

Abstract
Staphylococcus aureus CzrA and Mycobacterium tuberculosis NmtR are homologous zinc/cobalt-responsive and nickel/cobalt-responsive transcriptional repressors in vivo, respectively, and members of the ArsR/SmtB superfamily of prokaryotic metal sensor proteins. We show here that Zn(II) is the most potent negative allosteric regulator of czr operator/promoter binding in vitro with the trend Zn(II)>Co(II)≫Ni(II), whereas the opposite holds for the binding of NmtR to the nmt operator/promoter, Ni(II)>Co(II)>Zn(II). Characterization of the metal coordination complexes of CzrA and NmtR by UV/visible and x-ray absorption spectroscopies reveals that metals that form four-coordinate tetrahedral complexes with CzrA [Zn(II) and Co(II)] are potent regulators of DNA binding, whereas metals that form five- or six-coordinate complexes with NmtR [Ni(II) and Co(II)] are the strongest allosteric regulators in this system. Strikingly, the Zn(II) coordination complexes of CzrA and NmtR cannot be distinguished from one another by x-ray absorption spectroscopy, with the best fit a His-3-carboxylate complex in both cases. Inspection of the primary structures of CzrA and NmtR, coupled with previous functional data, suggests that three conserved His and one Asp from the C-terminal α5 helix donate ligands to create a four-coordinate complex in both CzrA and NmtR, with NmtR uniquely capable of expanding its coordination number in the Ni(II) and Co(II) complexes by recruiting additional His ligands from a C-terminal extension of the α5 helix.
About one-third of all proteins exploit specific metal ions to assist in macromolecular folding and/or function at the active site of metalloenzymes (1). All cells restrict the number of bioavailable metal atoms to avoid any excess that would otherwise compete with native metal ion sites that do not support biological activity (2). Essentially all cell types contain intracellular metal sensors that detect surplus metal ions and control the expression of genes encoding proteins that expel or sequester the extra ions (3). For some metals and some cell types, a complementary set of sensors detect deficiency and regulate genes encoding proteins that acquire more of the required ions (4, 5). It is currently poorly understood how such metal-sensing metalloregulators accurately discriminate between various metal ions.
SmtB/ArsR-family regulators are ubiquitous in bacterial genomes and bind to the operator/promoter (O/P) regions of gene(s) encoding proteins involved in metal export or sequestration, repressing transcription (for a review, see ref. 6). As the concentration of metal ion increases, the effector-binding sites of the regulators become occupied eliciting a conformational change that weakens the affinity for the O/P region, allowing transcription to proceed. Members of the SmtB/ArsR family include: As(III), Sb(III), Bi(III)-responsive ArsR (7), Zn(II)-responsive SmtB (8), Cd(II), Pb(II), Bi(III)-responsive CadC (9–11), Zn(II)-responsive ZiaR (12), Co(II), Zn(II)-responsive CzrA (13, 14), and, most recently, Ni(II), Co(II)-responsive NmtR (15).
Comparative structural and spectroscopic studies of six SmtB/ArsR family members reveal that individual members are characterized by one or both of two structurally distinct metal coordination sites (6, 11, 15–20). These two metal sites are designated α3N (or α3) and α5 (or α5C), named for the location of the metal-binding ligands within the known or predicted secondary structure of individual family members. The coordination environment and precise ligand set of the α3, α3N, and/or α5, α5C sites in the different SmtB/ArsR proteins differ and are presumed to contribute toward metal selectivity. A sequence comparison for proteins discussed herein is shown in Fig. 1 and highlights these sites.
Figure 1.

A multiple sequence alignment of Synechococcus PCC 7942 SmtB (P30340), S. aureus CzrA (O85142), and M. tuberculosis NmtR (NP_218262.1) generated by CLUSTAL W (36) (Swiss-Prot/TrEMBL accession nos. for CzrA and SmtB or the GenBank accession no. for NmtR are in parentheses). Residues that are identical (*), residues that are strongly similar (:), and residues that are weakly similar (.) are denoted below the alignment. Secondary structure elements are denoted above the alignment and based on the crystal structure of apo-SmtB (34) and chemical-shift indexing for CzrA (6). Conserved metal-binding residues located in the α5 site are boxed, as are proposed ligands to the α3N site in SmtB (C14, H18, C61, D64) (17, 18).
Here we report insights gained from the study of two additional family members, Staphylococcus aureus CzrA and Mycobacterium tuberculosis NmtR. CzrA and NmtR share 30% sequence identity and a high degree of similarity (60%) yet respond to distinct but partially overlapping metal profiles in vivo. S. aureus CzrA is a Co(II)/Zn(II)-specific sensor that regulates the expression of the czr operon, which encodes a Co(II)/Zn(II)-facilitated pump, CzrB, that effluxes metal out of the cell (13, 14). Electromobility-shift assays and in vivo expression studies indicate that Zn(II) is the strongest inducer of CzrA regulation, with Co(II) also capable of regulation but only at higher concentrations than Zn(II). Other metals, including Ni(II), have little to no effect on derepression of the czr operon (13, 14). The situation is precisely opposite for M. tuberculosis NmtR, with Ni(II) the strongest inducer of NmtR-dependent regulation of the nmt operon, which encodes a P-type ATPase metal efflux pump, NmtA (15). Co(II) is an intermediate inducer, whereas Zn(II) has no effect in vivo on derepression (15). We make the striking finding here that CzrA and NmtR achieve their distinct metal selectivities of negative regulation of DNA binding in vivo and in vitro largely on the basis of different coordination numbers and geometries, with CzrA forming functional four-coordinate metal complexes, whereas NmtR requires formation of five- or six-coordinate complexes to effect metalloregulation.
Materials and Methods
Chemicals.
All buffers were prepared by using Milli-Q deionized water. Mes, Hepes, and Tris buffer salts and ammonium sulfate were obtained from Sigma. All chromatography materials were obtained from Pharmacia Biotech. Ultrapure cobalt(II) chloride, nickel(II) chloride, and zinc(II) sulfate were obtained from Johnson Matthey (Raystan, U.K.).
Purification of CzrA and NmtR.
Recombinant CzrA and NmtR were expressed from pET-CzrA and -NmtR, respectively, in Escherichia coli BL21(DE3)/pLysS and were grown on 1.5% LB agar plates containing 0.1 mg/ml ampicillin and 34 μg/ml chloramphenicol at 37°C. The growth, expression, and purification of NmtR and CzrA were carried out by using the procedure described for SmtB (16), except that DTT was excluded from the purification because CzrA and NmtR do not contain cysteine residues. After purification, purified CzrA or NmtR was dialyzed against 6 liters of buffer S (10 mM Hepes/0.1 M NaCl, pH 7.0) with changes every 4 h for a total of 24 h at 4°C. Inspection of overloaded Coomassie-stained Tris-glycine SDS gels was used to estimate the purity to be >95%. N-terminal sequencing of CzrA and NmtR (five cycles) revealed that both proteins do not possess the expected N-terminal methionine residue. The first five cycles do match the expected N-terminal sequences of CzrA and NmtR after the methionine residue. The concentration of purified CzrA or NmtR was determined by using the calculated molar extinction coefficient at 280 nm of 4,470 M−1⋅cm−1 (21).
Atomic Absorption Spectroscopy.
The residual metal content of CzrA and NmtR was determined by using a Perkin–Elmer AAnalyst 700 atomic absorption spectrophotometer operating in flame mode. The total metal content of purified “metal-free” proteins was no more than 0.1 mol metal ion per mol protein monomer. For the determination of the concentration of metals used as titrants for optical titrations, each metal was detected by using Zn(II)-, Co(II)-, or Ni(II)-specific hollow cathode lamps. Zn(II) was detected at 213.9 nm (slit width, 0.7 nm), Co(II) at 240.7 nm (slit width, 0.2 nm), and Ni(II) at 232.0 nm (slit width, 0.2 nm).
Analytical Sedimentation Equilibrium Ultracentrifugation.
All experiments were run with a Beckman Coulter Optima XL-A analytical ultracentrifuge equipped with an An60 Ti rotor and two-channel 12-mm path length, charcoal-filled Epon Centerpieces, and quartz windows at 235 nm with a rotor speed of 30,000 rpm at 25.0°C. Apo-CzrA and -NmtR samples (5 μM) were prepared by dilution into 10 mM Hepes/0.4 M NaCl/0.05 mM EDTA, pH 7.0. Co(II), Ni(II), and Zn(II) protein samples were prepared by addition of 1 mol equiv of Co(II), Ni(II), or Zn(II) to the apoprotein in the absence of EDTA. The partial specific volumes (ν) of CzrA and NmtR were calculated to be 0.740 ml/g−1 and 0.736 ml/g−1, respectively, using the sednterp program (22). Sedimentation equilibrium data were fit by using origin software (Microcal, Northampton, MA) to a single ideal species model and a self-association model of a single ideal species, assuming a monomer–dimer equilibrium characterized by the association constant Kdimer, as described (19). The association constant was converted from absorbance to units of M−1 by using the calculated extinction coefficients ɛ235 = 22,543 M−1⋅cm−1 and ɛ235 = 25,246 M−1⋅cm−1 for CzrA and NmtR, respectively, and a path length l = 1.2 cm.
Fluorescence Anisotropy-Based DNA-Binding Experiments.
All fluorescence anisotropy experiments were carried out with an SLM 4800 (SLM-Aminco, Urbana, IL) spectrofluorometer operating in the steady-state mode fitted with Glan-Thompson (SLM Instruments, Urbana, IL) polarizers in the L format (11). The 57-mer double-stranded czr O/P oligonucleotide (Operon Technologies, Alameda, CA) used was fluorescein-labeled on one 5′-end with the sequence given below.
F-5′-TTAAATTAATATATGAACAAATATTCAGATGAAAGGA- TTGTCATATAATGTCAGAAC-3′
3′-AATTTAATTATATACTTGTTTATAAGTCTACTTTCCT- AACAGTATATTACAGTCTTG-5′
The 48-mer double-stranded nmt O/P oligonucleotide (Operon Technologies) used was also fluorescein-labeled on one 5′-end with the sequence given below.
F-5′-AGAAATAAATGAACAGTTGATCCTATATTCTGATATG- TTGGCCGGGTG-3′
3′-TCTTTATTTACTTGTCAACTAGGATATAAGACTATAC- AACCGGCCCAC-5′
All experiments were carried out with 40 nM double-stranded DNA in 10 mM Hepes and 0.4 M NaCl (pH 7.0) in a volume of 1.8 ml. Up to 650 μl of ≈25–35 μM apo-, Co(II)1-, Ni(II)1-, or Zn(II)1-protein stocks were added in known aliquots up to a total concentration of ≈6 μM protein. DNA-binding experiments were performed in the presence and absence of 50 μM EDTA for metal-free apo-protein and metal-complexed proteins, respectively. The binding equilibria of metal-complexed CzrA and NmtR were determined for protein titrants prepared 1:1 with metal (i.e., 1 molar equivalent of metal per monomer of protein or 2 molar equivalents per dimer).
Binding isotherms for CzrA were fit by using dynafit (23) to a model involving the sequential binding of five CzrA dimers (P2) to the DNA oligonucleotide (D), each defined by the indicated Ki, linked to the monomer–dimer equilibrium established for CzrA (see also ref. 17).
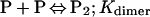 |
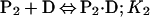 |
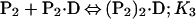 |
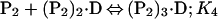 |
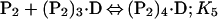 |
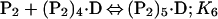 |
In these fits, Kdimer was fixed at the value determined by analytical equilibrium ultracentrifugation under identical solution conditions (Table 1). The characteristic anisotropies, ri, for each (P2)n⋅D complex, i.e., rP2⋅D, r(P2)2⋅D, r(P2)3⋅D, and r(P2)4⋅D, were determined from stoichiometric additions of wild-type CzrA to 10 μM dsDNA. These were found to be rP2⋅D = 0.121, r(P2)2⋅D = 0.128, r(P2)3⋅D = 0.135, and r(P2)4⋅D = 0.146 relative to a measured anisotropy value for the uncomplexed DNA (rD) of 0.117 and were treated as fixed parameters in resolution of Ki from all fits. If rD in a particular experiment was smaller or larger than rD = 0.117, then the fixed values of ri were adjusted accordingly. The value for r(P2)5⋅D was set to 0.15 in all fits when it was necessary to include it.
Table 1.
Sedimentation equilibrium ultracentrifugation analysis of CzrA and NmtR
| Sample, 5 μM | Model | Molarmass, g/mol | Kdimer,×105 M−1* | χ2, ×10−5† |
|---|---|---|---|---|
| apo-CzrA‡ | Single, ideal¶ | 19307 | 4.66 | |
| Dissociable dimer‖ | 11975 | 1.7 (±0.3) | 4.59 | |
| Co(II)1-CzrA§ | Single, ideal | 20358 | 5.42 | |
| Dissociable dimer | 11851 | 4.8 (±0.1) | 5.56 | |
| Ni(II)1-CzrA§ | Single, ideal | 20126 | 4.71 | |
| Dissociable dimer | 12051 | 6.3 (±0.2) | 5.34 | |
| Zn(II)1-CzrA§ | Single, ideal | 19784 | 4.85 | |
| Dissociable dimer | 11858 | 4.5 (±0.2) | 5.21 | |
| apo-NmtR‡ | Single, ideal | 20069 | 4.57 | |
| Dissociable dimer | 12549 | 1.9 (±0.4) | 5.83 | |
| Co(II)1-NmtR§ | Single, ideal | 21952 | 5.96 | |
| Dissociable dimer | 12849 | 3.9 (±0.4) | 7.56 | |
| Ni(II)1-NmtR§ | Single, ideal | 21539 | 5.45 | |
| Dissociable dimer | 12877 | 4.1 (±0.4) | 6.22 | |
| Zn(II)1-NmtR§ | Single, ideal | 19359 | 5.41 | |
| Dissociable dimer | 12597 | 3.7 (±0.6) | 6.96 |
The association constant as estimated with a monomer–dimer equilibrium model (model 2).
x2 = (∑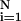 [f(Xi − Yi)]2/N − n), where N is the number of observations, n is the number of fitting parameters, f( ) is the fitting function, Xi and Yi are data points, and N − n equals the degrees of freedom.
[f(Xi − Yi)]2/N − n), where N is the number of observations, n is the number of fitting parameters, f( ) is the fitting function, Xi and Yi are data points, and N − n equals the degrees of freedom.
Conditions: 10 mM Hepes, 0.40 M NaCl, 0.1 mM EDTA, pH 7.0; 25°C.
Conditions: 10 mM Hepes, 0.40 M NaCl, pH 7.0; 25°C.
Model 1 fits the data to a single, ideal species with calculated dimer molecular weights of 23,715 g/mol for CzrA and 25,409 g/mol for NmtR (see Materials and Methods).
Model 2 fits the data to a reversible associating system assuming a monomer-dimer equilibrium. The calculated monomer molar masses are 11,857 g/mol for CzrA and 12,704 g/mol for NmtR (see Materials and Methods).
Anisotropy data for NmtR were also fit with dynafit (23) to a dissociable dimer model with a 1:1 binding stoichiometry (NmtR dimer to nmt O/P oligonucleotide) linked to a monomer–dimer equilibrium, i.e., the first two equilibria listed above. The binding isotherms (raw robs vs. [NmtR]total) were fit by using a fixed Kdimer derived from sedimentation equilibrium ultracentrifugation experiments (see Table 1).
Co(II)-Binding Experiments.
All metal-binding experiments were carried out in buffer S at ambient temperature by using a Hewlett–Packard model 8452A spectrophotometer. Apo-protein (0.8 ml of 150 μM) was prepared by dilution with buffer S. Experiments were performed and data were analyzed essentially as described (16).
X-Ray Absorption Spectroscopy.
Ni and Zn x-ray absorption spectroscopy data were collected at 10 K on NmtR and CzrA samples of 1.0–1.6 mM metal at Stanford Synchrotron Radiation Laboratory, beamline 9-3, with the Stanford Positron-Electron Accelerating Ring operating at 3.0 GeV. The beamline used focusing optics and a fully tuned monochromator containing a Si[220] crystal. A 30-element Ge solid-state x-ray fluorescence detector was used for data collection and used Soller slits and a Z–1 fluorescence filter for background rejection. All data collection parameters were as described (16). Extended x-ray absorption fine structure (EXAFS) analysis was performed with exafspak software (www-ssrl.slac.stanford.edu/exafspak. html) according to standard procedures (24). Multiple scattering models, calculated by using feff, version 8.2 (25, 26), were based on bis(aceto)-bis(imidazole)zinc(II) (27), tetra(imidazole)zinc(II) (28), hexakis(imidazole)nickel(II) chloride tetrahydrate (29), as described (30).
Results
Assembly State of CzrA and NmtR.
CzrA and NmtR were subjected to analytical equilibrium sedimentation ultracentrifugation at 5 μM protein monomer at pH 7.0, 25°C, and 0.4 M NaCl, solution conditions identical to those used to monitor DNA binding (below). The results of these experiments are presented in Table 1. The data reveal that both CzrA and NmtR are well described as dissociable homodimers under these conditions, with the magnitude of Kdimer generally in the 105 M−1 range for both apo-CzrA and -NmtR. This value is comparable to that previously found for the apo-zinc sensor, SmtB, but ≈10-fold less than that for apo-CadC (19). The magnitude of Kdimer in the presence of bound metal is consistently larger than that determined for the apoproteins, but the effect is relatively small (within a factor of two to three). This magnitude of Kdimer reveals that the homodimer is not strongly stabilized by metal binding in these proteins.
Allosteric Regulation of czr O/P- and nmt O/P-Binding of CzrA and NmtR, Respectively, by Co(II), Ni(II), and Zn(II).
Previously published gel mobility-shift experiments suggested that the addition of exogenously added Co(II) and Zn(II) to preformed apo-CzrA DNA complexes resulted in dissociation of the DNA-bound CzrA, but that addition of Ni(II) did not (13). In contrast, for NmtR, gel mobility-shift data suggested that the addition of Ni(II) and Co(II) to preformed apo-NmtR DNA complexes resulted in dissociation of the DNA-bound NmtR, but that Zn(II) had little effect at the same concentrations (15). However, the stoichiometry and affinity of CzrA and NmtR for their specific binding sites, as well as the mode of metalloregulation, could not be determined from those experiments.
Fig. 2A shows representative binding experiments carried out with apo-CzrA and a fluorescein-labeled 57-bp DNA oligonucleotide that contains the entire region of the czr O/P region footprinted by CzrA (13), under solution conditions of relatively high monovalent salt concentration (pH 7.0, 0.40 M NaCl, 25.0°C). The solid lines through each set of experimental data in Fig. 2A are nonlinear least-squares fits to the sequential dimer-binding model described in Materials and Methods with Kdimer constrained to the values given in Table 1. The large change in the anisotropy on CzrA binding suggests that CzrA oligomerizes or assembles on the DNA, forming complexes characterized by stoichiometries beyond that of a dimer binding to a single site on the DNA. This finding is consistent with the extensive footprint observed on the czr O/P (13). The same features characterize the binding of apo-SmtB to its O/P DNA (17). The binding affinity of the first three dimers of CzrA onto the DNA is essentially stoichiometric under these conditions and hence only a lower limit of 5 × 109 M−1 can be obtained for K2, K3, and K4. The binding affinity of the fourth dimer was found to be K5 = 3.4 (±0.5) × 108 M−1, whereas that of the fifth dimer was significantly weaker, K6 = 2.4 (±0.4) × 107 M−1.
Figure 2.

(A) Fluorescence anisotropy of CzrA binding to the 57-bp fluorescein labeled oligonucleotide containing the czr O/P region in the absence (⋄) and presence of Co(II) (○), Ni(II) (▾), and Zn(II) (□). (B) Fluorescence anisotropy of NmtR binding to the 48-bp fluroescein-labeled oligonucleotide containing the nmt O/P region in the absence (⋄) and presence of Co(II) (○), Ni(II) (▾), and Zn(II) (□). (Filled symbols use left and open symbols use right axes.) All experiments were performed at 25°C in 10 mM Hepes/0.4 M NaCl/0.1 mM EDTA (EDTA is not present in the metal–protein titrations), pH 7.0. The anisotropy, robs, is plotted as a function of total protein concentration in monomer. The solid lines through the data represent nonlinear least-squares fits to the binding models described in the text for CzrA and NmtR. For Co(II)1 CzrA, K2 = 6.8 (±0.8) × 106 M−1, K3 = 3.6 (±0.6) × 106 M−1, K4 = 1.3 (±0.3) × 106 M−1, K5 = 1.1 (±0.2) × 106 M−1; all other Ki values are given in the text.
To test the effect of regulatory metal ions on the association of CzrA for the fluorescein-labeled czr O/P region, metal:protein complexes were preformed and titrated into free DNA (12, 14, 15). Fig. 2A shows representative binding isotherms obtained for Co(II), Ni(II), and Zn(II) CzrA. Ni(II) CzrA has little effect on the association of CzrA with the DNA, because the data for apo-CzrA and Ni(II) CzrA are similar to one another. However, titration of Co(II) CzrA results in a reduced affinity of CzrA for DNA. The data were satisfactorily fit with the binding of four dimers, with the affinities of all four in the 106 M−1 range, or ≥1,000-fold reduced relative to apo-CzrA (Fig. 2A). Zn(II) CzrA results in even weaker association with the DNA, because only one Zn2 homodimer associates with an affinity of K2 = 4.7 (±0.3) × 105 M−1 or 105-fold lower affinity than apo-CzrA. These findings are in full agreement with previous studies that indicate that Zn(II) and Co(II) specifically and strongly negatively regulate czr O/P binding, with Zn(II) being the strongest inducer in vivo (13, 14).
Fig. 2B shows representative binding isotherms obtained for Co(II), Ni(II), and Zn(II) NmtR. The solid line through each set of experimental data represents a fit to a model that describes the binding of a fully dissociable NmtR homodimer to a single site on a 48-bp fluorescein-labeled DNA oligonucleotide encompassing the nmt O/P region (see refs. 11 and 19 for CadC). Kdimer for apo- and metal-bound NmtRs was constrained to an average value determined by analytical ultracentrifugation (Table 1), with the fits optimized for K2 and the value for the anisotropy (rmax) of the (NmtR)2–DNA complex (see Materials and Methods). Dimeric NmtR forms a complex with its DNA-binding site with K2 = 5.6 (±0.7) × 107 M−1, indicating strong linkage to the monomer–dimer equilibrium under these conditions. In striking contrast, the addition of a single equivalent of bound Ni(II) or Co(II) (Fig. 2B) results in negative regulation of nmt O/P binding, with K2 = 1.9 (±0.4) × 105 M−1 and K2 = 4.9 (±0.1) × 105 M−1, respectively, or about 200- and 100-fold reduced, respectively, relative to apo-NmtR. Relative to Ni(II) and Co(II), Zn(II) is the weakest negative regulator of nmt O/P binding but still induces some negative regulation of DNA binding K2 = 1.6 (±0.2) × 106 M−1, a result in qualitative agreement with previous gel mobility-shift assays (15).
Thus, the ability of metals to allosterically regulate specific O/P binding by CzrA and NmtR is precisely opposite one another with Zn(II)>Co(II)≫Ni(II) for CzrA, whereas for NmtR, Ni(II)>Co(II)>Zn(II) in vivo (13, 15) and in vitro. UV-visible electronic and x-ray absorption spectroscopies were used to determine the structural nature of the metal-sensing coordination complexes in each protein.
UV-Visible Absorption Spectroscopy of Co(II) CzrA and NmtR.
Shown in Fig. 3 are UV-visible absorption spectra that result on aerobic titration of metal-free apo-CzrA or apo-NmtR with Co(II) (pH 7.0, 0.1 M NaCl, 25°C). CzrA binds one molar equivalent of Co(II) per monomer with a spectrum characterized by intense d-d ligand field transitions at 570 nm, with ɛ570 ≈ 450 (±20) M−1⋅cm−1, clearly indicative of tetrahedral or distorted tetrahedral coordination geometry (16). The spectra of Co(II) SmtB, in which metal is restricted to bind to the α5 sites (18), is essentially identical to that of CzrA although less intense, also indicative of tetrahedral or distorted tetrahedral coordination geometries. In contrast, the low molar absorptivities of both the 1:1 and 2:1 Co(II):NmtR dimer complexes (15) are suggestive of five- or six-coordinate Co(II), rather than four (31).
Figure 3.

Electronic absorption spectrum of 150 μM Co(II) CzrA (solid), 150 μM Co(II)α5 SmtB (black dash-dot-dot-dot line), the 1:1 and 2:1 Co(II):dimer complexes of 500 μM NmtR (green and red solid, respectively), and the difference spectrum characteristic of the binding of the second Co(II) to the NmtR dimer (blue dash-dot line) (10). Conditions: pH 7.0, 10 mM Hepes with 100 mM NaCl (for CzrA and SmtB) or 400 mM NaCl (for NmtR).
X-Ray Absorption Spectroscopy.
Comparison of the EXAFS and Fourier transforms (FTs) for Ni(II)- or Zn(II)-bound forms of NmtR, CzrA, and SmtB [the last published previously (16) and reproduced here for comparison] reveals a distinction between the metal-binding sites of SmtB and those of NmtR and CzrA (Fig. 4). Noticeable differences in the EXAFS arise in the region near k ≈ 4 Å−1. This region is sensitive to variations in high-frequency oscillations that are commonly associated with the multiple-scattering contribution from the outer-shell atoms of the imidazole ring of histidine. An increase in the number of histidine ligands causes splitting of the main peak in this region, also visualized as increased intensity in the high-R region of the FT plot, specifically at ≈3 and 4 Å, which is diagnostic for histidine ligation. The absence of a 3-Å peak, in the presence of a 4-Å peak in the FT, has been assigned previously as the result of destructive interference between the outer-shell contributions from carboxylate and histidine ligands (16, 26). This is most clearly observed in the Zn FTs (Fig. 4) and supports the binding of a carboxylate-containing residue to the metal. The EXAFS is consistent with an increase in imidazole ligation in NmtR and CzrA, relative to SmtB. Another key difference between SmtB and NmtR/CzrA is the ≈2.3-Å shoulder in the main FT peak that appears in Zn(II) SmtB (Fig. 4). This shoulder arises from the contribution of a first-shell sulfur ligand contributed by cysteine (16).
Figure 4.

Ni (Upper) and Zn (Lower) k3-weighted EXAFS (Right Inset) and FTs (k = 2–12 Å−1) for NmtR (solid), CzrA (dotted), and SmtB [dashed (11)]. (Upper Left Inset) Comparison of the Ni K-edge spectra for NmtR (solid) and CzrA (dotted).
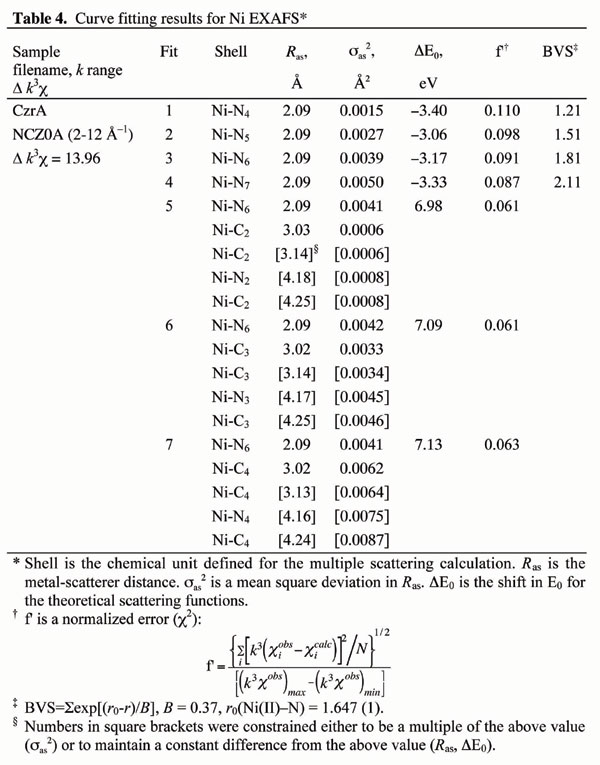
The results of extensive curve-fitting analysis of these samples (Tables 3–6, which are published as supporting information on the PNAS web site, www.pnas.org) are consistent with the observation that NmtR and CzrA have increased contribution from imidazole-containing ligands and no contribution from sulfur-containing ligands, consistent with the fact that neither CzrA nor NmtR have cysteine residues (Fig. 1). Best fits for both Ni(II) NmtR and Ni(II) CzrA (Tables 3 and 4; Fig. 5, which is published as supporting information on the PNAS web site) indicate a coordination environment containing three histidine ligands and three other oxygen- or nitrogen-containing ligands (Fits 1 and 2; Table 2). The approximate octahedral Ni(II) environment is also supported by the very low intensity of the 1s-3d transition in the preedge region (Fig. 4 Upper Left Inset). Similarly, Zn(II) NmtR and Zn(II) CzrA are best fit (Tables 5 and 6; Fig. 6, which is published as supporting information on the PNAS web site) with three histidine ligands, although in striking contrast to the Ni(II) complexes, the coordination geometry of the Zn(II)-binding site is most likely tetrahedral rather than octahedral (Fits 3 and 4; Table 1). This would be consistent with the tetrahedral coordination geometry associated with the Co(II) complex of CzrA, but in contrast to Co(II) NmtR, which clearly adopts a higher coordination number (Fig. 3) than Zn(II) NmtR.
Table 2.
Curve fitting results for EXAFS
| Sample filename (k range) Δk3χ | Fit | Shell | Ras, Å | σ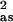 , Å2 , Å2
|
ΔE0,eV | f* |
|---|---|---|---|---|---|---|
| Ni(II) NmtR | 1 | Ni-N6 | 2.08 | 0.0045 | 7.20 | 0.061 |
| NNM0B (2–12 Å−1) | Ni-C3 | 3.02 | 0.0025 | |||
| Δk3χ = 13.89 | Ni-C3 | [3.13]† | [0.0026] | |||
| Ni-N3 | [4.17] | [0.0035] | ||||
| Ni-C3 | [4.24] | [0.0035] | ||||
| Ni(II) CzrA | 2 | Ni-N6 | 2.09 | 0.0042 | 7.09 | 0.061 |
| NCZ0A (2–12 Å−1) | Ni-C3 | 3.02 | 0.0033 | |||
| Δk3χ = 13.96 | Ni-C3 | [3.14] | [0.0034] | |||
| Ni-N3 | [4.17] | [0.0045] | ||||
| Ni-C3 | [4.25] | [0.0046] | ||||
| Zn(II) NmtR | 3 | Zn-O1‡ | 1.99 | 0.0017 | 5.62 | 0.069 |
| ZNM0B (2–12 Å−1) | Zn-C1 | [2.78] | [0.0024] | |||
| Δk3χ = 11.85 | Zn-O1 | [3.12] | [0.0027] | |||
| Ni-N3 | 1.99 | 0.0041 | ||||
| Ni-C3 | 2.96 | 0.0033 | ||||
| Ni-C3 | [3.03] | [0.0033] | ||||
| Ni-N3 | [4.15] | [0.0046] | ||||
| Ni-C3 | [4.16] | [0.0046] | ||||
| Zn(II) CzrA | 4 | Zn-O1‡ | 2.00 | 0.0005 | 5.49 | 0.083 |
| ZCZ0A (2–12 Å−1) | Zn-C1 | [2.79] | [0.0006] | |||
| Δk3χ = 11.93 | Zn-O1 | [3.15] | [0.0007] | |||
| Ni-N3 | 1.97 | 0.0024 | ||||
| Ni-C3 | 2.96 | 0.0040 | ||||
| Ni-C3 | [3.02] | [0.0040] | ||||
| Ni-N3 | [4.14] | [0.0055] | ||||
| Ni-C3 | [4.15] | [0.0055] |
Shell defines the absorber–scatterer pair (the subscript is Ns, the number of scatterers). Ras is the metal–scatterer distance. σ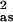 is a mean–square deviation in Ras. ΔE0 is the shift in E0 for the theoretical scattering functions.
is a mean–square deviation in Ras. ΔE0 is the shift in E0 for the theoretical scattering functions.
f′ is a normalized error (χ2): f′ = {∑i[k3(χ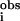 − χ
− χ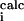 )]2/N}1/2/[(k3χobs)max − (k3χobs)min].
)]2/N}1/2/[(k3χobs)max − (k3χobs)min].
Numbers in square brackets were constrained either to be a multiple of the above value (σ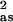 ) or to maintain a constant difference from the above value (Ras, ΔE0).
) or to maintain a constant difference from the above value (Ras, ΔE0).
The carboxylate ligand that yields these best fits displays a Zn–O–C angle of 108° and a coplanar Zn–O–C–O unit.
Discussion
The SmtB/ArsR family of metalloregulators is uniquely characterized by the presence of two metal-binding sites, neither of which is absolutely conserved across the family (6). Some SmtB/ArsR members possess only the α3N site and others possess just the α5 site, whereas some, including cyanobacterial zinc sensors SmtB and ZiaR, appear to possess both. Recent work from our laboratory has defined the coordination environments of the α3N and α5 metal-binding sites of SmtB (16–18) (Fig. 1). These studies reveal that the histidine/carboxylate-containing α5 site is responsible for binding Zn(II) in a tetrahedral coordination complex and allosterically regulating binding of SmtB to the smt operon (17, 18). The α3N site, which contains at least one cysteine thiolate ligand (Cys-14), not present in CzrA or NmtR (17) (see Fig. 1), plays some other role. These findings are fully compatible with characterization of mutant SmtBs in vivo (32). The α3N metal site possesses a higher affinity for Zn(II) in the free protein at equilibrium; this is the Zn(II) complex characterized in previous EXAFS studies and reproduced here for comparison (ref. 16; Fig. 4).
The protein determinants of metal ion specificity are not currently well defined. An attractive hypothesis is that proteins simply follow the trends predicted by traditional inorganic chemistry. Unfortunately, those trends have not provided a complete explanation for why certain metal ions are preferred by certain proteins. Sometimes the metal ion that binds with the highest affinity to the protein is not the metal ion that provides in vivo functionality, complicating the study of metal ion specificity (15). Our results provide a basis for bridging the gap between inorganic and biological chemistry that can be applied to the understanding of nature's selection process.
SmtB and CzrA that favor Zn(II) coordination at the α5 site use carboxylate and imidazole ligands to bind harder Lewis acids, such as Co(II), Ni(II), and Zn(II). NmtR, which discriminates toward Ni(II) and Co(II), also uses imidazoles. CadC and ArsR possess α3N and α3 metal sites that have all-cysteinyl coordination spheres that prefer softer Lewis acids, such as Cd(II), Pb(II), and Bi(III) (11, 19, 20). Interestingly, S. aureus pI258 CadC contains an intact α5 metal site that has been shown to bind Zn(II) and Co(II) directly; however, the binding of metal here is not regulatory for DNA binding in vitro (11) or in vivo (33).
Here, we provide spectroscopic evidence for the coordination environment of the metalloregulatory sites of S. aureus CzrA and M. tuberculosis NmtR in an attempt to determine how these proteins functionally discriminate between different transition metals. The spectroscopic properties of both CzrA and NmtR lend support to the model in which ligands located in the α5 site of the proteins contribute to binding Co(II), Ni(II), or Zn(II) (Fig. 1). The optical absorption spectra of Co(II)-substituted CzrA are consistent with a tetrahedral coordination model that utilizes three histidines and one carboxylate-containing residue to ligate the metal ion. Preliminary Co(II) XAS data support this coordination. XAS data for Zn(II) CzrA and Zn(II) NmtR also indicate a coordination model in which three histidines and one carboxylate residue donate ligands to the Zn(II) ion. By contrast, XAS data for Ni(II) NmtR and Ni(II) CzrA indicate a six-coordinate environment containing at least three histidine ligands and three other oxygen or nitrogen-containing ligands.
CzrA is selective for Co(II)/Zn(II), discriminating between Ni(II) and Co(II)/Zn(II) by creating a coordination environment in which the Ni(II) forms an octahedral complex, whereas Co(II)/Zn(II) binding is clearly tetrahedral. The XAS data are fully compatible with the α5 site schematized in the sequence alignment (Fig. 1), with Asp-84, His-86, His-97, and His-100 providing ligands to the metal. It seems likely that each pair of ligands (Asp-84/His-86 and His-97/His-100) is drawn from opposite monomers of the homodimer. This would create two identical C2-symmetric metal sites that straddle the α5 helices of each monomer, as originally hypothesized from the crystal structure of apo-SmtB (34). Recent crystallographic studies of the Zn2 CzrA homodimer are consistent with this hypothesis (M.A.P., C. Eicken, J. Sacchettini, and D.P.G., unpublished results). The DNA-binding data (Fig. 2) indicate that a tetrahedral coordination environment is required to effect negative allosteric regulation in vivo and in vitro. Thus, CzrA appears to discriminate between Co(II)/Zn(II) and Ni(II) strictly on the basis of the coordination geometry of the metal complex. How this complex expands to increase its coordination number in the Ni(II) complex is not known but presumably reflects the addition of solvent molecules. The binding of Ni(II) to the zinc sensor SmtB at low Ni(II):monomer stoichiometries can also be described by a similar mechanism (16). It has already been shown that Ni(II) binds weakly to both SmtB [≈104 weaker than Co(II)] (16) and CzrA (M.A.P. and D.G., unpublished results).
Conversely, NmtR appears to use an octahedral coordination environment to negatively regulate DNA binding with Co(II)/Ni(II) vs. Zn(II), despite the fact that Zn(II) and Ni(II) bind with comparable affinities to the protein (15). The distinguishing feature of NmtR relative to CzrA is the presence of a short C-terminal extension, which contains up to three additional potential hard Lewis base donors beyond the four ligands conserved between CzrA and NmtR (Asp-91, His-93, His-104, and His-107 in NmtR), including His-109, Asp-114, and His-116 (Fig. 1). Functional studies with substitution mutants reveal that His-109 and -116, as well as the four ligands in common with CzrA, are absolutely essential to effect Ni(II)-mediated derepression in vivo (15). This suggests a model in which Ni(II) and Co(II) are uniquely capable of recruiting one or two of these His residues into the coordination shell to create the six-coordinate species compatible with the optical absorption [Fig. 3 (15)] and XAS experiments presented here (Fig. 4). On the other hand, the Ni EXAFS FT peaks at 3 and 4 Å for NmtR and CzrA are very similar (Fig. 4), suggesting a similar histidine ligand count, although the precision of this determination can be affected by imidazole geometry or disorder (35).
NmtR requires hexacoordinate metal binding [as observed for Ni(II) and Co(II)] to affect allosteric derepression of transcription, whereas CzrA requires tetrahedral metal binding [as observed for Co(II) and Zn(II)] to achieve the same biological function. Thus Zn(II) NmtR adopts a less effective (tetrahedral) coordination, whereas Ni(II) CzrA is hexacoordinate and similarly ineffective. Nature has adapted the allosteric coupling of metal coordination (in the α5 helices) to DNA binding [to the α3-turn-αR region (6); Fig. 1] to take advantage of inorganic coordination preferences. The exact mechanism of this allosteric coupling and how the coupling must differ for CzrA and NmtR are still under investigation. However, the evidence presented here indicates that metal coordination number and geometry play a dominant role in determining which metal ions negatively allosterically regulate O/P binding in vitro.
Supplementary Material
Acknowledgments
We thank Dr. Nigel J. Robinson (University of Newcastle, Newcastle, U.K.) for comments on the manuscript and many helpful discussions. We thank Dr. Larry Dangott in the Protein Chemistry Laboratory at Texas A&M University for determination of the N-terminal sequences of CzrA and NmtR. We thank Dr. T. Ohta, University of Tsukuba (Tsukuba, Japan) for providing the S. aureus czr clone pH498 and Dr. J. Sacchettini (Texas A&M University) for genomic m. tuberculosis H37RV DNA. The XAS data were collected at Stanford Synchrotron Radiation Laboratory (SSRL), which is operated by the U.S. Department of Energy, Division of Chemical Sciences. The SSRL Biotechnology Program is supported by the National Institutes of Health (NIH), Biomedical Resource Technology Program, Division of Research Resources. These studies were supported in part by NIH Grants GM42569 (to D.P.G.) and GM42025 (to R.A.S.) and the Robert A. Welch Foundation (A-1295 to D.P.G.). M.A.P. was supported in part by an NIH Chemistry–Biology Interface Training Grant T32 GM 08523.
Abbreviations
- O/P
operator/promoter
- EXAFS
extended x-ray absorption fine structure
- FT
Fourier transform
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Rosenzweig A C. Chem Biol. 2002;9:673–677. doi: 10.1016/s1074-5521(02)00156-4. [DOI] [PubMed] [Google Scholar]
- 2.O'Halloran T V. Science. 1993;261:715–725. doi: 10.1126/science.8342038. [DOI] [PubMed] [Google Scholar]
- 3.Nies D H, Brown N L. In: Metal Ions in Gene Regulation. Silver S, Walden W, editors. New York: Chapman & Hall; 1998. pp. 77–103. [Google Scholar]
- 4.Hantke K. Curr Opin Microbiol. 2001;4:172–177. doi: 10.1016/s1369-5274(00)00184-3. [DOI] [PubMed] [Google Scholar]
- 5.Brocklehurst K R, Hobman J L, Lawley B, Blank L, Marshall S J, Brown N L, Morby A P. Mol Microbiol. 1999;31:893–902. doi: 10.1046/j.1365-2958.1999.01229.x. [DOI] [PubMed] [Google Scholar]
- 6. Busenlehner, L. S., Pennella, M. A. & Giedroc, D. P. (2003) FEMS Microbiol. Rev., in press. [DOI] [PubMed]
- 7.Xu C, Shi W, Rosen J Biol Chem. 1996;271:2427–2432. doi: 10.1074/jbc.271.5.2427. [DOI] [PubMed] [Google Scholar]
- 8.Huckle J W, Morby A P, Turner J S, Robinson N J. Mol Microbiol. 1993;7:177–187. doi: 10.1111/j.1365-2958.1993.tb01109.x. [DOI] [PubMed] [Google Scholar]
- 9.Endo G, Silver S. J Bacteriol. 1995;177:4437–4441. doi: 10.1128/jb.177.15.4437-4441.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun Y, Wong M D, Rosen B P. J Biol Chem. 2001;276:14955–14960. doi: 10.1074/jbc.M010595200. [DOI] [PubMed] [Google Scholar]
- 11.Busenlehner L S, Weng T C, Penner-Hahn J E, Giedroc D P. J Mol Biol. 2002;319:685–701. doi: 10.1016/S0022-2836(02)00299-1. [DOI] [PubMed] [Google Scholar]
- 12.Thelwell C, Robinson N J, Turner-Cavet J S. Proc Natl Acad Sci USA. 1998;95:10728–10733. doi: 10.1073/pnas.95.18.10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh V K, Xiong A, Usgaard T R, Chakrabarti S, Deora R, Misra T K, Jayaswal R K. Mol Microbiol. 1999;33:200–207. doi: 10.1046/j.1365-2958.1999.01466.x. [DOI] [PubMed] [Google Scholar]
- 14.Kuroda M, Hayashi H, Ohta T. Microbiol Immunol. 1999;43:115–125. doi: 10.1111/j.1348-0421.1999.tb02382.x. [DOI] [PubMed] [Google Scholar]
- 15.Cavet J S, Meng W, Pennella M A, Appelhoff R J, Giedroc D P, Robinson N J. J Biol Chem. 2002;277:38441–38448. doi: 10.1074/jbc.M207677200. [DOI] [PubMed] [Google Scholar]
- 16.VanZile M L, Cosper N J, Scott R A, Giedroc D P. Biochemistry. 2000;39:11818–11829. doi: 10.1021/bi001140o. [DOI] [PubMed] [Google Scholar]
- 17.VanZile M L, Chen X, Giedroc D P. Biochemistry. 2002;41:9776–9786. doi: 10.1021/bi020178t. [DOI] [PubMed] [Google Scholar]
- 18.VanZile M L, Chen X, Giedroc D P. Biochemistry. 2002;41:9765–9775. doi: 10.1021/bi0201771. [DOI] [PubMed] [Google Scholar]
- 19.Busenlehner L S, Cosper N J, Scott R A, Rosen B P, Wong M D, Giedroc D P. Biochemistry. 2001;40:4426–4436. doi: 10.1021/bi010006g. [DOI] [PubMed] [Google Scholar]
- 20.Busenlehner L S, Apuy J L, Giedroc D P. J Biol Inorg Chem. 2002;7:551–559. doi: 10.1007/s00775-001-0336-9. [DOI] [PubMed] [Google Scholar]
- 21.Pace C N, Vajdos F, Fee L, Grimsley G, Gray T. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laue T, Shaw B D, Ridgeway T M, Pelletier S L. In: Analytical Ultracentrifugation in Biochemistry and Polymer Science. Harding S E, Rowe A J, Horton J C, editors. Cambridge, U.K.: R. Soc. Chem.; 1991. pp. 90–125. [Google Scholar]
- 23.Kuzmic P. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 24.Scott R A. Methods Enzymol. 1985;117:414–459. doi: 10.1016/s0076-6879(85)17026-6. [DOI] [PubMed] [Google Scholar]
- 25.Poiarkova A V, Rehr J J. Phys Rev B. 1999;59:948–957. [Google Scholar]
- 26.Ankudinov A, Ravel B, Rehr J J, Conradson S D. Phys Rev B. 1998;58:7565–7576. [Google Scholar]
- 27.Horrocks W D, Jr, Ishley J N, Holmquist B, Thompson J S. J Inorg Biochem. 1980;12:131–141. doi: 10.1016/s0162-0134(00)80124-5. [DOI] [PubMed] [Google Scholar]
- 28.Bear C A, Duggen K A, Freeman H C. Acta Crystallogr B. 1975;31:2713–2715. [Google Scholar]
- 29.Konopelski J P, Reimann C W, Hubbard C R, Mighell A D, Santoro A. Acta Crystallogr B. 1976;32:2911–2913. [Google Scholar]
- 30.Yamaguchi K, Cosper N J, Stalhandske C, Scott R A, Pearson M A, Karplus P A, Hausinger R P. J Biol Inorg Chem. 1999;4:468–477. doi: 10.1007/s007750050333. [DOI] [PubMed] [Google Scholar]
- 31.Corwin D T, Jr, Fikar R, Koch S A. Inorg Chem. 1987;26:3079–3080. [Google Scholar]
- 32.Turner J S, Glands P D, Samson A C R, Robinson N J. Nucleic Acids Res. 1996;19:3714–3721. doi: 10.1093/nar/24.19.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong M D, Lin Y-F, Rosen B P. J Biol Chem. 2002;277:40930–40936. doi: 10.1074/jbc.M206536200. [DOI] [PubMed] [Google Scholar]
- 34.Cook W J, Kar S R, Taylor K B, Hall L M. J Mol Biol. 1998;275:337–347. doi: 10.1006/jmbi.1997.1443. [DOI] [PubMed] [Google Scholar]
- 35.Ferreira G C, Franco R, Mangravita A, George G N. Biochemistry. 2002;41:4809–4818. doi: 10.1021/bi015814m. [DOI] [PubMed] [Google Scholar]
- 36.Thompson J D, Gibson T J, Plewniak F, Jeanmougin F, Higgins D G. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}