

Abstract
Gas-phase photoelectron spectroscopy and density functional theory have been used to investigate the interactions between the sulfur π-orbitals of arene dithiolates and high-valent transition metals as minimum molecular models of the active site features of pyranopterin Mo/W enzymes. The compounds (Tp*)MoO(bdt) (compound 1), Cp2Mo(bdt) (compound 2), and Cp2Ti(bdt) (compound 3) [where Tp* is hydrotris(3,5-dimethyl-1-pyrazolyl)borate, bdt is 1,2-benzenedithiolate, and Cp is η5- cyclopentadienyl] provide access to three different electronic configurations of the metal, formally d1, d2, and d0, respectively. The gas-phase photoelectron spectra show that ionizations from occupied metal and sulfur based valence orbitals are more clearly observed in compounds 2 and 3 than in compound 1. The observed ionization energies and characters compare very well with those calculated by density functional theory. A “dithiolate-folding-effect” involving an interaction of the metal in-plane and sulfur-π orbitals is proposed to be a factor in the electron transfer reactions that regenerate the active sites of molybdenum and tungsten enzymes.
Coordination by the sulfur atoms of one or two ene-1,2-dithiolate (dithiolene) ligands of the novel substituted pyranopterin-dithiolate (“molybdopterin”; ref. 1) is a common structural feature of mononuclear molybdenum-containing enzymes (2–4). These enzymes catalyze a wide range of oxidation/reduction reactions in carbon, sulfur, and nitrogen metabolism. Fig. 1 shows the structure of the active site of sulfite oxidase, a representative example (5, 6) of the coordination of the pyranopterin-dithiolate (hereafter abbreviated S2pdt; ref. 7). These structural results raise fundamental questions about the role of the S2pdt coordination in the overall catalytic cycle of molybdenum enzymes (8). The unusual ability of ene-1,2-dithiolate ligands to stabilize metals in multiple oxidation states has been recognized since the compounds were first investigated (9). Proposed roles for the S2pdt ligand include functioning as an electron transfer conduit from the metal to other prosthetic groups (10) and as a modulator of the oxidation/reduction potential of the metal site (10). During catalysis, the metal center is proposed to pass through the M(VI/V/IV) oxidation states, i.e., the Mo d electron count changes from d0 to d1 to d2. Thus, studies of discrete metal dithiolate complexes encompassing these and related electron configurations may provide insight concerning metal thiolate bonding and reactivity in enzymes.
Figure 1.

The active site of chicken liver sulfite oxidase illustrating a pyranopterin dithiolate appended to the Mo center (5).
Previous structural studies of model molybdenum complexes of the type (Tp*)MoE(1,2-dithiolate) [where E is O or NO, Tp* is hydrotris(3,5-dimethyl-1-pyrazolyl)borate, and the 1,2-dithiolates are bdt (1,2-benzenedithiolate), bdtCl2 (3,6-dichlorobenzenedithiolate), and qdt (2,3-quinoxalinedithiolate)] have shown that the fold angle of the dithiolate metallacycle along the S⋅⋅⋅S vector (Fig. 2) varies in a way that depends on the occupation of a d orbital that is in the equatorial plane (11). For (Tp*)MoO(bdt), which has a formal 4d1 electron configuration, the fold angle is 21.3° (12, 13). However, for (Tp*)Mo(NO)(bdt), the fold angle is 41.1° (11). Although the metal center of (Tp*)Mo(NO)(bdt) is formally d4, the strong π-acceptor character of the NO ligand results in the d orbital in the equatorial plane being empty, whereas for (Tp*)MoO(bdt) this d orbital is half-filled (11). The larger fold angle for (Tp*)Mo(NO)(bdt) has been ascribed to a stabilizing interaction of the filled pπ orbitals on sulfur with the empty metal orbital in the equatorial plane on bending (11), which is illustrated schematically in Fig. 2. For other reported (Tp*)Mo(NO)(dithiolate) compounds, the fold angle is always 40–45° and is essentially independent of the electron-donating or electron-withdrawing nature of substituents on the dithiolate ligand (11). In contrast, for (Tp*)MoO(dithiolate) complexes the fold angle ranges from 6.9° in (Tp*)MoO(bdtCl2) (14) to 29.5° in (Tp*)MoO(qdt) (11), suggesting a much shallower potential surface for the interaction of the filled sulfur-pπ (Sπ) orbitals with the half-filled equatorial metal orbital (metal in-plane or Mip). For comparison, the calculated fold angles for dithiolates in protein structures (Table 1) range from 7–30° (5, 15–17).
Figure 2.

The bonding interaction of the symmetric combination (S ) of sulfur out-of-plane orbitals with the metal in-plane d orbital on folding of the dithiolate along the S⋅⋅⋅S vector indicated by the line.
) of sulfur out-of-plane orbitals with the metal in-plane d orbital on folding of the dithiolate along the S⋅⋅⋅S vector indicated by the line.
Table 1.
Fold angles measured for the dithiolate unit in structurally characterized representative pyranopterin Mo enzymes
The (Tp*)MoO(dithiolate) compounds have also been investigated by a variety of spectroscopic techniques, including EPR (12, 13), electronic absorption, resonance Raman, and magnetic CD spectroscopies (10, 18, 19). The general conclusion from these studies is that the singly occupied molecular orbital in these formally d1 centers has substantial sulfur character (10, 18, 19). From these studies it was proposed that pseudo-σ interactions between the in-plane metal orbital and sulfur in-plane lone pairs play an important role in electron transfer reactions that regenerate the active sites of enzymes (10). However, Fig. 2 suggests that sulfur out-of-plane orbitals could also have a major effect on these processes. To date, the role of fold angle on metal–sulfur interactions has not been quantitatively assessed.
A technique capable of assessing the metal and ligand character of orbitals is gas-phase photoelectron spectroscopy using ionization sources with different photon energies (20). Previously we have reported photoelectron spectra for (Tp*)MoE(tdt) (where E = O, S, or NO, and tdt is 3,4- toluenedithiolate) by using HeI, HeII and NeI photon sources that indicate strong mixing between Mo and S orbitals (21). Interestingly, the first ionization energies of (Tp*)Mo(NO)(tdt), a formally d4 metal center, and (Tp*)MoO(tdt), a formally d1 metal center, differ by only 0.05 eV, despite the variation in axial ligand and total number of metal d electrons. This similarity in ionization energies was ascribed to the “electronic buffer effect” of the dithiolate ligand (21). Subsequent photoelectron spectroscopic studies of other pairs of (Tp*)MoE(dithiolate) complexes (E = O, NO) have shown that the first ionization energies of the members of each pair are within ≈0.2 eV of one another. For a series of (Tp*)MoO(dithiolate) complexes with varying peripheral substitutions on the dithiolate ligand, the first ionization energies show a range of ≈0.7 eV and correlate with the lowest energy oxidation potential in solution (11, 22). Gas-phase photoelectron spectroscopy avoids possible complications of solvent and solid state effects, but a major drawback is that only neutral species are easily studied by this technique, and the (Tp*)MoO(dithiolate) system does not allow access to the important d0 and d2 electron configurations of the catalytic cycle. Additional analysis of the molybdenum-dithiolate interaction in the photoelectron spectra of (Tp*)MoO(dithiolate) is also difficult because the broad area of ionizations from the Tp* ligand is not well separated from the region containing Mo 4d and S 3p ionizations (11, 14, 21, 23). A third potential complication of interpretation of photoelectron spectra of (Tp*)MoO(dithiolate) complexes is the formation of both singlet and triplet states from ionization of such a d1 system, though we have seen no indication that this is a problem (24). Thus, we have further sought a simple system that can provide experimental gas-phase photoelectron data for formally d0 and d2 metal dithiolate complexes and which is amenable to density functional theory calculations with a minimum of simplifying assumptions.
The known bent-metallocene dithiolate compounds [Cp2M(dithiolate), M = Ti, Mo and Cp is η5-cyclopentadienyl] provide access to d0 (Ti) and d2 (Mo) electron configurations (25, 26). A general bonding description of Cp2MX2 compounds is well understood (27–29), and Lauher and Hoffmann (27) first explained that the variation in fold angle for Cp2M(dithiolate) compounds is caused by the occupancy of the metal d orbital in the equatorial plane (Mip) with respect to the dithiolate ligand. This orbital is empty for the folded d0 (Ti) case and filled for the more nearly planar d2 (Mo) case. For the (Tp*)MoO(dithiolate) molecules that we have previously studied there is also a metal-d orbital in the equatorial plane with respect to the dithiolate ligand that is, in this case, half-occupied for the d1 [Mo(V)] electron configuration. Fourmigué and coworkers (26, 30, 31) have studied Cp2M(dithiolate) and [CpM(dithiolate)2]−1,0 in the context of novel molecular materials. They have found that the d0 compounds, Cp2Ti(dithiolate), have large fold angles (e.g., 46.0° for Cp2Ti(bdt); ref. 26) in the solid state. The barrier to interconversion between the positive and negative extremes of fold angle in solution has been determined by NMR to be 14 kcal⋅mol−1 (25, 30), close to the computed value of 15 kcal⋅mol−1 (31). The observed folding for the Ti d0 systems could facilitate interaction of the filled Sπ orbitals with the empty Mip orbital, similar to the diagram shown in Fig. 2. In contrast, for the d2 metal system, Cp2Mo(bdt), the fold angle in the solid state is 9.0° (32). Fourmigué et al. (33) have found that the oxidation potentials of Cp2M(dithiolate) compounds are not significantly different for M = Mo and W, suggesting that the highest occupied molecular orbital (HOMO) has substantial sulfur character. Pilato et al. (34) have prepared Cp2Mo(dithiolate) compounds in which the dithiolate contains an appended pterin group as mimics for pyranopterin-dithiolate centers. Molecular orbital calculations and EPR data for [Cp2Mo(dithiolate)]+ cations indicate that the HOMO is significantly ligand based (33–38). A study of (η5-tBuC5H4)2Zr(Se2C6H4) (39) demonstrated that these systems are amenable to gas-phase photoelectron spectroscopy. To our knowledge, however, there has been no detailed investigation of the gas-phase photoelectron spectra of Cp2Mo(dithiolate) compounds to experimentally probe the molybdenum and sulfur character in the occupied valence orbitals of these systems to assess whether the valence molecular orbitals are primarily metal, primarily sulfur, or strongly mixed. Here we present gas-phase PES data for (Tp*)MoO(bdt) (1) and Cp2M(bdt) [M = Mo (2), Ti (3)]. The analysis of the experimental data are aided by density functional theory calculations on the neutral ground state of the molecules 1-3, and the ion states formed by photoionization.
Materials and Methods
Synthesis of Compounds.
The compounds (Tp*)MoO(bdt) (12), Cp2Mo(bdt) (32), and Cp2Ti(bdt) (40, 41) were synthesized according to published procedures and under anaerobic conditions by using either an inert atmosphere glove bag or standard Schlenk line techniques. The modified synthesis of Cp2Mo(bdt) involved a 1:3 mixture of water and organic solvent to give better yield of the final product. Electronic absorption (1,2-dichloroethane solutions on a modified Cary 14 with OLIS interface, 250–900 nm), EPR (frozen glasses at 77 K, in dry degassed toluene, at ≈9.1 GHz with a Bruker ESP 300) and infrared (KBr disks on a Nicolet Avatar ESP Fourier transform infrared, 4,000–400 cm−1) spectroscopies and mass spectrometry (FAB ionization in a 3-nitrobenzyl alcohol matrix on a JEOL HX110) were used to identify the compounds.
Photoelectron Spectroscopy.
Photoelectron spectra were recorded by using an instrument, procedures and calibration that have been described in more detail (21). During data collection, the instrument resolution (measured by using full-width half maximum of the argon 2P3/2 peak) was 0.020–0.030 eV. The sublimation temperatures were (10−4 Torr, monitored by using a “K” type thermocouple passed through a vacuum feedthrough and attached directly to the sample cell) 195–205°C for compound 1, 190–200°C for compound 2, and 175–185°C for compound 3.
In the figures the vertical length of each data mark represents the experimental variance of that point. The valence ionization bands are represented analytically with the best fit of asymmetric Gaussian peaks (42). The number of peaks used in a fit was based solely on the features of a given band profile. The peak positions are reproducible to about ±0.02 eV (≈3σ). The parameters describing an individual Gaussian peak are less certain when two or more peaks are close in energy and overlap.
Theoretical Methods.
The Amsterdam density functional theory package (ADF 2000.01) was used to study the electronic structures of the compounds 1–3 (43–47). The optimized geometry of 1 (Table 3, which is published as supporting information on the PNAS web site, www.pnas.org) was obtained beginning from the crystal structure geometry (Table 4, which is published as supporting information on the PNAS web site) (13), and optimized geometries of 2 and 3 (Tables 5 and 6, which are published as supporting information on the PNAS web site) were obtained beginning from geometric coordinates of a biscyclopentadienyl metal dithiolate system with a ligand fold angle of 1.2° and in C1 symmetry. The 3,5-dimethyl groups on Tp* were replaced by hydrogen atoms (Tp ligand) to simplify the computations. A generalized gradient approximation, with the exchange correction of Becke (48) and the correlation correction of Lee et al. (49), was used for all density functional calculations. Core levels (up to 3d for Mo, up to 1s for C, N, O, and S) were treated as frozen orbitals. No core levels for Ti were treated as frozen. The calculations used triple-ζ basis sets with Slater type orbitals and a polarization function for all elements besides Mo. Calculations on the ground-state molecules were performed in the spin-restricted mode. Spin-unrestricted calculations were performed on the relevant ion states at the fixed geometry of the neutral molecule (Table 7, which is published as supporting information on the PNAS web site). The difference between the total self-consistent field (SCF) energy of the ion and the total SCF energy of the neutral ground state is the ΔSCF estimate of the ionization energy. A linear correction was applied for comparison of the calculated and the observed energies, i.e., calculated ΔSCF energies were shifted by the difference between the experimentally obtained and the calculated first ionization energies. Similarly, orbital energy estimates of the ionization energies were shifted by the difference between the observed first ionization energy and the negative of the calculated eigenvalue of the HOMO.
Results and Discussion
Photoelectron Spectra.
The low energy valence regions of the gas-phase photoelectron spectra of 1-3 collected with both HeI and HeII photon sources are presented in Fig. 3. This energy region contains the ionizations that correspond to removal of electrons from the first two or three occupied molecular orbitals of these molecules. For the Ti-containing molecule (3), the spectra contain two ionizations that correspond to removal of electrons from the symmetric and antisymmetric (bands S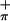 and S
and S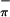 , respectively) combinations of sulfur-π orbital (Sπ) orbitals. For the Mo-containing molecules (1 and 2), an additional ionization is observed in this region that corresponds to removal of an electron from the half (1) or fully (2) occupied equatorial metal d orbital. The specific assignment of the ionizations can be made by comparison of the spectra to those of related molecules and by comparison of the relative ionization intensities as the source energy is varied (21). From previous experimental studies (20, 50) and calculations of atomic photoionization cross-sections (51), it is expected that ionizations from orbitals with significant Ti 3d or Mo 4d contributions will increase in intensity compared with ionizations of primarily S 3p character when data collected with a HeII photon source is compared with data collected with a HeI photon source. Mixing of metal and sulfur character in orbitals will result in smaller changes in relative intensities of ionizations being observed.
, respectively) combinations of sulfur-π orbital (Sπ) orbitals. For the Mo-containing molecules (1 and 2), an additional ionization is observed in this region that corresponds to removal of an electron from the half (1) or fully (2) occupied equatorial metal d orbital. The specific assignment of the ionizations can be made by comparison of the spectra to those of related molecules and by comparison of the relative ionization intensities as the source energy is varied (21). From previous experimental studies (20, 50) and calculations of atomic photoionization cross-sections (51), it is expected that ionizations from orbitals with significant Ti 3d or Mo 4d contributions will increase in intensity compared with ionizations of primarily S 3p character when data collected with a HeII photon source is compared with data collected with a HeI photon source. Mixing of metal and sulfur character in orbitals will result in smaller changes in relative intensities of ionizations being observed.
Figure 3.

Gas-phase photoelectron spectra of (Tp*)MoO(bdt) (1), Cp2Mo(bdt) (2), and Cp2Ti(bdt) (3) with HeI and HeII excitation.
For molecule 1, the lowest energy ionization band (labeled Mip in Fig. 3) at 7.04 eV is similar in appearance to the low energy band of related complexes having alkoxide ligands (23, 52) that has been assigned to ionization arising primarily from the half-filled metal-based orbital (21, 23, 52). The two ionizations between 7.25 eV to 8.00 eV can then be assigned to ionizations associated with symmetric (S ) and antisymmetric (S
) and antisymmetric (S ) combinations of Sπ orbitals based on previous assignment of analogous complexes (21, 22). More detailed assignment of the S
) combinations of Sπ orbitals based on previous assignment of analogous complexes (21, 22). More detailed assignment of the S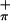 and S
and S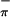 ionizations is not possible because of the significant overlap of these low-energy features and the beginning of the Tp* ligand based ionizations. The ratio of the areas of the Mip ionization and the sum of the overlapping S
ionizations is not possible because of the significant overlap of these low-energy features and the beginning of the Tp* ligand based ionizations. The ratio of the areas of the Mip ionization and the sum of the overlapping S and S
and S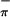 ionizations changes very little with change in photon source from HeI to HeII; the Mip/(S
ionizations changes very little with change in photon source from HeI to HeII; the Mip/(S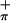 + S
+ S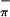 ) ratio in HeI is 0.66:1.00, that in HeII is 0.63:1.00. The observation of only small changes in the relative intensities of the first three bands with ionization source energy was suggested to be evidence of substantial mixing of metal and sulfur character in (Tp*)MoO(tdt) (21).
) ratio in HeI is 0.66:1.00, that in HeII is 0.63:1.00. The observation of only small changes in the relative intensities of the first three bands with ionization source energy was suggested to be evidence of substantial mixing of metal and sulfur character in (Tp*)MoO(tdt) (21).
The photoelectron spectra of the Ti molecule (3) show two overlapping ionization bands at 7.18 eV and 7.43 eV that must contain contributions from the S and S
and S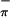 ionizations. The ratio of the areas of the low-energy band to the high-energy band increases significantly with change in photon source from HeI to HeII. The ratio in HeI is 1.00:1.02; that in HeII is 1.00:0.37. This behavior is consistent with the lower energy peak containing contributions from Ti. Therefore, the lower energy peak is assigned to S
ionizations. The ratio of the areas of the low-energy band to the high-energy band increases significantly with change in photon source from HeI to HeII. The ratio in HeI is 1.00:1.02; that in HeII is 1.00:0.37. This behavior is consistent with the lower energy peak containing contributions from Ti. Therefore, the lower energy peak is assigned to S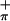 , which has the proper symmetry to interact with the empty metal orbital on folding. The higher energy peak in 3 is assigned to the S
, which has the proper symmetry to interact with the empty metal orbital on folding. The higher energy peak in 3 is assigned to the S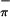 ionization, which does not mix significantly with the empty Ti d acceptor orbital (discussed later in the calculations section). The symmetric combination of the sulfur orbitals (S
ionization, which does not mix significantly with the empty Ti d acceptor orbital (discussed later in the calculations section). The symmetric combination of the sulfur orbitals (S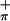 ) is more destabilized than the antisymmetric combination (S
) is more destabilized than the antisymmetric combination (S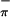 ) because of the greater interaction with the arene ring π orbital of appropriate symmetry (39). This analysis compares well with that of (η5-tBuC5H4)2Zr(Se2C6H4) (39), for which the first band was assigned as a combination of Se
) because of the greater interaction with the arene ring π orbital of appropriate symmetry (39). This analysis compares well with that of (η5-tBuC5H4)2Zr(Se2C6H4) (39), for which the first band was assigned as a combination of Se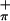 and Se
and Se ionizations.
ionizations.
Compound 2 shows three low-energy ionizations, one at 6.29 eV and an overlapping pair of peaks at 7.04 eV and 7.24 eV. The HeII data for 2 clearly show an increase in the relative intensity of the middle band, and perhaps a slight decrease in the relative intensity of the highest energy band. These results imply that the middle band (7.04 eV) is mainly metal-based, the lowest energy band (6.29 eV) is primarily S with some additional Mo 4d or C 2p character, and the highest energy band (7.24 eV) is S
with some additional Mo 4d or C 2p character, and the highest energy band (7.24 eV) is S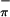 . The ratio of the areas of the S
. The ratio of the areas of the S /Mip/S
/Mip/S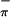 ionizations in HeI photon source is 1.00:1.03:0.82; that in HeII is 1.00:1.39:0.67. The energy of the metal based ionization (Mip) in 1 and 2 stays constant (at 7.04 eV) despite significant perturbations in the first coordination sphere and formal oxidation state of the metal.
ionizations in HeI photon source is 1.00:1.03:0.82; that in HeII is 1.00:1.39:0.67. The energy of the metal based ionization (Mip) in 1 and 2 stays constant (at 7.04 eV) despite significant perturbations in the first coordination sphere and formal oxidation state of the metal.
Computational Results.
The electronic structures of 1–3 were calculated by using C1 symmetry and geometry optimized molecular structures. The geometry optimizations are carried out on isolated molecules (45), corresponding to the conditions of the PES experiment. These molecular structures compare well with the reported structures determined from x-ray crystallography (Tables 8 and 9, which are published as supporting information on the PNAS web site). As a result of geometry optimization, the dithiolate fold angle changes from the crystallographic fold angle of 21.3° (12) to 31.0° in 1, from 9.0° (32) to 4.8° in 2, and from 46.0° (53) to 41.6° in 3.
Density functional theory calculations provide additional insight into the metal–dithiolate interactions and indicate that, on dithiolate folding, the mixing of metal d and sulfur pπ orbitals can be favored by their energy and symmetry match. The energies and general characters of the ionizations observed for molecules 1-3 by photoelectron spectroscopy match those calculated by the ΔSCF method and by comparison of orbital energies (Table 2 and Table 10, which is published as supporting information on the PNAS web site). The calculated orbital energies for 2 are low in comparison to the experimental values. This appears to be a function of the fold angle. Fig. 4 shows a Walsh diagram for the variation of the orbital eigenvalues for 2 with fold angle. The S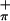 eigenvalue becomes less negative and Mip becomes more negative with increasing fold angle. The S
eigenvalue becomes less negative and Mip becomes more negative with increasing fold angle. The S eigenvalue and the onset of further ionizations remain relatively constant. The angle at which the orbital eigenvalues most closely resemble that determined by PES is ≈15°. Contour plots of the orbitals for 1-3 that correspond to the ionizations evaluated by photoelectron spectroscopy are shown in Fig. 5. The HOMO in 1 is primarily a metal in-plane orbital, whereas it is primarily a sulfur out-of-plane orbital in 2 and 3. The HOMO-1 orbital for 2 is a metal in-plane orbital. As mentioned earlier, the metal-based ionization has similar energy for 1 and 2. Thus, dithiolate appears to buffer the electron density at the metal center by affecting the overlap of Sπ orbitals with the metal center, thereby overcoming the striking differences of σ donor atoms (N and O) in 1 and π donor Cp ligands in 2. Folding of the ene-dithiolate can influence this overlap of Sπ (out-of-plane) and metal in-plane orbitals. The HOMO-2 orbital for 1 and 2 and the HOMO-1 orbital for 3 are primarily the antisymmetric sulfur (S
eigenvalue and the onset of further ionizations remain relatively constant. The angle at which the orbital eigenvalues most closely resemble that determined by PES is ≈15°. Contour plots of the orbitals for 1-3 that correspond to the ionizations evaluated by photoelectron spectroscopy are shown in Fig. 5. The HOMO in 1 is primarily a metal in-plane orbital, whereas it is primarily a sulfur out-of-plane orbital in 2 and 3. The HOMO-1 orbital for 2 is a metal in-plane orbital. As mentioned earlier, the metal-based ionization has similar energy for 1 and 2. Thus, dithiolate appears to buffer the electron density at the metal center by affecting the overlap of Sπ orbitals with the metal center, thereby overcoming the striking differences of σ donor atoms (N and O) in 1 and π donor Cp ligands in 2. Folding of the ene-dithiolate can influence this overlap of Sπ (out-of-plane) and metal in-plane orbitals. The HOMO-2 orbital for 1 and 2 and the HOMO-1 orbital for 3 are primarily the antisymmetric sulfur (S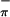 ) orbitals with little contribution from the metal. The symmetric S
) orbitals with little contribution from the metal. The symmetric S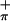 orbital has the right symmetry and energy to match the metal in-plane orbital on folding of the dithiolate unit, and the substantial mixing of the out-of-plane S
orbital has the right symmetry and energy to match the metal in-plane orbital on folding of the dithiolate unit, and the substantial mixing of the out-of-plane S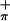 orbitals and metal in-plane orbital on folding is shown experimentally by the intensity changes observed in the HeI and HeII spectra of 2 and 3 (Fig. 3).
orbitals and metal in-plane orbital on folding is shown experimentally by the intensity changes observed in the HeI and HeII spectra of 2 and 3 (Fig. 3).
Table 2.
Experimental and calculated ΔSCF and orbital ionization energies (eV) for the ionizations from sulfur antisymmetric (S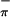 ), sulfur symmetric (S
), sulfur symmetric (S ), and metal in-plane (Mip) orbitals of 1–3
), and metal in-plane (Mip) orbitals of 1–3
| Compound method | (Tp*)MoO(bdt) (1)
|
Cp2Mo(bdt) (2)
|
Cp2Ti(bdt) (3)
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| PES (assignment) | ΔSCF† | Orbital energy† | PES (assignment) | ΔSCF | Orbital energy | PES (assignment) | ΔSCF | Orbital energy | |
| First ionization | 7.04 (Mip) | 7.04 (6.51) | 7.04 (4.35) | 6.29 (S ) ) |
6.29 (6.46) | 6.29 (3.96) | 7.18 (S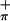 ) ) |
7.18 (6.67) | 7.18 (4.58) |
| Second ionization | 7.54 (S ) ) |
S 7.49 T 7.20 | 7.60 | 7.04 (Mip) | 6.75 | 6.58 | 7.43 (S ) ) |
7.37 | 7.36 |
| Third ionization | 7.71 (S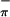 ) ) |
S 7.53 T 7.47‡ | 7.85 | 7.24 (S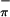 ) ) |
6.94 | 6.93 | — | — | — |
| Onset of further ionizations | 8.23 | Did not converge | 8.52 | 8.32 | 8.07 | 7.99 | 8.19 | 8.12 | 8.16 |
PES, experimental vertical ionization; orbital energy, negative of the orbital eigenvalue; S, singlet; T, triplet; SOMO, smallest occupied molecular orbit. ΔSCF ionization energies and orbital energies have been shifted so that first ΔSCF ionization energy and highest occupied orbital energy agree with the experimental first ionization energy. Values in parentheses are unshifted values of the HOMO/SOMO for ΔSCF, and the negative of the orbital eigenvalue of the HOMO/SOMO for orbital energy.
The methyl groups of the Tp* ligand were replaced with H atoms to simplify calculations.
SCF converged to three decimal places.
Figure 4.

Graph of orbital eigenvalues against fold angle for 2. The geometry optimized coordinates of the Cp2MoS2 core of 2 were kept fixed, and the benzene group was rotated relative to the MS2 plane while keeping the bond lengths constant.
Figure 5.

Calculated frontier orbitals (HOMO, HOMO-1, and HOMO-2) of 1–3. All contours have the same cutoff level (0.05).
Conclusions
The gas-phase photoelectron spectra of Cp2M(bdt), M = Ti (3) and Mo (2) clearly show that, under favorable conditions, the ionizations from primarily metal-based orbitals and primarily Sπ-based orbitals can be experimentally distinguished from one another. In addition, we have unambiguously demonstrated that the symmetric S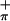 orbital, which can interact with the in-plane metal orbital on bending, is more easily ionized than the antisymmetric S
orbital, which can interact with the in-plane metal orbital on bending, is more easily ionized than the antisymmetric S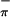 orbital. These results experimentally verify the bonding model originally put forth by Lauher and Hoffmann (27) for bent-metallocene dithiolate compounds such as 2 and 3. Our analysis and assignment of the low-energy ionizations of compounds 2 and 3 also provide a framework for investigation and direct assignment of the Sπ and metal-based ionizations in related metal dithiolate systems that may be more strongly mixed than 2 and 3 or that may have less favorable separation of their metal-sulfur region from the ionizations of other ligands.
orbital. These results experimentally verify the bonding model originally put forth by Lauher and Hoffmann (27) for bent-metallocene dithiolate compounds such as 2 and 3. Our analysis and assignment of the low-energy ionizations of compounds 2 and 3 also provide a framework for investigation and direct assignment of the Sπ and metal-based ionizations in related metal dithiolate systems that may be more strongly mixed than 2 and 3 or that may have less favorable separation of their metal-sulfur region from the ionizations of other ligands.
The variation in the fold angle of the dithiolate ligand reflects the electron occupation in the equatorial in-plane metal orbital, as has been noted (11, 27). Folding of the dithiolate ligand enables the Sπ electron density to modulate the electron density in the equatorial plane of the metal, and thus folding can play a very significant role in the metal–sulfur anisotropic bonding interaction. In the case of 3, which has formally a Ti(IV) d0 metal center, the dithiolate ligand can be thought of as a six-electron donor. Each of the thiolate σ-orbitals provides two electrons, and two additional electrons come from the S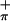 orbital. Hence, folding the dithiolate ligand effectively stabilizes 3 as an 18-electron complex. This “dithiolate folding effect” is in contrast to the nitrosyl group where the transformation from linear to bent coordination of the ligand transfers a pair of electrons from the metal to a localized orbital on the ligand (54).
orbital. Hence, folding the dithiolate ligand effectively stabilizes 3 as an 18-electron complex. This “dithiolate folding effect” is in contrast to the nitrosyl group where the transformation from linear to bent coordination of the ligand transfers a pair of electrons from the metal to a localized orbital on the ligand (54).
Implications for Enzymes.
For molybdenum and tungsten enzymes the “dithiolate-folding-effect” of the S2pdt ligand may be important in stabilizing the multiple oxidation states that result from electron transfer or oxygen atom transfer (OAT). For example, in sulfite oxidase (Fig. 1), reduction of the metal in-plane orbital due to the release of an equatorial oxygen atom during OAT should make the dithiolene unit of S2pdt more planar compared with the fully oxidized enzyme. In this regard, we note that the calculated dithiolate fold angles for the two chemically equivalent, but crystallographically distinct, molybdenum centers in sulfite oxidase of 6.6° and 7.0° (Table 1) are smaller than that for the oxidized center of aldehyde oxidoreductase (16.6°), whose oxidized structure has been determined to high resolution (1.28 Å) (15). The exact oxidation state of sulfite oxidase in the crystal structure is not known, but there is evidence for photoreduction of the molybdenum center in the synchrotron beam during data collection (5, 55). The fold angle studies on model systems support the view that the reported structure of sulfite oxidase (5) is of a reduced form of the enzyme. Dithiolate folding can also modify the electropositive nature of the metal center by varying the overlap of sulfur lone pairs with the metal in-plane orbital. The Sπ orbitals of dithiolate can therefore serve as an essential instrument for the buffering of electron density at the metal center by either involving themselves in strong mixing with an empty metal orbital, as shown here for the d0 Ti complex (3), or by localization of electron density on the Sπ orbitals in the presence of a filled metal orbital, as shown here for the d2 Mo complex (2). Dithiolate folding may also provide a mechanism for the sulfur-π orbitals of the S2pdt ligand to act as an effective electron transfer pathway from the in-plane orbitals on the metal center to other redox partners via the pyranopterin. Finally, we note that for molybdenum and tungsten enzymes the energies involved in substrate binding, docking with electron transfer partners, and dynamic motions of the protein skeleton, may modulate the dithiolate fold angle and, consequently, the electron distribution, overall reduction potential and reactivity of the metal center.
Supporting Information
Geometry optimized and crystal structure coordinates for 1–3, selected computational input, and results are available in Tables 3–11, which are published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
Support by National Institutes of Health Grant GM-37773 (to J.H.E.) and National Science Foundation Grant CHE-9618900 (to D.L.L.) is gratefully acknowledged.
Abbreviations
- HOMO
highest occupied molecular orbital
- SCF
self-consistent field
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates for 2 have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom (CSD reference no. 201539).
References
- 1.Rajagopalan K, Johnson J L. J Biol Chem. 1992;267:10199–10202. [PubMed] [Google Scholar]
- 2.Hille R. Chem Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 3.Sigel A, Sigel H. Molybdenum and Tungsten: Their Roles in Biological Processes, Metals Ions in Biological Systems. Vol. 39. New York: Dekker; 2002. [Google Scholar]
- 4.Garner D C, Banham R, Cooper S J, Davies E S, Stewart L J. In: Handbook on Metalloproteins. Bertini I, Sigel A, Sigel H, editors. New York: Dekker; 2001. pp. 1023–1090. [Google Scholar]
- 5.Kisker C, Schindelin H, Pacheco A, Wehbi W, Garrett R M, Rajagopalan K V, Enemark J H, Rees D C. Cell. 1997;91:973–983. doi: 10.1016/s0092-8674(00)80488-2. [DOI] [PubMed] [Google Scholar]
- 6.Kisker C. In: Handbook of Metalloproteins. Messerschmidt A, Huber R, Weighardt K, Poulos T, editors. Vol. 2. New York: Wiley; 2001. pp. 1121–1135. [Google Scholar]
- 7.Enemark J H, Cosper M M. In: Molybdenum and Tungsten: Their Roles in Biological Processes, Metal Ions in Biological Systems. Sigel A, Sigel H, editors. Vol. 39. New York: Dekker; 2002. pp. 621–654. [PubMed] [Google Scholar]
- 8.Schindelin H, Kisker C, Rees D. J Biol Inorg Chem. 1997;2:773–781. [Google Scholar]
- 9.Eisenberg R. Prog Inorg Chem. 1970;12:295–369. [Google Scholar]
- 10.Inscore F E, McNaughton R, Westcott B, Helton M E, Jones R, Dhawan I K, Enemark J H, Kirk M L. Inorg Chem. 1999;38:1401–1410. [Google Scholar]
- 11.Joshi H K, Inscore F E, Schirlin J T, Dhawan I K, Carducci M D, Bill T G, Enemark J H. Inorg Chim Acta. 2002;337:275–286. [Google Scholar]
- 12.Dhawan I K, Pacheco A, Enemark J H. J Am Chem Soc. 1994;116:7911–7912. [Google Scholar]
- 13.Dhawan I K, Enemark J H. Inorg Chem. 1996;35:4873–4882. doi: 10.1021/ic9605276. [DOI] [PubMed] [Google Scholar]
- 14.Inscore F E, Joshi H K, McElhaney A E, Enemark J H. Inorg Chim Acta. 2002;331:246–256. [Google Scholar]
- 15.Rebelo J, Dias J, Huber R, Moura J, Romão M. J Biol Inorg Chem. 2001;6:791–800. doi: 10.1007/s007750100255. [DOI] [PubMed] [Google Scholar]
- 16.Enroth C, Eger B T, Okamoto K, Nishino T, Nishino T, Pai E F. Proc Natl Acad Sci USA. 2000;97:10723–10728. doi: 10.1073/pnas.97.20.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H K, Temple C, Rajagopalan K, Schindelin H. J Am Chem Soc. 2000;122:7673–7680. [Google Scholar]
- 18.Carducci M D, Brown C, Solomon E I, Enemark J H. J Am Chem Soc. 1994;116:11856–11868. [Google Scholar]
- 19.McMaster J, Carducci M D, Yang Y-S, Solomon E I, Enemark J H. Inorg Chem. 2001;40:687–702. doi: 10.1021/ic0005846. [DOI] [PubMed] [Google Scholar]
- 20.Green J C. Acc Chem Res. 1994;27:131–137. [Google Scholar]
- 21.Westcott B L, Gruhn N E, Enemark J H. J Am Chem Soc. 1998;120:3382–3386. [Google Scholar]
- 22.Helton M E, Gruhn N E, McNaughton R L, Kirk M L. Inorg Chem. 2000;39:2273–2278. doi: 10.1021/ic9912878. [DOI] [PubMed] [Google Scholar]
- 23.Westcott B L, Enemark J H. Inorg Chem. 1997;36:5404–5405. [Google Scholar]
- 24.Wang X-B, Inscore F E, Yang X, Cooney J J A, Enemark J H, Wang L-S. J Am Chem Soc. 2002;124:10182–10191. doi: 10.1021/ja0265557. [DOI] [PubMed] [Google Scholar]
- 25.Guyon F, Lenoir C, Fourmigué M, Larsen J, Amaudrut J. Bull Soc Chim Fr. 1994;131:217–226. [Google Scholar]
- 26.Fourmigué M. Coord Chem Rev. 1998;178–180:823–864. [Google Scholar]
- 27.Lauher J W, Hoffmann R. J Am Chem Soc. 1976;98:1729–1742. [Google Scholar]
- 28.Green J C. Struct Bonding (Berlin) 1981;43:37–112. [Google Scholar]
- 29.Green J C. Chem Soc Rev. 1998;27:263–272. [Google Scholar]
- 30.Guyon F, Fourmigué M, Audebert P, Amaudrut J. Inorg Chim Acta. 1995;239:117–124. [Google Scholar]
- 31.Domercq B, Coulon C, Fourmigué M. Inorg Chem. 2001;40:371–378. doi: 10.1021/ic000550y. [DOI] [PubMed] [Google Scholar]
- 32.Kutoglu A, Köpf H. J Organomet Chem. 1970;25:455–460. [Google Scholar]
- 33.Fourmigué M, Domercq B, Jourdain I V, Molinié P, Guyon F, Amaudrut J. Chem Eur J. 1998;4:1714–1723. [Google Scholar]
- 34.Pilato R S, Eriksen K, Greaney M, Gea Y, Taylor E, Goswami S, Kilpatrick L, Spiro T, Rheingold A, Stiefel E I. Am Chem Soc Symp Ser. 1993;535:83–97. [Google Scholar]
- 35.Fourmigué M, Lenoir C, Coulon C, Guyon F, Amaudrut J. Inorg Chem. 1995;34:4979–4985. [Google Scholar]
- 36.Kaiwar S P, Hsu J K, Vodacek A, Yap G, Liable-Sands L M, Rheingold A L, Pilato R S. Inorg Chem. 1997;36:2406–2412. doi: 10.1021/ic961428v. [DOI] [PubMed] [Google Scholar]
- 37.Fomitchev D V, Lim B S, Holm R. Inorg Chem. 2001;40:645–654. doi: 10.1021/ic001046w. [DOI] [PubMed] [Google Scholar]
- 38.Domercq B, Coulon C, Feneyrou P, Dentan V, Robin P, Fourmigué M. Adv Funct Mater. 2002;12:359–366. [Google Scholar]
- 39.Guimon C, Pfister-Guillouzo G, Meunier P, Gautheron B, Tainturier G, Pouly S. J Organomet Chem. 1985;284:299–312. [Google Scholar]
- 40.Klapötke T, Köpf H. Z Anorg Allg Chem. 1988;558:217–222. [Google Scholar]
- 41.Lowe N D, Garner C D. J Chem Soc Dalton Trans. 1993;14:2197–2207. [Google Scholar]
- 42.Lichtenberger D L, Copenhaver A S. J Electron Spectrosc Rel Phenom. 1990;50:335–352. [Google Scholar]
- 43.Baerends E J, Ellis D E, Ros P. Chem Phys. 1973;2:41–51. [Google Scholar]
- 44.Fonseca Guerra C, Snijders J G, te Velde G, Baerends E J. Theor Chem Acc. 1998;99:391–399. [Google Scholar]
- 45.Te Velde G, Bickelhaupt F M, Baerends E J, Fonseca Guerra C, Van Gisbergen S J A, Snijders J G, Ziegler T. J Comput Chem. 2001;22:931–967. [Google Scholar]
- 46.te Velde G, Baerends E J. J Comput Phys. 1992;99:84–98. [Google Scholar]
- 47.Versluis L, Ziegler T. J Chem Phys. 1988;88:322–329. [Google Scholar]
- 48.Becke A D. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 49.Lee C, Yang W, Parr R G. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 50.Glass R S, Gruhn N E, Lichtenberger D L, Lorance E, Pollard J R, Birringer M, Block E, DeOrazio R, He C, Shan Z, Zhang X. J Am Chem Soc. 2000;122:5065–5074. [Google Scholar]
- 51.Yeh J, Lindau I. At Data Nucl Data Tables. 1985;32:1–155. [Google Scholar]
- 52.Chang C S J, Rai-Chaudhuri A, Lichtenberger D L, Enemark J H. Polyhedron. 1990;9:1965–1973. [Google Scholar]
- 53.Kutoglu A. Z Anorg Allg Chem. 1972;390:195–209. [Google Scholar]
- 54.Enemark J H, Feltham R D. Coord Chem Rev. 1974;13:339–406. [Google Scholar]
- 55.George G N, Pickering I J, Kisker C. Inorg Chem. 1999;38:2539–2540. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.