidizing complex (PSII-WOC)], which catalyzes the extraction of four electrons and four protons from two water molecules mediated by an inorganic core, Mn4OxCa1Cly, and a tyrosyl radical (8, 9). Only a limited number of model Mn complexes having mononuclear (10, 11) or binuclear manganese-oxo cores have contributed to understanding the kinetic steps in pcet chemistry (3, 12). These and related studies led to different results with some systems following a pathway of initial H abstraction (3, 13, 14) [with large kinetic isotope effect (KIE) in the range of 30–40 (15)], in contrast to other cases that exhibited an electrochemical step followed by a proton-transfer step with minimal H/D isotope effect (1, 16). Herein we report kinetics parameters for the initial pcet reaction between phenothiazine (pzH) and the manganese-oxo–cubane complexes, Mn4O4(Ph2PO2)6 (1) and [Mn4O4(Ph2PO2)6]+(ClO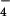 ) (1+ClO
) (1+ClO ) (Scheme S1), and relate them to data for the PSII-WOC. Both complexes are structurally characterized and contain the Mn4O4-cubane core surrounded by six facially bridging diphenylphosphinate (Ph2PO
) (Scheme S1), and relate them to data for the PSII-WOC. Both complexes are structurally characterized and contain the Mn4O4-cubane core surrounded by six facially bridging diphenylphosphinate (Ph2PO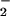 ) ligands (17, 18). In previous work we determined the reaction stoichiometries and estimated the O—H bond enthalpies for these reactions (Scheme S1; refs. 19 and 20).
) ligands (17, 18). In previous work we determined the reaction stoichiometries and estimated the O—H bond enthalpies for these reactions (Scheme S1; refs. 19 and 20).
Scheme 1.

Reduction of Mn4O4(Ph2PO2)6 and [Mn4O4(Ph2PO2)6]+ by pzH occurs by H-atom vs. hydride transfer, respectively.
The cubane core in 1+ has been proposed to be a structural model for the highest oxidation state of the PSII-WOC (S4) (21, 22). Support for this proposal comes from the ability of complex 1 to produce O2 after removal of one of the Ph2PO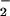 ligands via a photochemical reaction that is highly selective to the Mn4O4-cubane topology (23, 24).
ligands via a photochemical reaction that is highly selective to the Mn4O4-cubane topology (23, 24).
Materials and Methods
Mn4O4(O2PPh2)6 and [Mn4O4(O2PPh2)6]+(ClO ) were synthesized as described (18, 20). pzH (Aldrich) was used after recrystallization in CH3CN. Mono-deuterio-pzH (pzD) was obtained by stirring a solution of pzH in CH3CN with excess 2H2O (99%) for 4 h and stripping solvent under vacuum. This operation was repeated six times. The IR spectrum showed the disappearance of the N—H band at 3,341 cm−1 and the formation of the corresponding N—D band at 2,490 cm−1. The yield based on integrated band areas is >90%.
) were synthesized as described (18, 20). pzH (Aldrich) was used after recrystallization in CH3CN. Mono-deuterio-pzH (pzD) was obtained by stirring a solution of pzH in CH3CN with excess 2H2O (99%) for 4 h and stripping solvent under vacuum. This operation was repeated six times. The IR spectrum showed the disappearance of the N—H band at 3,341 cm−1 and the formation of the corresponding N—D band at 2,490 cm−1. The yield based on integrated band areas is >90%.
For calibration purposes, the pzH+• cation radical, the pz+ cation, and the pz• neutral radical all were synthesized, and their molar absorptivities were confirmed from prior reports (see Fig. 7, which is published as supporting information on the PNAS web site, www.pnas.org). EPR spectra of pz• and pzH+• were used to further identify these species (19). pzH+• was prepared by the method of Billion (25, 26). Synthesis, isolation, and characterization of pz+(ClO ) was performed by stoichiometric reaction between 1+(ClO
) was performed by stoichiometric reaction between 1+(ClO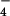 ) and pzH as described elsewhere (20). The properties matched those of pz+(ClO
) and pzH as described elsewhere (20). The properties matched those of pz+(ClO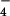 ) prepared by a literature method (27). The pz• radical was prepared in solution by deprotonation of the pzH+• radical with one equivalent of triethylamine (20). Reactions were performed by using the highest-quality dichloromethane available (OmniSolv, spectrograde, EM Science; residual water = 0.0006%).
) prepared by a literature method (27). The pz• radical was prepared in solution by deprotonation of the pzH+• radical with one equivalent of triethylamine (20). Reactions were performed by using the highest-quality dichloromethane available (OmniSolv, spectrograde, EM Science; residual water = 0.0006%).
A Hewlett–Packard 8452A spectrophotometer was used for measurements of absorbance spectra. Kinetic studies were carried out on a thermostated Hi-Tech SF-61DX2 double-mixing stopped-flow spectrophotometer at 25 or 6.7°C. The progress of the reactions was followed by monitoring the UV-Vis spectrum over the interval 350–600 nm as a function of time beginning at 70 ms.
Complex 1 is sparingly soluble only in dichloromethane, which greatly restricted the range of temperatures that could be examined: at the upper end by the need to avoid solvent evaporation due to the low boiling point (40°C) and at the lower end by solubility (<0.2 mM at 6.7°C). Replicate data sets were recorded and averaged to ensure reliability within this limited range. The enthalpy and entropy of activation were estimated by using the Eyring equation (28). Analysis of the initial kinetic data were done graphically by using the method of initial rates and numerically by using the kinetic modeling program GEPASI 3.21 with standard mass-action rate constants (29, 30).
Results
H-Atom Kinetics.
Fig. 1 illustrates the absorbance changes during the titration of complex 1 with 0–8 equivalents of pzH. Kinetic measurements were made at 384 nm, where the pz• radical has a sharp peak and there is little interference from pzH+•, pz+, pzH, or Mn4O4L6 at the concentrations used in the present experiments. The absorbance spectra for the former three species are shown in Fig. 7. The measured extinction coefficients at this wavelength are ɛpz• = 10.90 mM−1⋅cm−1 (pz•), ɛpzH+• = 1.00 mM−1⋅cm−1 (pzH+•), ɛpz+ = 0.50 mM−1⋅cm−1 (pz+), ɛpzH = 0.08 mM−1⋅cm−1 (pzH), ɛ1 = 3.84 mM−1⋅cm−1 (1), and ɛ3 = 3.12 mM−1⋅cm−1 (3).
Figure 1.

UV-Vis spectra of the titration of 1 with pzH. Bands at 316 and 384 nm correspond to free pzH and pz•, respectively. The overlay spectra are from 0– 4 equivalents of pzH in successive 0.5 equivalents, and the upper two traces are for 5 and 8 equivalents of pzH.
Under pseudo first-order conditions with an excess of 1 only a single exponential process was observed as shown in Fig. 2A. The initial slope of plots of ln(A∝ − At) vs. time was used to obtain the observed pseudo first-order rate constant k with [1]/[pzH] = 3.3–50. The k
with [1]/[pzH] = 3.3–50. The k values were reproduced to ±5% and are plotted in Fig. 2B vs. the initial concentration of 1. The rate constant k
values were reproduced to ±5% and are plotted in Fig. 2B vs. the initial concentration of 1. The rate constant k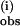 is independent of the pzH concentration at all concentrations examined. The bimolecular rate constant obtained from the slope k(i) = 21,700 ± 1,000 M−1⋅s−1 at 25°C. The same procedure was used for the reaction of 1 and pzD, also shown in Fig. 2B. This also revealed a first-order concentration dependence on 1, and the value for the apparent bimolecular rate constant was the same within the errors, k(i) = 22,600 ± 1,000 M−1⋅s−1. The resulting KIE is k
is independent of the pzH concentration at all concentrations examined. The bimolecular rate constant obtained from the slope k(i) = 21,700 ± 1,000 M−1⋅s−1 at 25°C. The same procedure was used for the reaction of 1 and pzD, also shown in Fig. 2B. This also revealed a first-order concentration dependence on 1, and the value for the apparent bimolecular rate constant was the same within the errors, k(i) = 22,600 ± 1,000 M−1⋅s−1. The resulting KIE is k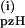 /k
/k = 0.96.
= 0.96.
Figure 2.

(A) Plot of ln(A∝ − At), measured at λ = 384 nm (pz• radical) vs. time for the first kinetic phase (i) measured in excess 1. Initial concentration ratios (pzH/1) were 1:3.33, 1:10, 1:20, 1:33.3, and 1:50 (at [pzH] = 0.003 mM). (B) Dependence of the initial slope of the rate of formation of the pz• radical, k , vs. the concentration of 1. Using [pzH] = 0.003 mM (□) the slope yields the bimolecular rate constant of 2.17 ± 0.1 × 104 M−1⋅s−1 (linear correlation coefficient, 0.999) Using [pzD] = 0.003 mM (♦) the slope yields the bimolecular rate constant of 2.26 ± 0.1 × 104 M−1⋅s−1 (linear correlation coefficient, 0.999; T = 25°C).
, vs. the concentration of 1. Using [pzH] = 0.003 mM (□) the slope yields the bimolecular rate constant of 2.17 ± 0.1 × 104 M−1⋅s−1 (linear correlation coefficient, 0.999) Using [pzD] = 0.003 mM (♦) the slope yields the bimolecular rate constant of 2.26 ± 0.1 × 104 M−1⋅s−1 (linear correlation coefficient, 0.999; T = 25°C).
The pz• radical-formation kinetics exhibit a second resolved phase when the concentration of pzH exceeds that of complex 1. This is shown by the solid curves in Fig. 3 beginning at an initial ratio of pzH/1 = 2:1. At higher ratios the slower phase gets faster and its amplitude increases as shown by the data for ratios at 3:1, 4:1, and 92:1. This slower nonexponential phase represents the additional steps that produce the remaining three pz• radicals that make up the total stoichiometry of four pz• radicals for all initial ratios ≥4:1 pzH/1 (Scheme S1; ref. 23).
Figure 3.

Dependence of the pz• radical concentration vs. time at excess pzH. Initial concentration ratios (1:pzH) were 1:1, 1:2, 1:3, 1:4, and 1:92. The solid lines correspond to the experimental data, and the dotted lines correspond to numerical simulations using the model shown in Scheme S2. The largest residual = 2.8% deviation between the model and the data. [1] = 0.03 mM for all the experiments except for 1/pzH = 1:92, for which [1] = 0.0013 mM. Data were obtained at 384 nm.
The rate data were fitted to two pseudo first-order rate processes by subtraction of the initial exponential phase by linear extrapolation to time 0 of the plot of ln(A∝ − At) vs. time (Fig. 8, which is published as supporting information on the PNAS web site). This yielded k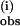 from the slope. The remaining best-fit linear slope gave k
from the slope. The remaining best-fit linear slope gave k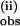 . The resulting values of k
. The resulting values of k and k
and k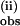 are plotted in Fig. 4 A and B, respectively, vs. the initial concentration of pzH using excess pzH.
are plotted in Fig. 4 A and B, respectively, vs. the initial concentration of pzH using excess pzH.
Figure 4.

H/D KIE on the rate constants measured with excess pzH or pzD. Dependence of the pseudo first-order rate constants on concentration of pzD or pzH for the first (k ) (A) and second (k
) (A) and second (k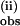 ) (B) phase. [L6Mn4O4] = 0.0025 mM; T = 25°C. A constant was subtracted from the k(i) rate constant (1 s−1) to account for a mixing artifact.
) (B) phase. [L6Mn4O4] = 0.0025 mM; T = 25°C. A constant was subtracted from the k(i) rate constant (1 s−1) to account for a mixing artifact.
The value of k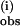 obtained under the two ranges of pzH concentrations [Fig. 2 (pzH < L6Mn4O4) and Fig. 4 (pzH > L6Mn4O4)] yields approximately the same value for k(i) except that the value obtained at low pzH concentrations is more accurate because it does not require deconvolution of the slower phase. The initial exponential phase becomes a smaller fraction of the overall decay at excess pzH concentrations and saturates, becoming independent of the concentration of pzH. This saturation suggests that complex formation or product release limits the rate of reaction at high pzH concentrations.
obtained under the two ranges of pzH concentrations [Fig. 2 (pzH < L6Mn4O4) and Fig. 4 (pzH > L6Mn4O4)] yields approximately the same value for k(i) except that the value obtained at low pzH concentrations is more accurate because it does not require deconvolution of the slower phase. The initial exponential phase becomes a smaller fraction of the overall decay at excess pzH concentrations and saturates, becoming independent of the concentration of pzH. This saturation suggests that complex formation or product release limits the rate of reaction at high pzH concentrations.
Fig. 4B shows that k slows appreciably with increasing concentration of pzH and eventually saturates, becoming independent of pzH concentration. The largest value of the bimolecular rate constant [k(ii)] was obtained from the initial (linear) slope of k
slows appreciably with increasing concentration of pzH and eventually saturates, becoming independent of pzH concentration. The largest value of the bimolecular rate constant [k(ii)] was obtained from the initial (linear) slope of k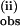 in Fig. 4B between 0.05 and 0.12 mM pzH. This yielded the value k(ii) = 1.6 × 103 M−1⋅s−1. The initial and second rate constants, obtained between 0 and 2 equivalents of pzH, are listed in Table 1. At higher pzH concentrations the assumption of a biexponential fit becomes increasingly poor. The derived bimolecular rate constant k(ii) is slower and eventually reaches saturation, becoming independent of the pzH concentration. This yields a first-order rate constant of 600 s−1 at pzH saturation. k(ii) monitors the rate of pz• radical formation by reaction of pzH with partially reduced species (1H, 2, and 2H) and therefore reflects an average over multiple steps.
in Fig. 4B between 0.05 and 0.12 mM pzH. This yielded the value k(ii) = 1.6 × 103 M−1⋅s−1. The initial and second rate constants, obtained between 0 and 2 equivalents of pzH, are listed in Table 1. At higher pzH concentrations the assumption of a biexponential fit becomes increasingly poor. The derived bimolecular rate constant k(ii) is slower and eventually reaches saturation, becoming independent of the pzH concentration. This yields a first-order rate constant of 600 s−1 at pzH saturation. k(ii) monitors the rate of pz• radical formation by reaction of pzH with partially reduced species (1H, 2, and 2H) and therefore reflects an average over multiple steps.
Table 1.
Rate constants and thermochemical data for the oxidation of pzH at 298 K

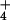
Fig. 4 also plots the rate of pz• radical formation as a function of (excess) concentration of mono-deuterated pzD, replacing pzH. Two kinetic phases are again observed. The initial phase corresponding to k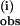 is indistinguishable from the rate constant obtained with pzH over the same range of concentrations (Fig. 4A). From the initial slopes of these plots one obtains the bimolecular rate constant k(i), which gives the KIE = k(i)(pzH)/k(i)(pzD) = 1.0. By contrast, the second phase corresponding to k
is indistinguishable from the rate constant obtained with pzH over the same range of concentrations (Fig. 4A). From the initial slopes of these plots one obtains the bimolecular rate constant k(i), which gives the KIE = k(i)(pzH)/k(i)(pzD) = 1.0. By contrast, the second phase corresponding to k was measurably slower with pzD than with pzH, ranging from KIE = k
was measurably slower with pzD than with pzH, ranging from KIE = k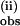 (pzH)/k
(pzH)/k (pzD) = 1.4 at ≈2 equivalents of pzH to a maximum value of 1.9 at ≈4 equivalents or more of pzH (Fig. 4B).
(pzD) = 1.4 at ≈2 equivalents of pzH to a maximum value of 1.9 at ≈4 equivalents or more of pzH (Fig. 4B).
The rate of reaction of 1 with pzH was also studied at 6.7°C over the same range of concentrations as described above. The same kinetic behavior was found. Again, a single exponential phase was observed at excess of 1. k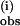 was computed from replicate measurements and is plotted in Fig. 5A as a function of the concentration of 1 and compared with that obtained at 25°C. From the observed linear relationship of the plot at 6.7°C a slope of 18,200 ± 900 M−1⋅s−1 is obtained for the bimolecular rate constant k(i), corresponding to a 16% decrease vs. 25°C. Only a limited range of temperatures could be examined in dichloromethane because of its low boiling point and the exceptionally low solubility of 1 (the only solvent in which 1 is appreciably soluble). An activation enthalpy and activation entropy for k(i) were estimated in the temperature range of 25–6.7°C from the intercepts of a plot of ln(k(i)/T) vs. 1/T using the Eyring equation, k(i) = (kT/h)exp(ΔS‡/R)exp(ΔH‡/RT), yielding ΔH‡ = −1.2 kcal/mol and ΔS‡ = −32 cal/mol⋅K and ΔG‡ = 8.3 kcal/mol at 298 K (Fig. 9, which is published as supporting information on the PNAS web site).
was computed from replicate measurements and is plotted in Fig. 5A as a function of the concentration of 1 and compared with that obtained at 25°C. From the observed linear relationship of the plot at 6.7°C a slope of 18,200 ± 900 M−1⋅s−1 is obtained for the bimolecular rate constant k(i), corresponding to a 16% decrease vs. 25°C. Only a limited range of temperatures could be examined in dichloromethane because of its low boiling point and the exceptionally low solubility of 1 (the only solvent in which 1 is appreciably soluble). An activation enthalpy and activation entropy for k(i) were estimated in the temperature range of 25–6.7°C from the intercepts of a plot of ln(k(i)/T) vs. 1/T using the Eyring equation, k(i) = (kT/h)exp(ΔS‡/R)exp(ΔH‡/RT), yielding ΔH‡ = −1.2 kcal/mol and ΔS‡ = −32 cal/mol⋅K and ΔG‡ = 8.3 kcal/mol at 298 K (Fig. 9, which is published as supporting information on the PNAS web site).
Figure 5.

(A) Comparison of k measured under pseudo first-order conditions with excess 1 at T = 25°C (□) and 6.7°C (♦) ([pzH] = 0.003 mM). See Fig. 2 for details. The dependence on pzH concentration at excess pzH concentrations of k
measured under pseudo first-order conditions with excess 1 at T = 25°C (□) and 6.7°C (♦) ([pzH] = 0.003 mM). See Fig. 2 for details. The dependence on pzH concentration at excess pzH concentrations of k (B) and the second phase k
(B) and the second phase k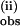 (C) at both 25 and 6.7°C. In B and C, [Mn4O4L6] = 0.0025 mM. A constant was subtracted from the k
(C) at both 25 and 6.7°C. In B and C, [Mn4O4L6] = 0.0025 mM. A constant was subtracted from the k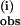 rate constant (1 s−1) to account for a mixing artifact in B.
rate constant (1 s−1) to account for a mixing artifact in B.
Analogous to the data at 25°C, the data at 6.7°C also exhibit a slower nonexponential phase in the presence of 2 or more equivalents of pzH, from which k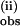 was obtained as a function of pzH concentration. k
was obtained as a function of pzH concentration. k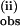 decreases at the lower temperature by ≈15–20% (Fig. 5 B and C). The similar temperature dependences of k
decreases at the lower temperature by ≈15–20% (Fig. 5 B and C). The similar temperature dependences of k and k
and k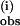 suggest that similar activation processes are involved. However, the temperature dependence of k
suggest that similar activation processes are involved. However, the temperature dependence of k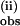 was not analyzed further, because this phase represents multiple radical-forming steps.
was not analyzed further, because this phase represents multiple radical-forming steps.
Hydride Transfer to 1+.
Kinetic absorbance measurements of the reaction of pzH with 1+ were performed while monitoring wavelengths corresponding to the peak absorptions for pzH, pz+, pzH+•, and pz• (spectra are shown in Fig. 7). These measurements showed that formation of the pz+ cation is the only observable product between 0 and 1 equivalent of pzH. Between 2 and 4 equivalents a second product appears corresponding to exclusive formation of the neutral pz• radical. The overall product stoichiometry of the reaction with 4 or more equivalents of pzH was found to be 1 pz+ and 3 pz• (Scheme S1) (20). The kinetics of the first step with excess 1+ are depicted in Fig. 6A, monitoring at the peak absorbance of pz+ (422 nm). The initial rate was examined in the range of 0.01–0.03 and found to be first-order in the concentration of 1+ with a bimolecular rate constant of 2.0 ± 0.03 × 104 M−1⋅s−1 (Fig. 5B). Because no other intermediates were detected, the data suggest that hydride transfer may occur via a concerted process, although transient intermediates with lifetimes shorter than 0.1 s would not be detectable. Remarkably, this rate constant is within a factor of 25%, the same as for the initial H-transfer step between pzH and 1.
Figure 6.

(A) Time dependence of the formation of the pz+ cation (absorbance peak at 422 nm) for the reaction of pzH with excess 1+(ClO ) at 278 K. (B) Dependence of the pseudo first-order rate constant on the initial concentration of 1+(ClO
) at 278 K. (B) Dependence of the pseudo first-order rate constant on the initial concentration of 1+(ClO ). (Inset) Initial concentration ratios of pzH/1+ (1:3.33, 1:5, and 1:10).
). (Inset) Initial concentration ratios of pzH/1+ (1:3.33, 1:5, and 1:10).
Discussion
The linear dependence of both k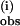 and k
and k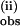 on pzH concentration indicates that these bimolecular processes are first-order in pzH, with at least a 10-fold faster rate of oxidation of the first pzH molecule than the second. The second kinetic phase is observed only above concentrations of pzH that exceed that of 1. The second through fourth pz• radicals form with progressively slower rate constants, leading to the overall stoichiometry shown in Scheme S1.
on pzH concentration indicates that these bimolecular processes are first-order in pzH, with at least a 10-fold faster rate of oxidation of the first pzH molecule than the second. The second kinetic phase is observed only above concentrations of pzH that exceed that of 1. The second through fourth pz• radicals form with progressively slower rate constants, leading to the overall stoichiometry shown in Scheme S1.
The saturation of the observed rate constants, both k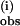 and k
and k , with increasing pzH concentration (Fig. 4) provides evidence for the formation of a precursor complex between pzH/pz• and 1/1H on the first and subsequent steps, respectively. The bimolecular rate constant k(i) is ≈106-fold slower than the predicted bimolecular diffusive encounter frequency for molecules of this size (28), suggesting that steric constraints may be limiting the rate of radical formation. The 16% decrease in k(i) between 25 and 6.7°C arises from a small enthalpy decrease in the transition state, ΔH‡ = −1.2 kcal/mol, and an appreciable entropy of activation, ΔS‡ = −32 cal/mol⋅K. The small negative ΔH‡ indicates that the rate-determining step in formation of the first pz• radical has no enthalpy barrier and actually falls in the range observed for weak van der Waals interactions (31). This value is compatible with formation of a complex between the planar aromatic pzH molecule and the hydrophobic pocket formed by the adjacent phenyl rings from three Ph2PO
, with increasing pzH concentration (Fig. 4) provides evidence for the formation of a precursor complex between pzH/pz• and 1/1H on the first and subsequent steps, respectively. The bimolecular rate constant k(i) is ≈106-fold slower than the predicted bimolecular diffusive encounter frequency for molecules of this size (28), suggesting that steric constraints may be limiting the rate of radical formation. The 16% decrease in k(i) between 25 and 6.7°C arises from a small enthalpy decrease in the transition state, ΔH‡ = −1.2 kcal/mol, and an appreciable entropy of activation, ΔS‡ = −32 cal/mol⋅K. The small negative ΔH‡ indicates that the rate-determining step in formation of the first pz• radical has no enthalpy barrier and actually falls in the range observed for weak van der Waals interactions (31). This value is compatible with formation of a complex between the planar aromatic pzH molecule and the hydrophobic pocket formed by the adjacent phenyl rings from three Ph2PO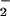 ligands that surround each of the core oxygen atoms in 1. ΔS‡ dominates the free energy of activation (−TΔS‡ = 9.5 kcal/mol at 298 K), indicating a transition state that is significantly more ordered than the reactants. The source of entropy decrease in the transition state is postulated as due to the loss of phenyl group rotation from the three adjacent Ph2PO
ligands that surround each of the core oxygen atoms in 1. ΔS‡ dominates the free energy of activation (−TΔS‡ = 9.5 kcal/mol at 298 K), indicating a transition state that is significantly more ordered than the reactants. The source of entropy decrease in the transition state is postulated as due to the loss of phenyl group rotation from the three adjacent Ph2PO ligands that surround each corner oxygen. Such a pathway would suggest that a requirement for pcet to 1 is very close contact, possibly involving direct van der Waals contact between the N—H bond in pzH and the μ3-oxo of 1.
ligands that surround each corner oxygen. Such a pathway would suggest that a requirement for pcet to 1 is very close contact, possibly involving direct van der Waals contact between the N—H bond in pzH and the μ3-oxo of 1.
Precursor complex formation is also indicated by (i) the longer lifetime of the pz• radical that is observed in the reaction mixtures containing reduced 1, compared with a faster bimolecular decay of free pz• radicals (in the absence of 1) and (ii) enhanced spin relaxation of the pz• radical seen by EPR spectroscopy in the presence of the reduced (paramagnetic) cluster vs. the free pz• radical (19).
Numerical Modeling.
Numerical simulations of the experimental kinetics were performed at each concentration by using standard mass-action rate constants without inclusion of the number of oxo sites in 1. A two-step model gives an approximate fit for ratios of 1/pzH between 1:1 and 1:2 but is increasingly poor above this ratio where the remaining 2 equivalents of pz• radical form. The fit of the data to a four-step model given by Scheme S2 is shown in Fig. 3 by the dotted lines for each of five initial pzH concentrations. Similarly good fits were obtained for the absorbance traces observed by using pzD (Fig. 10, which is published as supporting information on the PNAS web site). The largest residual corresponds to a 2.8% (pzH) and 2.7% (pzD) deviation between the model and the data for the five initial pzH concentrations examined. The numerically derived rate constants for the first two radical-forming steps are in excellent agreement with k(i) and k(ii) obtained graphically. The numerically derived second, third, and fourth rate constants cover a range that is bounded by the values of the graphically determined rate constant k(ii) obtained for concentration ratios (pzH/1) >1:1 (i.e., between 1:1 and 92:1). However, the agreement between the model and the data should not necessarily be considered support for the later steps of the kinetic model; other, more complex models may also fit the data. Hence, the kinetic parameters following the first two steps of the model have not been analyzed further.
Scheme 2.

Steps in the pcet reductive dehydration of 1.
KIEs.
The absence of a KIE on k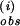 supports the identification of the rate-limiting initial radical-forming step between 1 and pzH as a process in which the N—H bond is not appreciably weakened in the transition state. This conclusion is consistent with the proposed complex-formation step in which attainment of a reactive precursor complex is rate-limiting before pcet (i.e., conformational gating). The pcet process is not rate-limiting and appears to occur by concerted H-atom transfer.
supports the identification of the rate-limiting initial radical-forming step between 1 and pzH as a process in which the N—H bond is not appreciably weakened in the transition state. This conclusion is consistent with the proposed complex-formation step in which attainment of a reactive precursor complex is rate-limiting before pcet (i.e., conformational gating). The pcet process is not rate-limiting and appears to occur by concerted H-atom transfer.
Other pcet pathways are not supported. For example, another explanation to consider is that formation of the initial pz• radical within the precursor complex is rate-limiting but has no discernible KIE. For this to be true requires that there is no appreciable change in the N—H bond strength (i.e., N—H vibrational frequency) in the transition state that controls the rate of the pcet process. This would be the case if the redox and proton-transfer steps are uncoupled such that electron transfer is rate-limiting (early transition state), followed by a faster (kinetically unobservable) proton-transfer step from the pzD+• cation radical. This interpretation is consistent with the high proton affinity of 1− (pKa > 22, 1−/1H) but is inconsistent with the observation that the pz− anion does not react with 1 by electron transfer (20). The latter reaction is thermodynamically unfavorable, whereas reduction by net H atom is thermodynamically allowed. An alternative interpretation to produce a small KIE is that the precursor complex already has transferred the proton, [1H+pz−], before a rate-limiting electron-transfer step. However, this latter process is very unlikely considering the highly unfavorable difference in pKa values (pKa < 3 for 1H+ vs. pKa = 22 for pzH) (20, 32).
The small KIE for k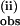 , increasing from 1.4 to 1.9 between 2 and 4 equivalents of pzH (pzD) (Fig. 4), suggests that there could be an increase in the N—H bond character present in the transition state(s) of the remaining redox steps. This behavior could be expected if the driving force for the H-atom transfer is lowered after partial reduction or if steric constraints are progressively relaxed after reduction such that atom transfer comprises a greater fraction of the transition state (5).
, increasing from 1.4 to 1.9 between 2 and 4 equivalents of pzH (pzD) (Fig. 4), suggests that there could be an increase in the N—H bond character present in the transition state(s) of the remaining redox steps. This behavior could be expected if the driving force for the H-atom transfer is lowered after partial reduction or if steric constraints are progressively relaxed after reduction such that atom transfer comprises a greater fraction of the transition state (5).
The KIEs for k(i) and k(ii) are small in comparison with other pcet reactions involving cleavage of N—H bonds. For example, formation of an N—H bond in bis-imidazoline (Hbim) by net transfer of an H atom to Fe(III)(Hbim)(H2bim)2 from a C—H bond in dihydroanthracene has a CH/CD isotope effect of 4 at the lower end (33). A large H/D KIE of 41 is observed for the redox reaction involving cleavage of the N—H hydrazido bond in the Os(IV) complex (tpy)OsCl2[HNN(CH2)4O] by quinone to form the hydrosemiquinone (34).
Table 1 compares the rate constants and H/D KIE for H-atom abstraction from pzH by a diverse group of radical oxidants (alkyl, alkoxyl, and peroxyl radicals) and for 1. These rate constants range from 3- to 105-fold faster than for 1. The rate constants are also compared with the bond-dissociation enthalpy (BDE) of the X—H bond that is formed. These were obtained from the NIST database of gas-phase proton affinity and electron affinity data by using the exact expression, −BDE = EA + PA + 13.6 eV, where IP = 13.6 eV is the gas-phase ionization potential of an H atom. The rate constants for 1 and the radical oxidants in Table 1 do not exhibit a linear correlation with BDE even though within each family of radical oxidants there is usually a good linear correlation with the BDE of the substrate (3). The BDE for dissociation of the O—H bond in 1H (>94 kcal/mol) was used for this comparison (20). The poor correlation of rate constants from different X—H groups with the BDE indicates that an additional factor(s) other than the homolytic bond-dissociation energy of X—H contributes to determining the rates. Several other factors may contribute to the different rates for these reactions including specific orientations and energetic requirements for precursor complex formation and specific solvation effects (5).
Hydride vs. H-Atom Transfer.
The similarity in the rate constants for the initial H-atom transfer between pzH and 1 vs. hydride transfer between pzH and 1+ came as a surprise (Table 1), because these reactions involve differences in both reactant charges and BDEs. The BDE difference for these reaction types is estimated at ≤33 kcal/mol, which is attributable to the 1.4-V electrochemical potential for oxidation of 1 to 1+. The invariance of the rate for these different reaction types indicates that steric inaccessibility to the oxygen atoms in the Mn4O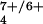 cores is likely responsible for controlling the rates of both hydride and H transfer, respectively, as well as suppressing the H/D KIE (for 1/1H). From the analysis given above we are led to the conclusion that the use of BDEs to rationalize the rates of exothermic pcet reactions is not always applicable.
cores is likely responsible for controlling the rates of both hydride and H transfer, respectively, as well as suppressing the H/D KIE (for 1/1H). From the analysis given above we are led to the conclusion that the use of BDEs to rationalize the rates of exothermic pcet reactions is not always applicable.
Relevance to Photosynthetic Water Oxidation.
The directions of the proposed pcet reactions attributed to the spontaneous S-state transitions are reversed compared with the pcet reactions shown here for the cubane complex. This reversal is not due primarily to the somewhat stronger OH bond energy in tyrosine (86.5 kcal/mol; ref. 35) vs. the slightly weaker NH bond energy in pzH (79.3–82.3 kcal/mol; refs. 4 and 32) or to an electronic difference between these similar π aromatic radicals. Rather, it means that a considerably weaker BDE of the substrate water O—H bond is required than for the O—H bond of complex 1H (>94 kcal/mol vs. 119 kcal/mol for free water). Some cationic dimanganese(III,IV) complexes containing water or hydroxide ligated to an Mn2O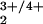 core do have sufficiently weak O—H bonds to allow direct H-atom transfer to the tyrosyl radical (35). However, the cumulative evidence is against the existence of one or more isolated Mn2O
core do have sufficiently weak O—H bonds to allow direct H-atom transfer to the tyrosyl radical (35). However, the cumulative evidence is against the existence of one or more isolated Mn2O cores comprising the PSII-WOC and rather indicates a tetramanganese-oxo core (Mn4OxCa1Cly) that is directly activated by Ca2+ and Cl− (21, 36, 37). Thus, the water-splitting chemistry and possibly the substrate O—H BDE in the WOC has not been suitably represented yet by the available Mn-only model complexes, all of which lack activation by Ca2+.
cores comprising the PSII-WOC and rather indicates a tetramanganese-oxo core (Mn4OxCa1Cly) that is directly activated by Ca2+ and Cl− (21, 36, 37). Thus, the water-splitting chemistry and possibly the substrate O—H BDE in the WOC has not been suitably represented yet by the available Mn-only model complexes, all of which lack activation by Ca2+.
Rate measurements for each of the four S-state transitions involving the reduction of a photo-generated tyrosyl radical by the WOC-substrate complex leading up to O2 production on S4 indicate successively decreasing first-order rate constants in the range of 2 × 104 to 1 × 103 s−1, activation energies of 1.2, 2.5, 9, and 5 kcal/mol (38), and KIEs in the range of kH/kD ≈ 1.3–1.4 to 1.3–2.9 (39) and 1.5–2.3 (40). Mechanisms have been proposed in which these rate constants, KIEs, and activation energies have been offered as evidence for concerted H transfer in the rate-limiting process for each S-state transition (8, 9, 41–43). The present data challenge this interpretation.
The exothermic H atom-transfer reactions to both 1 and 1H from pzH yield rate constants that are comparable to the fastest S-state transitions and have comparably small or smaller KIEs. The first H-atom transfer has a small negative activation enthalpy (−1.2 kcal/mol) compared with the weakly positive values for S0 → S1 and S1 → S2 transitions and considerably smaller than seen for the later S-state transitions. We conclude from this comparison that the kinetic parameters for the first two S-state transitions do not exhibit characteristics that can be attributed unambiguously to a concerted H atom-transfer process but may involve more weakly coupled pcet reactions such as occurs with distributed hydrogen-bond networks (44). Kinetic experiments probing the reactions of YZ after removal of the Mn cluster have also suggested that pcet rather than concerted H-atom transfer is a better description of the early S-state transitions (45).
Recent x-ray diffraction studies of PSII-WOC single crystals reveal that the shortest distance between the closest electron density that could possibly be a Mn atom of the inorganic core and the phenoxyl oxygen atom of Yz is 7–7.5 Å (46, 47). This relatively long distance eliminates direct H transfer and requires either the existence of an obligatory base for transferring the proton (i.e., pcet) or a large-scale conformational change to reduce the distance to within the deBroglie wavelength of the H atom. A large structural change appears to occur within the PSII-WOC but only on the S2 → S3 → S4 transitions, although it is not yet structurally characterized (38).
Supplementary Material
Acknowledgments
This work is in memory of Jerry Babcock. We thank Dr. Shelby Hatch and Tom Carrell for technical assistance and Lynn Mendenko for housing. This work was supported by National Institutes of Health Grant GM-39932, the Spanish Ministerio de Educación y Ciencia for a postdoctoral fellowship (to M.M.), and the Ecole Normale Superieure de Lyon and the French government for fellowship support (to E.B.).
Abbreviations
- pcet
proton-coupled electron transfer
- PSII-WOC
photosystem-II water-oxidizing complex
- KIE
kinetic isotope effect
- pzH
phenothiazine
- pzH+•
the cation radical
- pz+
the cation and pz• the neutral radical
- Ph2PO

diphenylphosphinate
- pzD
mono-deuterio-pzH
- BDE
bond-dissociation enthalpy
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Binstead R A, Meyer T J. J Am Chem Soc. 1987;109:3287–3297. [Google Scholar]
- 2.Holm R H, Donahue J P. Polyhedron. 1993;12:571–589. [Google Scholar]
- 3.Mayer J. Acc Chem Res. 1998;31:441–450. [Google Scholar]
- 4.Lucarini M, Pedrielli P, Pedulli G F, Valgimigli L, Gigmes D, Tordo P. J Am Chem Soc. 1999;121:11546–11553. [Google Scholar]
- 5.Roth J P, Yoder J C, Won T-J, Mayer J M. Science. 2001;294:2524–2526. doi: 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]
- 6.Hammes-Schiffer S. Chemphyschem. 2002;3:33–42. doi: 10.1002/1439-7641(20020118)3:1<33::AID-CPHC33>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 7.Soudackov A, Hammes-Schiffer S. J Am Chem Soc. 1999;121:10598–10607. [Google Scholar]
- 8.Tommos C, Babcock G T. Acc Chem Res. 1998;31:18–25. [Google Scholar]
- 9.Hoganson C W, Babcock G T. Science. 1997;277:1953–1955. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- 10.Gupta R, MacBeth C E, Young V G, Borokik A S. J Am Chem Soc. 2002;124:1136–1137. doi: 10.1021/ja016741x. [DOI] [PubMed] [Google Scholar]
- 11.Gardner K A, Kuehnert L L, Mayer J M. Inorg Chem. 1997;36:2069–2078. doi: 10.1021/ic961297y. [DOI] [PubMed] [Google Scholar]
- 12.Kramarz K W, Norton J R. Prog Inorg Chem. 1994;42:1–65. [Google Scholar]
- 13.Green J, Dalton H. J Biol Chem. 1989;264:17698–17703. [PubMed] [Google Scholar]
- 14.Caroll J M, Norton J R. J Am Chem Soc. 1992;114:8744–8745. [Google Scholar]
- 15.Iordanova N, Hammes-Schiffer S. J Am Chem Soc. 2002;124:4848–4856. doi: 10.1021/ja017633d. [DOI] [PubMed] [Google Scholar]
- 16.Manchandra R, Thorp H H, Brudvig G W, Crabtree R H. Inorg Chem. 1991;30:494–497. [Google Scholar]
- 17.Rüttinger W, Campana C, Dismukes G C. J Am Chem Soc. 1997;119:6670–6671. [Google Scholar]
- 18.Ruettinger W F, Ho D M, Dismukes G C. Inorg Chem. 1999;38:1036–1037. doi: 10.1021/ic981145y. [DOI] [PubMed] [Google Scholar]
- 19.Ruettinger W F, Dismukes G C. Inorg Chem. 2000;39:1021–1027. doi: 10.1021/ic9911421. , and correction (2000) 39, 4186. [DOI] [PubMed] [Google Scholar]
- 20. Carrell, T. G., Bourles, E., Lin, M. & Dismukes, G. C. (2003) Inorg. Chem. 42, in press. [DOI] [PubMed]
- 21.Carrell T G, Tyryshkin A, Dismukes G C. J Biol Inorg Chem. 2002;7:2–22. doi: 10.1007/s00775-001-0305-3. [DOI] [PubMed] [Google Scholar]
- 22.Vincent J B, Christou G. Inorg Chim Acta. 1987;136:L41–L43. [Google Scholar]
- 23.Ruettinger W, Yagi M, Wolf K, Bernasek S, Dismukes G C. J Am Chem Soc. 2000;122:10353–10357. [Google Scholar]
- 24.Yagi M, Wolf K V, Baesjou P J, Bernasek S L, Dismukes G C. Angew Chem Int Ed Engl. 2001;40:2925–2928. [PubMed] [Google Scholar]
- 25.Billon J-P. Bull Soc Chim Fr. 1961;306:1923–1929. [Google Scholar]
- 26. Gilbert, B. C., Hanson, P., Norman, R. O. C. & Sutcliffe, B. T. (1966) Chem. Commun., 161–164.
- 27.Billon J-P. Ann Chim (France) 1962;7:190–196. [Google Scholar]
- 28.Laidler K J. Chemical Kinetics. New York: Harper and Row; 1987. [Google Scholar]
- 29.Mendes P. Trends Biochem Sci. 1997;22:361–363. doi: 10.1016/s0968-0004(97)01103-1. [DOI] [PubMed] [Google Scholar]
- 30.Mendes P, Kell D B. Bioinformatics. 1998;14:869–883. doi: 10.1093/bioinformatics/14.10.869. [DOI] [PubMed] [Google Scholar]
- 31.Berry R S, Rice S, Ross J R. Physical Chemistry. New York: Wiley; 1980. [Google Scholar]
- 32.Bordwell F G, Zhang X-M, Cheng J-P. J Org Chem. 1993;58:6410–6416. [Google Scholar]
- 33.Roth J P, Mayer J M. Inorg Chem. 1999;38:2760–2761. doi: 10.1021/ic990251c. [DOI] [PubMed] [Google Scholar]
- 34.Huynh M H V, Meyer T J, White P S. J Am Chem Soc. 1999;121:4530–4531. [Google Scholar]
- 35.Baldwin M, Pecoraro V L. J Am Chem Soc. 1996;118:11325–11326. [Google Scholar]
- 36.Cinco R M, Robblee J H, Rompel A, Fernandez C, Yachandra V K, Sauer K, Klein M P. J Phys Chem B. 1998;102:8248–8256. doi: 10.1021/jp981658q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cinco R M, McFarlane Holman K L, Robblee J H, Yano J, Pizarro S A, Bellacchio E, Sauer K, Yachandra V K. Biochemistry. 2002;41:12928–12933. doi: 10.1021/bi026569p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renger G, Christen G, Karge M, Eckert H-J, Irrgang K-D. J Biol Inorg Chem. 1998;3:360–366. [Google Scholar]
- 39.Lydakis-Simantiris N, Dorlet P, Ghanotakis D F, Babcock G T. Biochemistry. 1998;37:6427–6435. doi: 10.1021/bi972980e. [DOI] [PubMed] [Google Scholar]
- 40.Karge M, Irrgang K-D, Renger G. Biochemistry. 1997;36:8904–8913. doi: 10.1021/bi962342g. [DOI] [PubMed] [Google Scholar]
- 41.Manchandra R, Brudvig G W, Crabtree R H. Coord Chem Rev. 1995;144:1–38. [Google Scholar]
- 42.Westphal K L, Tommos C, Cukier R I, Babcock G T. Curr Opin Plant Biol. 2000;3:236–242. [PubMed] [Google Scholar]
- 43.Hoganson G W, Lydakis-Symantiris N, Tang X-S, Tommos C, Warncke K, Babcock G T, Diner B A, McCracken J, Styring S. Photosynth Res. 1995;46:177–184. doi: 10.1007/BF00020428. [DOI] [PubMed] [Google Scholar]
- 44.Tommos C, Babcock G T. Biochim Biophys Acta Bioenerg. 2000;1458:199–219. doi: 10.1016/s0005-2728(00)00069-4. [DOI] [PubMed] [Google Scholar]
- 45.Kuhne H, Brudvig G W. J Phys Chem B. 2002;106:8189–8196. [Google Scholar]
- 46.Zouni A, Witt H T, Kern J, Fromme P, Krauss N, Saenger W, Orth P. Nature. 2001;409:739–743. doi: 10.1038/35055589. [DOI] [PubMed] [Google Scholar]
- 47.Zouni A, Kern J, Loll B, Fromme P, Witt H, Orth P, Krauβ N, Saenger W, Biesiadka J. Proceedings of the 12th International Congress on Photosynthesis. Melbourne: CSIRO; 2001. [Google Scholar]
- 48.Lias S G. J Phys Chem Ref Data. 1988;17:21. [Google Scholar]
- 49.Burton A, Ingold K U, Walton J C. J Org Chem. 1996;61:3779–3782. doi: 10.1021/jo952162x. [DOI] [PubMed] [Google Scholar]
- 50. Burrows, H. D., Kemp, T. J. & Welbourn, M. J. (1973) J. Chem. Soc. Perkin Trans. 2, 969–974.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.