

Abstract
WT and leucine → valine distal pocket mutants of nitrophorin 2 (NP2) and their NO complexes have been investigated by spectroelectrochemistry. NO complexes of two of the mutants exhibit more positive reduction potential shifts than does the WT protein, thus indicating stabilization of the Fe(II)–NO state. This more positive reduction potential for NP2-L132V and the double mutant is consistent with the hypothesis that smaller valine residues may allow the heme to regain planarity instead of being significantly ruffled, as in WT NP2. Thus, ruffling may stabilize the Fe(III)–NO state, which is required for facile NO dissociation. NMR spectroscopic investigations show that the sterically demanding 2-methylimidazole ligand readily binds to all three distal pocket mutants to create low-spin Fe(III) complexes having axial ligands in nearly perpendicular planes; it also binds to the WT protein in the presence of higher concentrations of 2-methylimidazole, but yields a different ligand plane orientation than is present in any of the three distal pocket mutants. NOESY spectra of NP2–ImH mutants exhibit chemical exchange cross peaks, whereas WT NP2–ImH shows no chemical exchange. Chemical exchange in the case of the distal leucine → valine mutants is caused by ImH ligand orientational dynamics. The two angular orientations of the ImH ligand could be determined from the 1H chemical shifts of the heme methyls, and the rate of interconversion of the two forms could be estimated from the NOESY diagonal and cross peak intensities. Keq is 100 or larger and favors an orientation similar to that found for the WT NP2–ImH complex.
The nitrophorins (NPs) are a group of NO-carrying heme proteins found in the saliva of bloodsucking insects. One such insect is Rhodnius prolixus (the “kissing bug”), which in its adult phase has four salivary NPs (1) and at least two additional ones discovered recently (2) at earlier stages of insect development. These interesting heme proteins sequester NO, produced by an NO synthase (NOS) similar to vertebrate constitutive NOS that is present in the salivary glands (3–5), keeping it stable for long periods of time by binding it as an axial ligand to a ferriheme center (6). Upon injection into the tissues of the bug's victim, NO dissociates and diffuses through the tissues to the nearby capillaries to cause vasodilation and thereby allow more blood to be transported to the site of the wound. At the same time, histamine, whose role is to cause swelling, itching, and the beginning of the immune response, is released by mast cells and platelets of the victim. This histamine binds to the heme sites of the Rhodnius NPs, hence preventing the insect's detection for a period of time of time (7). These two properties of the NPs of R. prolixus contribute to the transmission of the protozoan Trypanasoma cruzi, the vector of Chagas' disease (8–10), to the victim via the feces of the insect, left behind at the site of the bite (1). Chagas' disease is a serious health problem in tropical Latin America, and both the insect and the disease have now reached the United States (8–10).
The Rhodnius proteins, which have been named NP 1–4 in order of their abundances in the insect saliva, have been investigated by a number of spectroscopic techniques (1, 11, 12), spectroelectrochemistry (1, 12, 13), and stopped-flow kinetics (13, 14), and the solid-state structures of one or more ligand complexes of NP1 (12, 15), NP2 (16), and NP4 (17–19) have been determined by x-ray crystallography. The structures are unique for heme proteins, in that the heme is located at the open end of a β-barrel (20), rather than in the more commonly observed largely α-helical globin or four-helix bundle folds. The ferriheme prosthetic group is bound to the protein via a histidine ligand, leaving the sixth coordination site available to bind NO or other ligands.
Of the four Rhodnius NPs, NP2 is especially important, in that in addition to its NO-releasing and histamine-binding roles, it is the only one of the four proteins that has anticoagulant activity. NP2 acts in this role by inhibiting the intrinsic factor Xase complex, and this activity is independent of the presence of heme (22, 23).
Crystal structures of the Rhodnius NPs have shown clearly that the two leucines in the distal heme cavity, Leu-123 and Leu-133 for NP1 and NP4, Leu-122 and Leu-132 for NP2 and NP3, are in contact with the heme (12, 15–19) and may in fact contribute to ruffling of the porphyrinate ring (19), which could affect the FeIII/FeII reduction potential and thus the reactivity of the ferriheme center toward NO (19). Placement of these and another key distal pocket residue, Ile-120, in the distal pocket of NP2 is shown in two views in Fig. 1; Leu-122 is above the β-pyrrole carbon to which the 6-propionate group of the heme is attached and is near the γ-meso carbon, whereas Leu-132 is placed over the β-pyrrole carbon to which the 2-vinyl group is attached and is near the α-meso carbon. These two meso-carbons are clearly below the mean plane of the heme, which is thus significantly ruffled.
Figure 1.

Two views of the distal heme pocket residues L132, L122 (dark gray), and Ile-120 (light gray).
Normally, ferriheme–nitrosyl complexes are unstable with respect to reduction to Fe(II)–NO in the presence of excess NO (24), but the NPs must maintain the ferric-NO state to remain capable of NO transport and efficient release. This is because the Kd of ferriheme–NO complexes are in the micromolar range whereas those of Fe(II)–NO complexes are in the pico- to femtomolar range (13). How is stabilization of Fe(III)–NO accomplished? Although carboxylate groups near the heme are one factor that helps to stabilize the Fe(III) state (12, 25), another may be heme ruffling (19). It has been argued that for low-spin ferrihemes ruffling of the heme should disfavor reduction from Fe(III) to Fe(II), and hence ruffled hemes have more negative reduction potentials than planar hemes (19, 26, 27). This is probably because low-spin Fe(II) strongly disfavors a ruffled heme core (26, 28). Thus, it appears that provision of a ruffled heme at least aids in maintaining the iron in the ferric state and in the case of the NP–NO complexes may help prevent autoreduction of the Fe(III)–NO complex. As mentioned above, steric contacts with the two leucine residues in the distal pocket appear to force the heme to be ruffled (19).
The two distal leucines occur just at the end of the G and at the beginning of the H β-strands of each of the proteins (14–19), which places them on either side of the NO and histamine binding sites; Leu-122 is fairly near the surface of the protein, whereas Leu-132 is buried in the heme pocket. It is clear that Leu-122 and Leu-132 control to some degree the ligand binding properties of the NPs, for we found in preliminary investigations that we could only bind the bulky 2-methylimidazole (2MeImH) ligand to WT NP2 at extremely high concentrations of the ligand. We have thus investigated the role of the distal residue side chains, including those of Leu-122 and Leu-132 of NP2, by preparing the single and double Leu → Val mutants and studying the ligand-binding and redox properties of these mutant proteins. We find that mutation of Leu-132 shifts the heme-NO reduction potential more positive and hence destabilizes the desired Fe(III)–NO with respect to the Fe(II)–NO oxidation state. We also find that changing only one of these leucines to a valine is enough to increase the ability of the heme to bind more sterically demanding axial ligands (such as 2MeImH), and that chemical exchange cross peaks are then observed in the NOESY spectra of some axial ligand complexes of these NP2 mutants. As will be shown below, this chemical exchange arises from the creation of sufficient space to allow a second possible orientation of the planar ligand.
Experimental Procedures
Protein Sample Preparation.
Cloning and expression plasmids used were those as reported (16), with some optimization of codons; expression in Escherichia coli and purification of WT and mutant NP2 apoprotein were carried out similarly to the procedures discussed (16). Leu → Val site-directed mutant genes were prepared according to standard genetic engineering methods and expressed as reported (16). WT and mutant NP2 proteins were stored in lyophilized form at −80°C until use.
Spectroelectrochemical Measurements.
Methods used for spectroelectrochemical measurements have been detailed (12, 13). Reduction potentials of NP2 and its NO complex at pH 5.5 and 7.5 have been reported (13). For the NP2 distal pocket mutants, protein solutions (≈0.5 mM) for electrochemical studies were prepared as described in ≈0.1 M phosphate buffer at pH 5.5 and 7.5 by using 1 mM methyl viologen (Aldrich) and 0.2 mM Ru(NH3)6Cl3 (Alfa Products, Ward Hill, MA) as electrochemical mediators (12, 13).
NMR Sample Preparation.
NMR samples consisted of 3- to 6-mM solutions of each protein in D2O containing 30 mM phosphate buffer at pH 7.0 (uncorrected for the deuterium isotope effect). To obtain the low-spin complexes, the high-spin NP2 WT or mutant proteins were titrated with ImH or 2MeImH until the proton NMR signals in the 70–19 ppm region disappeared. Concomitantly, these signals were replaced by signals in the 22–10 (ImH complex) or 19–10 ppm region (2MeImH complex). [In the case of the WT protein, a very high concentration of 2MeImH (≈1 M) was required to produce the low-spin complex with reasonably sharp signals.] The samples used for saturation transfer studies at 35°C were prepared by titrating in 2MeImH until the signals in the 19–10 ppm region had sufficient intensity to be clearly observed, whereas the majority of the sample still exhibited the high-spin Fe(III) peaks. The percent of low-spin complex in these samples was in the range of 20–30%.
NMR Data Collection.
NMR data were collected over the temperature range 10–37°C with the chemical shift referenced to residual water. NOESY and heteronuclear multiple quantum coherence (HMQC) spectra were obtained on a Bruker DRX-500 spectrometer operating at 500.03-MHz proton Larmor frequency. The 1H-13C HMQC experiments were recorded by using the 5-mm inverse-detection probe with decoupling during the acquisition. A recycle time of 200 ms and refocusing time of 2.5 ms (29) were used. The water-eliminated Fourier transform-(WEFT)-NOESY experiments used 160-ms relaxation delay and 160-ms recovery delay. The mixing times for the NOESY experiments were 5–30 ms. All 2D spectra were collected with 1,024 or 2,048 data points in t2 and with 256–512 blocks in t1 with 400–800 scans per block. The rate constants were estimated by using the expressions derived from the modified Bloch equations, which use the diagonal and cross peak intensities measured from NOESY experiments (30).
Electron Paramagnetic Resonance (EPR) Data Collection.
EPR spectra were obtained on frozen protein solutions at 4.2 K on a Bruker ESP-300E EPR spectrometer operating at X-band by using 0.2-mW microwave power, 100-kHz modulation frequency, and 2- to 4-G modulation amplitude. A Systron-Donner (Sylmar, CA) frequency counter was used to measure the microwave frequency.
Results and Discussion
Effect of Distal Pocket Residue Mutation on the Reduction Potential of NP2.
Table 1 shows the reduction potentials of the L122V, L132V, and double mutant of NP2 in the absence of added ligand and the presence of NO, as compared with those of the WT protein (13). For the high-spin aqua complexes at pH 7.5, in the absence of added ligand, the reduction potentials of the L132V single and the L122V,L132V double mutant are more negative than for the WT protein, whereas the L122V single mutant has a less negative reduction potential than WT NP2. The pH dependence of the reduction potentials is quite interesting. Whereas the WT and L122V mutants have a relatively small pH dependence over the range of 5.5–7.5 (−23 and −21 mV, respectively), as observed for all four WT NPs (12, 13), the L132V mutant has a −65-mV dependence, and the L122,132V double mutant has a −140-mV change in potential over the same pH range. In fact, between pH 5.5 and 6.5 the potential of the double mutant changes by −114 mV, indicative of a proton-coupled reduction reaction with loss of two protons over this single pH unit. The structures of these mutant proteins have not yet been determined, and thus we cannot at this time define the source of the two-proton dependence of the reduction potential between pH 5.5 and 6.5. Nevertheless, at the pH relevant to NO loss in the tissues (≈7.5), the reduction potential of the high-spin L132V single and L122,132V double mutants are the most negative of the series of proteins studied (Table 1), 60 and 63 mV more negative than that of WT NP2. A more negative reduction potential means that the Fe(III) oxidation state is more strongly stabilized over that of Fe(II), thus making the protein more difficult to reduce.
Table 1.
Reduction potentials of NP2 distal pocket mutants as compared to the WT protein (13)
| Protein ligand | Reduction potential, mV vs. SHE (Slope, mV)
|
Ec − E°, mV | |
|---|---|---|---|
| H2O (no added ligand) | NO | ||
| NP2 WT (13) | |||
| pH 5.5 | −287 (59) | +49 (60) | +336 |
| pH 7.5 | −310 (62) | +8 (61) | +318 |
| NP2 L122V | |||
| pH 5.5 | −258 ± 2 (59 ± 4) | +64 ± 2 (59 ± 2) | +322 |
| pH 7.5 | −279 ± 2 (62 ± 2) | +25 ± 2 (62 ± 2) | +304 |
| NP2 L132V | |||
| pH 5.5 | −295 ± 2 (61 ± 3) | +56 ± 2 (58 ± 2) | +351 |
| pH 7.5 | −360 ± 2 (60 ± 2) | +46 ± 2 (57 ± 2) | +406 |
| NP2 L122V, L132V | |||
| pH 5.5 | −223 ± 3 (59 ± 2) | +112 ± 2 (59 ± 2) | +335 |
| pH 6.5 | −337 ± 2 (59 ± 2) | +77 ± 2 (59 ± 2) | +414 |
| pH 7.5 | −363 ± 2 (61 ± 3) | +45 ± 1 (63 ± 1) | +408 |
The Nernst equation defines the difference between the reduction potential of the protein in the absence and presence of a ligand in terms of the ratio of the formation constants of the ligand complexes KIII/KII for the two oxidation states involved in the redox couple,
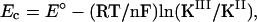 |
1 |
where Ec and E° are the reduction potentials in the presence and absence of ligand, respectively (12, 13). This method has been used to analyze the reduction potentials presented in Table 1 as differences between the potential of the aqua complex and that of the added ligand complex. For the NO complexes at pH 7.5, the potential changes Ec − E° become more positive in the order L122V < WT < L132V = L122V,L132V, meaning that the Fe(II)–NO form is stabilized over the Fe(III)–NO form to a greater degree in that order, 90 mV more stabilized for the L132V single and L122,132V double mutants, more so than is apparent from the Fe(III)–NO reduction potentials alone. A 90-mV more positive potential change equates to a factor of 32 decrease in the ratio of the equilibrium constants.
Removal of one of the methylene groups of L132 or both of the leucines by conversion of the residue(s) to valine likely allows the heme to become more planar, and that greater planarity destabilizes the Fe(III)–NO form relative to Fe(II)–NO by a factor of 32. Yet we find that the presence of Leu-132 in the distal pocket stabilizes the desired Fe(III)–NO oxidation state, whereas Leu-122 has practically no effect. The fact that Leu-132 is buried whereas Leu-122 is near the surface of the protein argues against these potential changes being caused by greater exposure of the heme in the mutants to the aqueous medium, as found in some cases (31–33). Nevertheless, the reason for the pH dependence of the reduction potential of the aqua complexes of the L132V and double mutant proteins must be determined in future work.
NMR Spectroscopy of Distal Pocket Mutants of NP2.
The NPs are stable as ferriheme proteins, and thus the odd-electron molecule NO, when bound to the Fe(III) heme center, produces a diamagnetic complex. In contrast, the NO-free forms of the NPs are high-spin (S = 5/2) paramagnetic Fe(III) complexes (1). Binding of even-electron donor ligands such as imidazoles to the NO-free forms of these proteins produces low-spin Fe(III) (S = 1/2) complexes (34). Unpaired electron(s) on the metal of both S = 5/2 and S = 1/2 Fe(III) proteins act as “beacons” that “illuminate” the protons in the vicinity of the metal, by causing shifts (called hyperfine, isotropic, or paramagnetic shifts) of the resonances from those observed in a diamagnetic protein. These shifts can allow much to be learned about the intimate details of the iron binding center (35, 36), including the orientation of planar axial ligands (37, 38).
Early in this investigation it was found that of the four similar-structured proteins NPs 1–4, NP2 provides by far the simplest NMR spectra (1). Hence, we recently reported the assignment of all of the heme resonances of NP2 in its low-spin N-methylimidazole and ImH complexes, as well as nearly complete assignment of the heme resonances of this protein in its aqua complex, the high-spin form (34). The same was not immediately possible for the other three NPs, because each of them shows significant amounts of both isomers that result from the two possible orientations of the unsymmetrical protohemin molecule in the protein pocket. Protohemin can insert itself randomly into the chiral protein pocket in one of two ways that interchange the positions of the vinyl and methyl groups at the 1,4 and 2,3 β-pyrrole positions, hence providing double the number of unique heme substituent resonances for the chiral heme protein (39–43). In most cases, the two rotational isomers are not of equal stability, so that over a period of hours, the heme “turns itself over” to adopt the preferred equilibrium orientation ratio. In fact, it is much more likely that the hemin leaves and reenters the heme binding cavity in random orientation many times until it approaches the equilibrium ratio of the two orientations for that particular protein, than that it is able to turn itself over within the heme cavity. Thus, hemin binding to a b-type heme protein is a constantly dynamic process, even though the concentration of free hemin is too small to be detected. There is probably little functional significance to this heme rotational isomerism, which results in only very small differences in reduction potentials of the two cytochrome b5 heme rotational isomers (39), for example (however, see ref. 43), but its presence can certainly complicate the interpretation of the NMR spectral data for heme b-containing proteins.
Proton NMR Spectra of the High-Spin (S = 5/2) (NO-Free) Forms of Leu → Val Distal Pocket Mutants of NP2.
The hyperfine-shifted part of the NMR spectra of the high-spin form of WT NP2 and its Leu → Val distal pocket mutants is shown in Fig. 2, where it can be seen that unique resonances, most of which have intensities corresponding to one or three protons, are observed that are similar in general appearance to those of previously studied high-spin Fe(III) proteins (36), especially horseradish peroxidase (36, 44, 45) and cytochrome c′ (36, 46–48). The assigned heme methyl and propionate resonances of NP2 (34) and its distal pocket mutants are labeled. Assignment of the spectra of the Leu → Val mutant proteins was accomplished as before (34) via saturation transfer difference spectroscopy, in this case from the high-spin form of each protein to its 2MeImH complex. The heme methyl assignments are presented in Table 2. The heme in NP2 and its three distal pocket mutants is clearly bound to His-57. The heme thus experiences different contact and dipolar shifts at different heme methyl and other substituent positions because of the orientation of the proximal histidine ImH plane (34, 37, 38). In fact, based on our previous finding (34, 37) of the relationship between the pattern of heme methyl resonances and the orientation of the proximal histidine imidazole plane, the exact overlap of the 8 and 5 M resonances in the WT protein defines the angle as exactly 135° (34), 136° for the L132V mutant, 137° for the L122V mutant, and 138–139° for the L122V,L132V double mutant, by using the NII-Fe-NIV axis of the heme to define the angle. Whether this small apparent rotation of the proximal His-57 imidazole plane with distal residue mutation results from a long-range interaction between distal and proximal residues or from a slight change in the “seating” of the hemin in the mutant protein pockets cannot be unambiguously determined without solving the 3D structures of the mutants, although it seems most likely that a slight change in the “seating” of the hemin may be involved.
Figure 2.

Downfield region of the hyperfine-shifted proton resonances of the NMR spectra of the high-spin form of NP2 and its Leu → Val distal pocket mutants. (A) NP2 WT. (B) L132V mutant. (C) L122V mutant. (D) L122V,L132V double mutant. Data were recorded at 25°C.
Table 2.
Heme methyl shifts (ppm, referenced to HDO peak) of high-spin WT and distal pocket mutants, recorded at pH 7.0, 15°C
| WT | L132V | L122V | L122,132V | |
|---|---|---|---|---|
| 1M | 59.2 | 60.0 | 58.8 | 59 |
| 3M | 50.1 | 52.8 | 50.3 | 53.2 |
| 5M | 64.3 | 67.1 | 63.4 | 65.8 |
| 8M | 64.3 | 67.7 | 64.6 | 67.6 |
| Order | 8=5>1>3 | 8>5>1>3 | 8>5>1>3 | 8>5>1>3 |
| Average shift | 59.5 | 61.9 | 59.3 | 61.4 |
| Spread | 14.2 | 14.9 | 14.3 | 14.4 |
| Angle, ° | 135 | 136 | 137 | 138–139 |
Proton NMR Spectra of the Low-Spin (S = 1/2) (ImH or 2MeImH Complex) Forms of the Leu → Val Distal Pocket Mutants of NP2.
Fig. 3A shows the HMQC spectrum of the ImH complex of the L122V mutant of NP2, where it can be seen that this complex has two heme methyl resonances at 13.6 ppm, another at 12.7 ppm, and the fourth at 2.9 ppm; α- and β-propionate and β-vinyl resonances can also be identified. Fig. 3B shows the HMQC spectrum of the 2MeImH complex of the same mutant. In this case heme methyl resonances are seen at 18.4, 17.8, 15.3, and 10.0 ppm; propionate and vinyl resonances are again observed.
Figure 3.

(A) HMQC spectrum of the ImH complex of the L122V mutant of NP2, recorded at 30°C. The heme 5,3 M (1H 13.6, 13C −28.4, −34.5 ppm), 1 M (1H 12.7, 13C −22.3 ppm), 8 M (1H 2.9, 13C −7.8 ppm), and methylene 6-Hα and 7-Hα group doublets (−38.9, −26.2 ppm, respectively) are seen in the upfield carbon region. 6-Hβ and 7-Hβ are observed as doublets at 128.9 and 103.1 ppm (13C). (B) HMQC spectrum of the 2MeImH complex of the same mutant, with heme methyls assigned.
Fig. 4 shows the 1D and WEFT-NOESY spectra of the ImH and 2MeImH complexes of the L122V mutant of NP2. The strategy for assignment of the heme resonances used both WEFT-NOESY and HMQC spectra and was carried out as described (34). Methyl assignments for the two complexes of this mutant, as well as the other two Leu → Val mutants and the WT NP2 are summarized in Table 3.
Figure 4.

1D and WEFT-NOESY spectra of the ImH (A) and 2MeImH (B) complexes of the L122V mutant of NP2. (A) Spectra were recorded at 30°C, pH 7, at 500 MHz. (B) Spectra were recorded at 15°C, pH 7, also at 500 MHz. From these spectra, together with the HMQC spectra of Fig. 3, the assignment of all heme resonances could be made (heme methyl shifts given in Table 3). (A) Chemical exchange cross peaks are also seen at 23.4, 2.9 ppm (8 M), 22.7, 13.6 ppm (5 M), 19.1, 12.7 ppm (1 M) for the major ⇄ minor exchange process and are marked by *. For NP2–L122V–ImH, from the volume intensities of the diagonal and cross peaks and the mixing time used (30) (τm = 30 ms), the rate constants calculated are kf = 54 s−1 and kr = 0.5 s−1 at 30°C, with Keq = kf/kr ≈100. The order of heme methyl resonances for the second isomer (8 > 5 > 1 > 3) is reproduced if the orientation of the ImH nodal plane is ≈51°. (B) Chemical exchange cross peaks are seen at 12.6, 2.4 ppm for the free ligand/bound ligand exchange process.
Table 3.
Heme methyl shifts (ppm, referenced to HDO peak) of ImH and 2MeImH complexes of WT and distal pocket mutants of NP2, recorded at pH 7.0 in D2O/phosphate buffer
| NP2 WT
|
NP2-L132V
|
NP2-L122V
|
NP2-L122,132V
|
|||||
|---|---|---|---|---|---|---|---|---|
| ImH | 2MeImH | ImH (minor) | 2MeImH | ImH (minor) | 2MeImH | ImH (minor) | 2MeImH | |
| 1M | 11.5 | —* | 14.0 (19.1) | 18.5 | 12.7 (19.1) | 16.7 | 14.8 (18.2) | 16.3 |
| 3M | 15.6 | —* | 12.4 | 9.6 | 13.6 | 10.0 | 11.2 | 9.0 |
| 5M | 12.5 | 21.4 | 16.0 (21.9) | 18.2 | 13.6 (22.7) | 17.8 | 16.1 (19.6) | 16.5 |
| 8M | 1.1 | 21.6 | 3.9 (24.3) | 15.2 | 2.9 (23.4) | 15.3 | 6.2 (23.8) | 16.0 |
| Order | 3>5>1>8 | 8>5 | 5>1>3>8 (8>5>1>3) | 1>5>8>3 | 3=5>1>8 (8>5>1>3) | 5>1>8>3 | 5>1>3>8 (8>5>1>3) | 5>1>8>3 |
| Average shift, ppm | 10.2 | — | 11.5 | 15.4 | 10.7 | 15.0 | 12.1 | 14.5 |
| Spread, ppm | 14.5 | — | 12.1 | 8.9 | 10.7 | 7.8 | 9.9 | 7.5 |
| Angle, ° | 155 | ≈46 | 165 (51) | 27–28 | 158.5 (51) | 32 | 168–170 (56) | 35 |
ImH, recorded at 30°C. 2MeImH, recorded at 15°C. Shifts of minor chemical exchange isomer are indicated in parentheses.
Could not be assigned.
From the heme methyl assignments summarized in Table 3, we then determined the orientation of the nodal plane of the ferriheme π orbital used in spin delocalization in the ImH complex in each case by using the program heme methyl shift patterns (available at www.chem.arizona.edu/∼shokhirn/nikolai/programs/prgsciedu.html). The results are presented in Table 3. For the ImH complexes, the smallest angle (155°) is observed for WT NP2, followed by the L122V mutant (159°), followed by the L132V mutant (165°), and the largest angle being found for the double mutant (168°). As analyzed (34) it appears that the exogenous ImH ligand is more important than His-57 in determining the orientation of the nodal plane of the π orbital of the porphyrin ring that leads to the observed pattern of methyl shifts, and thus this nodal plane defines the orientation of the exogenous ligand. Thus, the dihedral angles between His-57 and exogenous ImH ligand planes are 20° for WT, 21.5° for the L122V mutant, 29° for the L132V mutant, and 30° for the double mutant. In line with this, rhombic EPR spectra (gz,y,x = 3.02, 2.26, 1.37) characteristic of approximately parallel axial ligand planes are observed for the ImH complexes of the WT and single mutant proteins (not shown).
Whereas WT NP2-ImH does not show any chemical exchange on the NMR time scale, mutation of either one or both of the leucine residues in the distal heme pocket creates a second possible orientation of the ImH ligand plane, and chemical exchange is observed in the WEFT-NOESY spectra. Such an example is observed in Fig. 4A, where the NMR resonances of the minor species of L122V NP2–ImH are not readily observed in the 1D spectrum recorded at 30°, but they are clearly detected in the WEFT-NOESY spectra by observation of chemical exchange cross peaks between the heme methyl resonance at 2.9 ppm (8 M), 13.6 ppm (5 M), and 12.7 ppm (1 M) ppm and unobserved peaks at 23.4, 22.7, and 19.1 ppm, respectively. Thus, even though the concentration of the minor form is too low (≈1% that of the major form) to allow the signals to be observed in the 1D 1H NMR spectra, the chemical shifts of three of the four methyl groups of the minor ImH orientation complex could be determined from the chemical exchange cross peaks in the NOESY spectra and are included in Table 3. The rate constants kf and kr for major ⇄ minor interconversion of L122V NP2–ImH could be estimated from the diagonal and cross peak volumes (30) and are reported in the Fig. 4 legend. For the other two mutants, chemical exchange cross peaks were also observed, but they were so weak as to preclude quantitation of the rate and equilibrium constants. Interestingly, whereas both single mutant proteins have pure normal rhombic EPR spectra, as mentioned above, the EPR spectrum of the double mutant NP2–ImH complex shows both a normal rhombic and a large gmax [g value of low-spin Fe(III) >3.2] EPR signal (3.03, 2.25, ≈1.3, and 3.29, respectively), indicative of the existence in the frozen solution of some molecules having approximately parallel and others having approximately perpendicular axial ligand planes. The intensity of the large gmax signal is larger than would have been expected, based on the ratio of the two species estimated (<1:100) at ambient temperatures, indicating that the perpendicular ligand complex of the double mutant protein increases in stability at the expense of the parallel as the temperature is decreased (although it remains the minor species).
In contrast to the major forms of the ImH complexes, the 2MeImH complexes of the mutant proteins have heme methyl shift patterns that suggest an angle of ≈27–35° for the orientation of the nodal plane of the π orbital of the porphyrin ring that is used for spin delocalization. There is no way to achieve this angle except by taking into account the fact that 2MeImH prefers to bind with ligands in perpendicular planes lying over the meso-carbons (49). It thus appears that in this case also the orientation of the nodal plane of the heme is controlled by the exogenous ligand alone, and the observed nodal plane angle of 27–35° is that of the 2MeImH ligand. Hence, taking into account the known orientation of His-57 (Table 3), a dihedral angle of 71–76° is predicted for the ligand planes of the three mutants. These dihedral angles are similar to that of a model heme complex (70°) that gives a large gmax EPR signal (50). Indeed, all three mutants give clean large gmax EPR spectra, with g = 3.43–3.47, indicative of apparent perpendicular orientation of the 2MeImH and His-57 imidazole planes (21, 49).
Summary.
In this work we have found that making the side chain of distal Leu-132 smaller by substituting it for valine in either the single or double (L122,132V) mutant creates enough space in the distal pocket to allow the Fe(II)–NO complex to be stabilized relative to Fe(III)–NO by a factor of 32 in equilibrium constant ratio, thus suggesting that ruffling of the porphyrin ring is an important factor in stabilizing the Fe(III)–NO center. These same mutations also create enough space in the distal pocket to allow the relatively small ImH ligand to take on two specific orientations that differ markedly, with the first being similar to that of the ImH ligand in the WT NP2 protein, having a small dihedral angle between the His-57 imidazole plane and that of the exogenous ImH, and the second being almost perpendicular to the His-57 imidazole plane, with dihedral angles of 85°, 84°, and 82° for the L132V, L122V, and double mutants, respectively, as minor species. At ambient temperatures, the equilibrium strongly favors the orientation having a small dihedral angle (Keq ≈100 or larger).
Acknowledgments
We thank Dr. Arnold Raitsimring for recording the EPR spectra. The financial support of National Institutes of Health Grants HL54826 and GM58727 is gratefully acknowledged.
Abbreviations
- NP
nitrophorin
- 2MeImH
2-methylimidazole
- HMQC
heteronuclear multiple quantum coherence
- WEFT
water-eliminated Fourier transform
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Walker F A, Montfort W R. In: Advances in Inorganic Chemistry. Mauk A G, Sykes A G, editors. Vol. 51. San Diego: Academic; 2001. pp. 295–358. [Google Scholar]
- 2.Moreira M F, Coelho H S L, Zingali R B, Oliveira P L, Masuda H. Insect Biochem Mol Biol. 2003;33:23–28. doi: 10.1016/s0965-1748(02)00163-7. [DOI] [PubMed] [Google Scholar]
- 3.Ribeiro J M C, Nussenzveig R H. FEBS Lett. 1993;330:165–168. doi: 10.1016/0014-5793(93)80265-v. [DOI] [PubMed] [Google Scholar]
- 4.Nussenzveig R, H, Bentley D L, Ribeiro J M C. J Exp Biol. 1995;198:1093–1098. doi: 10.1242/jeb.198.5.1093. [DOI] [PubMed] [Google Scholar]
- 5.Yuda M, Hirai M, Miura K, Matsumura H, Ando K, Chinzei Y. Eur J Biochem. 1996;242:807–812. doi: 10.1111/j.1432-1033.1996.0807r.x. [DOI] [PubMed] [Google Scholar]
- 6.Ribeiro J M C, Hazzard J M H, Nussenzveig R, Champagne D, Walker F A. Science. 1993;260:539–541. doi: 10.1126/science.8386393. [DOI] [PubMed] [Google Scholar]
- 7.Ribeiro J M C, Walker F A. J Exp Med. 1994;180:2251–2257. doi: 10.1084/jem.180.6.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neva F A. In: Cecil Textbook of Medicine. 18th Ed. Wyngaarden J B, Smith L H, editors. Philadelphia: Saunders; 1988. pp. 1865–1869. [Google Scholar]
- 9.Kirchhoff L V. In: Harrison's Principles of Internal Medicine. 12th Ed. Wilson J D, Braunwald E, Isselbacher K J, Petersdorf R G, Martin J B, Fauci A S, Root R K, editors. New York: McGraw–Hill; 1991. pp. 791–793. [Google Scholar]
- 10.Kirchhoff L V. N Engl J Med. 1993;329:639–644. doi: 10.1056/NEJM199308263290909. [DOI] [PubMed] [Google Scholar]
- 11.Maes E M, Walker F A, Montfort W R, Czernuszewicz R S. J Am Chem Soc. 2001;123:11664–11672. doi: 10.1021/ja0031927. [DOI] [PubMed] [Google Scholar]
- 12.Ding X D, Weichsel A, Andersen J F, Shokhireva T K, Balfour C, Pierik A J, Averill B A, Montfort W R, Walker F A. J Am Chem Soc. 1999;121:128–138. [Google Scholar]
- 13.Andersen J F, Ding X D, Balfour C, Champagne D E, Walker F A, Montfort W R. Biochemistry. 2000;39:10118–10131. doi: 10.1021/bi000766b. [DOI] [PubMed] [Google Scholar]
- 14.Andersen J F, Champagne D E, Weichsel A, Ribeiro J M C, Balfour C A, Dress V, Montfort W R. Biochemistry. 1997;36:4423–4428. doi: 10.1021/bi9628883. [DOI] [PubMed] [Google Scholar]
- 15.Weichsel A, Andersen J F, Champagne D E, Walker F A, Montfort W R. Nat Struct Biol. 1997;5:304–309. doi: 10.1038/nsb0498-304. [DOI] [PubMed] [Google Scholar]
- 16.Andersen J F, Montfort W R. J Biol Chem. 2000;275:30496–30503. doi: 10.1074/jbc.M002857200. [DOI] [PubMed] [Google Scholar]
- 17.Andersen J F, Weichsel A, Balfour C A, Champagne D E, Montfort W R. Structure (London) 1998;6:1315–1327. doi: 10.1016/s0969-2126(98)00131-2. [DOI] [PubMed] [Google Scholar]
- 18.Weichsel A, Andersen J F, Roberts S A, Montfort W R. Nat Struct Biol. 2000;7:551–554. doi: 10.1038/76769. [DOI] [PubMed] [Google Scholar]
- 19.Roberts S A, Weichsel A, Qiu Y, Shelnutt J A, Walker F A, Montfort W R. Biochemistry. 2001;40:11327–11337. doi: 10.1021/bi0109257. [DOI] [PubMed] [Google Scholar]
- 20.Montfort W R, Weichsel A, Andersen J F. Biochim Biophys Acta. 2000;1482:110–118. doi: 10.1016/s0167-4838(00)00165-5. [DOI] [PubMed] [Google Scholar]
- 21.Walker F A. Coord Chem Rev. 1999;185–186:471–534. [Google Scholar]
- 22.Ribeiro J M C, Schneider M, Guimaraes J A. Biochem J. 1995;308:243–249. doi: 10.1042/bj3080243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Ribeiro J M C, Guimaraes J A, Walsh P N. Biochemistry. 1998;37:10681–10690. doi: 10.1021/bi973050y. [DOI] [PubMed] [Google Scholar]
- 24.Hoshino M, Maeda M, Konishi R, Seki H, Ford P C. J Am Chem Soc. 1996;118:5702–5707. [Google Scholar]
- 25.Varadarajan R, Zewert T E, Gray H B, Boxer S G. Science. 1989;243:69–72. doi: 10.1126/science.2563171. [DOI] [PubMed] [Google Scholar]
- 26.Safo M K, Nesset M J M, Walker F A, Debrunner P G, Scheidt W R. J Am Chem Soc. 1997;119:9438–9448. [Google Scholar]
- 27.Nessett M J M, Shokhirev N V, Enemark P D, Jacobson S E, Walker F A. Inorg Chem. 1996;35:5188–5200. [Google Scholar]
- 28.Polam J R, Shokhireva T Kh, Raffii K, Simonis U, Walker F A. Inorg Chim Acta. 1997;263:109–117. [Google Scholar]
- 29.Bertini I, Luchinat C, Macinai R, Martinuzzi S, Pierattelli R, Viezzoli R S. Inorg Chim Acta. 1998;269:125–134. [Google Scholar]
- 30.Ernst R R, Bodenhausen G, Wokaun A. Principles of Nuclear Magnetic Resonance in One and Two Dimensions. Oxford, U.K.: Clarendon; 1992. [Google Scholar]
- 31.Churg A K, Warshel A. Biochemistry. 1986;25:1675–1681. doi: 10.1021/bi00355a035. [DOI] [PubMed] [Google Scholar]
- 32.Gunner M R, Honig B. Proc Natl Acad Sci USA. 1991;88:9151–9155. doi: 10.1073/pnas.88.20.9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tezcan F A, Winkler J R, Gray H B. J Am Chem Soc. 1998;120:13383–13388. [Google Scholar]
- 34.Shokhireva T Kh, Shokhirev N V, Walker F A. Biochemistry. 2003;42:679–693. doi: 10.1021/bi026765w. [DOI] [PubMed] [Google Scholar]
- 35.Walker F A. In: The Porphyrin Handbook. Kadish K M, Smith K M, Guilard R, editors. Vol. 5. San Diego: Academic; 2000. pp. 81–183. [Google Scholar]
- 36.La Mar G N, Satterlee J D, De Ropp J S. In: The Porphyrin Handbook. Kadish K M, Smith K M, Guilard R, editors. Vol. 5. San Diego: Academic; 2000. pp. 185–298. [Google Scholar]
- 37.Shokhirev N V, Walker F A. J Biol Inorg Chem. 1998;3:581–594. [Google Scholar]
- 38.Bertini I, Luchinat C, Parigi G, Walker F A. J Biol Inorg Chem. 1999;4:515–519. doi: 10.1007/s007750050337. , and correction (1999) 4, 846. [DOI] [PubMed] [Google Scholar]
- 39.Walker F A, Emrick D, Rivera J E, Hanquet B J, Buttlaire D H. J Am Chem Soc. 1988;110:6234–6240. doi: 10.1021/ja00226a045. [DOI] [PubMed] [Google Scholar]
- 40.La Mar G N, Smith K M, Gersonde K, Sick H, Overkamp M. J Biol Chem. 1980;255:66–70. [PubMed] [Google Scholar]
- 41.La Mar G N, de Ropp J S, Smith K M, Langry K C. J Biol Chem. 1981;256:237–243. [PubMed] [Google Scholar]
- 42.La Mar G N, Davis N L, Parish D W, Smith K M. J Mol Biol. 1983;168:887–896. doi: 10.1016/s0022-2836(83)80080-1. [DOI] [PubMed] [Google Scholar]
- 43.La Mar G N, Satterlee J D, De Ropp J S. In: The Porphyrin Handbook. Kadish K M, Smith K M, Guilard R, editors. Vol. 5. San Diego: Academic; 2000. pp. 218–220. [Google Scholar]
- 44.De Ropp J S, La Mar G N. J Am Chem Soc. 1991;113:4348–4350. [Google Scholar]
- 45.De Ropp J S, Mandal P, Brauer S L, La Mar G N. J Am Chem Soc. 1997;119:4732–4739. [Google Scholar]
- 46.La Mar G N, Jackson J T, Dugad L B, Cusanovich M A, Bartsch R G. J Biol Chem. 1990;265:16173–161780. [PubMed] [Google Scholar]
- 47.Bertini I, Gori G, Luchinat C, Vila A J. Biochemistry. 1993;32:776–783. doi: 10.1021/bi00054a006. [DOI] [PubMed] [Google Scholar]
- 48.Clark K, Dugad L B, Bartsch R G, Cusanovich M A, La Mar G N. J Am Chem Soc. 1996;118:4654–4664. [Google Scholar]
- 49.Walker F A, Huynh B H, Scheidt W R, Osvath S R. J Am Chem Soc. 1986;108:5288–5297. [Google Scholar]
- 50.Ogura H, Yatsunyk L, Medforth C J, Smith K M, Barkigia K M, Renner M W, Melamed D, Walker F A. J Am Chem Soc. 2001;123:6564–6578. doi: 10.1021/ja004053s. [DOI] [PubMed] [Google Scholar]