

Abstract
Superoxide reductases (SORs) contain a characteristic square-pyramidal [Fe(NHis)4(SCys)] active site that catalyzes reduction of superoxide to hydrogen peroxide in several anaerobic bacteria and archaea. Some SORs, referred to as two-iron SORs (2Fe-SORs), also contain a lower-potential [Fe(SCys)4] site that is presumed to have an electron transfer function. However, the intra- and inter-subunit distances between [Fe(SCys)4] and [Fe(NHis)4(SCys)] iron centers within the 2Fe-SOR homodimer seem too long for efficient electron transfer between these sites. The possible role of the [Fe(SCys)4] site in 2Fe-SORs was addressed in this work by examination of an engineered Desulfovibrio vulgaris 2Fe-SOR variant, C13S, in which one ligand residue of the [Fe(SCys)4] site, cysteine 13, was changed to serine. This single amino acid residue change destroyed the native [Fe(SCys)4] site with complete loss of its iron, but left the [Fe(NHis)4(SCys)] site and the protein homodimer intact. The spectroscopic, redox and superoxide reactivity properties of the [Fe(NHis)4(SCys)] site in the C13S variant were nearly indistinguishable from those of the wild-type 2Fe-SOR. Aerobic growth complementation of a superoxide dismutase (SOD)-deficient Escherichia coli strain showed that the presence of the [Fe(NHis)4(SCys)] site in C13S 2Fe-SOR was apparently sufficient to catalyze reduction of the intracellular superoxide to nonlethal levels. As is the case for the wild-type protein, C13S 2Fe-SOR did not show any detectable SOD activity, i.e., destruction of the [Fe(SCys)4] site did not unmask latent SOD activity of the [Fe(NHis)4(SCys)] site. Possible alternative roles for the [Fe(SCys)4] site in 2Fe-SORs are considered.
Keywords: oxidative stress‖2Fe-SOR‖1Fe-SOR‖pulse-radiolysis
In several obligately anaerobic bacteria and archaea a unique class of non-heme iron proteins called superoxide reductases (SORs) catalyze reaction 1,
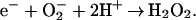 |
1 |
SORs have been shown to play an alternative and/or complementary role to that of superoxide dismutases (SODs) in oxidative stress protection (1–8). SORs have recently attracted a great deal of attention because of their novel activity and unique square-pyramidal ferrous [Fe(NHis)4(SCys)] active site (9–16). Due at least in part to its high reduction potential [≥200 mV vs. normal hydrogen electrode (NHE)], this ferrous site is remarkably stable to oxidation in air, yet it reacts with superoxide in an essentially diffusion-controlled fashion, as diagrammed in Fig. 1A (17–20). At least one transient species forms during this reaction, which has a characteristic visible absorption feature at ≈600 nm, and which has been formulated as a ferric-peroxo or ferric-hydroperoxo species. This species decays on the millisecond time scale at room temperature in a superoxide-independent process to the “resting” ferric state by loss of the peroxo ligand as H2O2. A carboxylate ligand from a conserved glutamate residue enters the coordination sphere in the resting ferric state (21). This glutamate, however, appears to play little or no role in catalysis of reaction 1, nor does its substitution by an alanine lead to reaction of superoxide with the ferric site, i.e., to SOD-type activity (19, 20, 22).
Figure 1.

(A) Scheme for the SOR reaction with superoxide. (B) Ribbon drawing of the D. desulfuricans 2Fe-SOR homodimer (PDB ID 1DFX) (24) with darker and lighter shaded subunits. Iron centers are represented as spheres. The drawing was generated by using RASMOL (25).
SORs can be subclassified into 1Fe-SORs, which contain the [Fe(NHis)4(SCys)] active site as the only cofactor, and 2Fe-SORs, which, as shown in Fig. 1B, contain an additional N-terminal polypeptide domain with a [Fe(SCys)4] site, the role of which is presumed to be electron transfer. In fact, the reduction potential of the [Fe(SCys)4] site, ≈2 mV vs. NHE (23) renders it thermodynamically capable of reducing the ferric [Fe(NHis)4(SCys)] site. The crystal structure of the 2Fe-SOR homodimer from Desulfovibrio desulfuricans (24), however, shows both intra- (≈22 Å) and intersubunit (≈32 Å) through-space distances between [Fe(SCys)4] and [Fe(NHis)4(SCys)] iron centers that seem beyond the range for efficient intraprotein electron transfer (26). Thirty-eight- and 93-residue polypeptides corresponding to the N-terminal [Fe(SCys)4]- and C-terminal [Fe(NHis)4(SCys)]-containing domains, respectively, of Desulfovibrio vulgaris 2Fe-SOR were genetically engineered and expressed separately in Escherichia coli (27). The isolated 2Fe-SOR domains largely retained their constituent iron sites with native-like absorption spectra and reduction potentials, but the recombinant 2Fe-SOR fragments, either separately or in combination, were reported to show no evidence for reaction with superoxide. These results imply that a functional 2Fe-SOR requires both domains and perhaps both constituent iron sites in a single native polypeptide. On the other hand, the fact that 1Fe-SORs are apparently fully functional with only an [Fe(NHis)4(SCys)] site (5, 28) implies that the [Fe(SCys)4] site is not essential for the superoxide reductase activity of 2Fe-SORs. In this work we address these alternative possibilities by examination of an engineered D. vulgaris 2Fe-SOR variant in which one cysteine residue of the [Fe(SCys)4] site was changed to serine. This single amino acid residue change was found to completely destroy the native [Fe(SCys)4] site, but to leave the [Fe(NHis)4(SCys)] site and the protein homodimer intact.
Materials and Methods
Reagents and General Procedures.
Reagents and buffers were the highest grade commercially available and were used as received. Enzymes were purchased from Sigma. All reagent solutions and media were prepared in water purified with a Millipore Ultrapurification system to a resistivity of ≈18 MΩ, to minimize trace metal-ion contamination. Standard procedures for molecular biology manipulations were followed (29). The correctness of plasmid gene sequences was verified by nucleotide sequencing at the University of Georgia Molecular Genetics Instrumentation Facility. Recombinant rubredoxin and 2Fe-SOR both from D. vulgaris were prepared as described (17, 20, 30). The N-terminal 2Fe-SOR, a protein fragment corresponding to residues 1–38 of D. vulgaris 2Fe-SOR, was expressed and purified by using a protocol similar to that described by Ascenso et al. (27), which is described in Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org. Except where noted, all E. coli strains harboring plasmids were cultured in media containing 100 mg/liter ampicillin.
Preparation of C13S 2Fe-SOR.
A plasmid encoding the D. vulgaris 2Fe-SOR, but replacing the codon for cysteine13 with that for serine, was generated from the plasmid, pRbo (17), by using two complimentary oligonucleotide primers containing the desired C13S codon change (underlined): 5′-CAAATGCATCCACTCTGGCAACATCGTCG-3′ and 5′-CGACGATGTTGCCAGAGTGGATGCATT-3′ and the Quik- Change mutagenesis kit (Stratagene) following procedures described in the product manual. The resulting plasmid, pC13Rbo, was transformed into E. coli strain QC774 (sodAsodB) (31). Procedures for overexpression, isolation, and purification of the C13S 2Fe-SOR variant from E. coli QC774[pC13SRbo] were similar to those previously described for isolation of the corresponding wild-type 2Fe-SOR (17, 20) and are described in the supporting information. Approximately 5 mg of pure C13S 2Fe-SOR was isolated per liter of culture. The colorless, as-isolated protein was stored at −80°C.
Metal and Protein Analyses.
Metal contents of proteins were determined by inductively coupled plasma-atomic emission at the University of Georgia Chemical Analysis Laboratory. Protein concentrations were determined by using the Bio-Rad protein assay with BSA as standard for wild-type 2Fe-SOR for 2Fe-SOR C13S and N-terminal 2Fe-SOR. Native molecular weights were determined by calibrated elution times from a HiPrep 16/60 Sephacryl S-100 column (Amersham Pharmacia). Concentrations of non-heme iron proteins were determined spectrophotometrically by using the previously reported molar absorptivities: rubredoxin, ɛ490 = 8,700 M−1⋅cm−1 (30); 2Fe-SORpink, ɛ502 = 4,300 M−1⋅cm−1 (protein monomer basis) (20); C13S 2Fe-SOR, ɛ645 = 1,900 M−1⋅cm−1 (protein monomer basis, determined in this work for the chemically oxidized protein from the iron analysis).
Oxidations of [Fe(NHis)4(SCys)] Sites.
Recombinant D. vulgaris wild-type and C13S 2Fe-SORs containing ferric [Fe(NHis)4(SCys)] sites were prepared by adding a 5-fold molar excess of potassium hexachloroiridate from a buffered stock solution. The excess oxidant was then removed by repetitive concentration/redilution in a Centricon YM5 filter (Amicon) or by passage through a 5-ml HiTrap desalting column (Amersham Pharmacia).
Reduction Potentials.
A spectrophotometric, dye-mediated, electrochemical titration method similar to those described for other SORs was used (32–34). All measurements were conducted at room temperature (≈23°C) and pH 7. Further experimental details are supplied as supporting information and in the text and Fig. 3 legend.
Figure 3.

Nernst plot of fractional 650-nm absorbance change vs. potential data (corrected to NHE) for wild-type and C13S 2Fe-SOR at pH 7 and room temperature. (See supporting information for other experimental conditions). C13S 2Fe-SOR was titrated with either sodium dithionite (filled squares) or potassium hexachloroiridate (filled circles). Open circles are analogously obtained data for the wild-type 2Fe-SOR. The solid curve is a least-squares fit of the C13S 2Fe-SOR data to the Nernst equation assuming a one-electron process with the midpoint potential cited in the text.
Growth Complementation.
The aerobic growth rates of the superoxide-deficient E. coli strain QC774 (31) harboring plasmids individually for wild-type D. vulgaris 2Fe-SOR (pRbo) (17), C13S 2Fe-SOR (pC13SRbo) (this work), N-terminal 2Fe-SOR (pRbo1–38) (see supporting information), E. coli Mn-SOD (pDT1–5) (31) or the empty control plasmid, pCYB1 (New England Biolabs) were conducted as follows. Each strain was cultured aerobically at 37°C to stationary phase in M63 minimal medium supplemented with 0.02% Casamino acids and 1 × 10−4 % vitamin B1. Eight 3-μl aliquots of each of the above cultures were used to inoculate eight 200-μl volumes of aerobic M63 medium without added supplements or antibiotics in individual wells of a 96-well plate. The cultures in the 96-well plate, which had initial OD600 values from 0.05 to 0.07, were then incubated with shaking at 37°C, and growths were monitored by periodic measurements of OD600 in a microplate reader (Molecular Devices).
Pulse Radiolysis.
The rates of reactions of as-isolated D. vulgaris wild-type 2Fe-SORpink and as-isolated C13S 2Fe-SOR with superoxide were measured at 25°C by pulse radiolysis using the 2-MeV Van de Graaff accelerator at Brookhaven National Laboratory, as described (17, 20). Additional experimental details are provided in the Fig. 5 legend. The data were analyzed by using the Brookhaven National Laboratory Pulse Radiolysis Program.
Figure 5.

Time courses for the formation (A) and decay (B) of the 600-nm absorbing transient species and spectra constructed from the absorbance transients (C) after pulse radiolysis of as-isolated C13S 2Fe-SOR in 100 μM Tris, 10 mM sodium formate, pH 7.8, at 25°C. The trace in A was obtained after pulse radiolysis of the C13S 2Fe-SOR solution (200 μM in iron sites) to generate 1.0 μM superoxide and was best fit to a first-order rate constant of 1.9 × 105 sec−1. The trace in B was measured for the same solution as for A after pulse radiolysis to generate 2.0 μM superoxide and was best fit to a first-order rate constant of 100 sec−1. The absorption spectra in C were constructed from pulse radiolysis absorbance vs. time traces analogous to those shown in A and B for a solution of C13S 2Fe-SOR (100 μM in iron sites) pulsed with 1.5–2.0 μM superoxide. Open circles, initial absorbances before the pulse; filled circles, absorbances at ≈50 μs after the pulse; filled squares, initial absorbances after the pulse by using millisecond detector; open squares, 100 msec after the pulse.
SOR Assay.
This coupled assay measures SOR-dependent consumption of NADPH by superoxide, and was conducted as described (30). Experimental conditions are given in the text and Fig. 6 legend.
Figure 6.

Dependence of NADPH/superoxide oxidoreductase activity on wild-type, C13S, and N-terminal 2Fe-SORs. Rates of NADPH consumption were monitored at room temperature in a 1-ml cuvette as decreases in absorbance at 340 nm (NADPH ɛ340 = 6,220 M−1⋅cm−1) in solutions containing (in the added order) 500 μM xanthine, 100 μM NADPH, 186 units/ml catalase, 1 μM Rub, 1 μM spinach ferredoxin:NADP+ oxidoreductase, and either wild-type or C13S 2Fe-SOR (1 μM in [Fe(NHis)4(SCys)] sites) or N-terminal 2Fe-SOR (1 μM in iron sites). After recording a “baseline” NADPH consumption rate for 50 sec, a precalibrated amount of xanthine oxidase was added to produce a flux of 12 μM superoxide per min, and NADPH consumption was monitored for several minutes. Absorbance spikes caused by the various additions and mixing are omitted, and the traces obtained with each SOR are offset vertically by an arbitrary amount for clarity.
Results and Discussion
Physical, Spectroscopic, and Redox Characterization of C13S 2Fe-SOR.
Based on amino acid sequence alignment and high homology with the structurally characterized D. desulfuricans 2Fe-SOR (24), cysteine 13 in D. vulgaris 2Fe-SOR supplies a ligand to the [Fe(SCys)4] site. The recombinant D. vulgaris wild-type 2Fe-SOR and C13S 2Fe-SOR variant, when expressed in E. coli grown in minimal medium supplemented with iron, were determined to contain 2.1 ± 0.3 and 1.2 ± 0.3 mol iron/mol protein monomer, respectively. These values indicated full occupancy of both [Fe(SCys)4] and [Fe(NHis)4(SCys)] sites for wild-type 2Fe-SOR, but only approximately one iron per C13S 2Fe-SOR monomer. Zinc and other transition metal contents were negligible (<0.05 mol metal/mol monomer) for both proteins. Gel filtration verified that both wild-type and C13S 2Fe-SOR are dimeric. Under the assay conditions used in this work, wild-type 2Fe-SOR is known to remain dimeric (35).
Fig. 2 shows the near UV-visible absorption spectra of both as-isolated C13S 2Fe-SOR, labeled C13Sclear, and hexachloroiridate-oxidized C13S 2Fe-SOR, labeled C13Sblue. An absorbance ratio, A280/A645 = 17, for this chemically oxidized form was used as a standard of protein purity. The “clear” and “blue” subscripts in Fig. 2 reflect the respective colors of the protein solutions and also correspond to ferrous and ferric oxidation states, respectively, of the [Fe(NHis)4(SCys)] site. These oxidation state assignments are based on the close similarity of the C13Sclear and C13Sblue spectra in Fig. 2 to the corresponding as-isolated and chemically oxidized 1Fe-SORs from several sources (5, 36, 37). The 645-nm absorption feature in the C13Sblue spectrum (ɛ645 = 1,900 M−1⋅cm−1) is also very similar to that of the wild-type 2Fe-SOR (17), which is labeled wild typegray-pink in Fig. 2 Inset. The wild-type 2Fe-SOR is isolated with ferric [Fe(SCys)4] and ferrous [Fe(NHis)4(SCys)] sites, a form referred to as 2Fe-SORpink (32). This form can be oxidized to 2Fe-SORgray, in which both iron sites are ferric. The wild typegray-pink difference absorption spectrum shown in the inset to Fig. 2, thus, subtracts the contribution of the ferric [Fe(SCys)4] site, and the remaining absorption, also centered at ≈645 nm, is, therefore, caused by the ferric [Fe(NHis)4(SCys)] site. On the basis of magnetic circular dichroism and resonance Raman spectroscopic studies on a 1Fe-SOR, the ≈645 nm absorption feature has been assigned to a π-type cysteine sulfur → ferric charge transfer transition (37, 38). The absorption corresponding to the shoulder at ≈320 nm in the C13Sclearspectrum has been similarly assigned as the analogous higher-energy π-type cysteine sulfur → ferrous charge transfer transition of the reduced [Fe(NHis)4(SCys)] site. The absorption corresponding to the shoulder at ≈330 nm in the C13Sblue spectrum in Fig. 2 also appears in all reported ferric 1Fe-SOR spectra and has not been definitively assigned.
Figure 2.

Absorption spectra of D. vulgaris C13S 2Fe-SOR in 50 mM 3-(N-morpholino)propanesulfonic acid, pH 7.5. Shown are spectra of as-isolated C13S 2Fe-SOR (thin black trace labeled “C13Sclear”) and hexachloroiridate-oxidized C13S 2Fe-SOR (thick black trace labeled “C13Sblue”). (Inset) The ≈645-nm absorption feature in the C13S 2Fe-SORblue spectrum (thick black trace) compared with that of the wild-type 2Fe-SORgray-pink difference absorption spectrum (dashed trace).
The stronger visible and near-UV absorptions of the ferric [Fe(SCys)4] site, with ɛ502 = 4,300 M−1⋅cm−1 and ɛ375 ≈ 8,000 M−1⋅cm−1 (27), are, thus, absent in both as-isolated and chemically oxidized C13S 2Fe-SOR.§ Engineered substitutions of a serine for a cysteine ligand residue are known to result in stable ferric [Fe(SCys)3(OSer)] sites in rubredoxins and rubrerythrin with absorption features in the visible region that are blue-shifted from those of the [Fe(SCys)4] sites (39, 40). The absorption spectra of the C13S variant, however, show no evidence of such blue-shifted features.¶ Furthermore, formation of the [Fe(SCys)3(OSer)] sites in rubredoxin and rubrerythrin required in vitro incorporation of iron, and we have been unable to incorporate iron into the isolated C13S 2Fe-SOR above the ≈1 per subunit cited above by using the methods described for these other serine-substituted proteins.
The Nernst plots in Fig. 3 were obtained by measuring 650-nm absorption intensities of wild-type or C13S 2Fe-SORs at a series of poised redox potentials. Based on these data, the [Fe(NHis)4(SCys)] site of C13S 2Fe-SOR underwent a reversible one-electron redox process with a measured midpoint reduction potential of + 285 ± 3 mV vs. NHE. This potential was very similar to that determined for the wild-type 2Fe-SOR, +272 ± 7 mV, under the same conditions. Midpoint reduction potentials of [Fe(NHis)4(SCys)] sites in SORs have been reported over the range of +90 to +430 mV vs. NHE (23, 41), depending on the technique used, but most of the reported values cluster within the range of 200–250 mV (27, 32–34, 36, 37).
The spectroscopic, analytical and redox results together show that the D. vulgaris C13S 2Fe-SOR variant contains a [Fe(NHis)4(SCys)] site as its only cofactor, and that this site in the C13S variant has very similar spectroscopic and redox properties to those in other SORs. Most notably, these characteristic properties of the [Fe(NHis)4(SCys)] site in the C13S variant are not significantly perturbed from those of the corresponding wild-type 2Fe-SOR. The fact that the C13S 2Fe-SOR remains soluble and dimeric also suggests that the polypeptide structure has not been drastically altered by the loss of the [Fe(SCys)4] site.
Complementation of E. coli (sodAsodB) by C13S 2Fe-SOR.
E. coli sodAsodB strains lack the genes encoding both MnSOD and FeSOD and are unable to grow in aerobic minimal media unless supplemented with amino acids (31). This phenotype is attributed to superoxide damage to iron–sulfur cluster-containing enzymes, some of which are part of the branched-chain amino acid pathway, and to Fenton chemistry resulting from elevated “free” iron levels in the cytoplasm (42). The E. coli genome contains no SOR homolog. Pianzzola et al. (1) first showed that plasmid-borne expression of 2Fe-SORs, including that from D. vulgaris, can restore the aerobic growth phenotype to sodAsodB strains of E. coli. This complementation was subsequently shown to be caused by lowering of intracellular superoxide levels (2), presumably by SOR's catalysis of reaction 1 using an unidentified endogenous source of electrons (4). Fig. 4 shows that plasmid-borne expression of D. vulgaris C13S 2Fe-SOR also restores aerobic growth to the E. coli sodAsodB strain QC774 in minimal medium without supplements and is indistinguishable from the behavior of the wild-type 2Fe-SOR in this regard. As reported previously for the wild-type protein, the background “leaky” expression was sufficient for complementation, i.e., no inducer of overexpression was necessary (7). A plasmid containing a gene encoding the N-terminal fragment, residues 1–38 of D. vulgaris 2Fe-SOR, did not restore the aerobic growth phenotype to strain QC774 under the same conditions. These results show that a 2Fe-SOR lacking its native [Fe(SCys)4] site is capable of lowering the superoxide levels below lethality in E. coli. Note that the spectroscopic and redox characterizations of C13S 2Fe-SOR described above were performed on protein isolated from the same sodAsodB strain, also grown in aerobic minimal medium (supplemented with iron, as described in the supporting information).
Figure 4.

Aerobic growth versus time of E. coli QC774 (sodAsodB) expressing plasmid-borne genes encoding E. coli MnSOD (pDT1–5), D. vulgaris 2Fe-SOR (pRbo), C13S 2Fe-SOR (pC13Srbo), N-terminal 2Fe-SOR (pRbo1–38), or the control plasmid, pCYB1, with no inserted gene. Freshly inoculated cultures in M63 medium were incubated with shaking at 37°C, and growths were monitored as OD600 periodically for 24 h; data points for the first 12 h are shown (with connecting lines extrapolating toward the 24-h data). Symbols are defined within the figure and represent the averages (range ± 12%) for eight duplicate cultures.
Pulse Radiolysis.
Results of pulse radiolysis experiments, shown in Fig. 5 indicate that as-isolated C13S 2Fe-SOR, which contains a predominantly ferrous [Fe(NHis)4(SCys)] site, reacts with superoxide in a manner very similar to that of the wild-type 2Fe-SORpink as outlined in Fig. 1. Fits to the kinetic traces in Fig. 5 A and B resulted in a calculated second order rate constant of 1 × 109 M−1⋅sec−1 for formation of a transient intermediate and 100 sec−1 for its decay. Fig. 5C shows a spectrum at 50 μsec after the pulse (at maximum absorbance of the transient) constructed from kinetic traces obtained at several wavelengths between 470 and 700 nm. This transient species with λmax ≈ 595 nm and ɛ595 nm = 3,700 M−1⋅cm−1 is nearly superimposable on the 100-μsec spectrum of the intermediate previously reported for the wild-type 2Fe-SORpink (17, 20). No other intermediate species were detected. The analogously constructed spectrum in Fig. 5C obtained 100 msec after the pulse (after apparently complete decay of the intermediate) has λmax ≈ 645 nm, ɛ595 = 2,700 M−1⋅cm−1, which matches well with the spectrum of the chemically oxidized C13S 2Fe-SOR shown in Fig. 2, except for a somewhat higher calculated molar absorptivity. The extinction coefficients in Fig. 5C were calculated assuming that the substoichiometric amounts of superoxide generated in each pulse were quantitatively reduced by 1:1 mol/mol reaction with ferrous [Fe(NHis)4(SCys)] sites of the 2Fe-SOR. This somewhat higher calculated intensity was also previously reported for the 100-msec spectra obtained from pulse radiolysis of wild-type 2Fe-SOR and can attributed to experimental uncertainty (20).
SOD Activity.
Ambiguities arise in interpreting apparent SOD activities of SORs when measured by using the traditional SOD assay (3), which is based on the inhibition of reduction of cytochrome c caused by scavenging of superoxide (43). However, when pulse radiolysis is used at pH 7.8, the dismutation rate of superoxide can be measured directly by the decrease in absorbance of superoxide at 260 nm (ɛ260 = 2,100 M−1⋅cm−1) (44). Any increase over the natural dismutation rate in the presence of a catalyst can be unambiguously interpreted as SOD activity. Classical SODs enhance superoxide dismutation rates by at least several thousand-fold under these conditions (17, 45, 46). However, no significant increases over the spontaneous dismutation rates were observed at two different temperatures in the presence of either wild-type or C13S 2Fe-SOR (see Fig. 8, which is published as supporting information on the PNAS web site). Destruction of the [Fe(SCys)4] site, therefore, does not unmask latent SOD activity of D. vulgaris 2Fe-SOR.
NADPH/Superoxide Oxidoreductase Activity.
An in vitro superoxide reductase assay has been reported in which NADPH reduces superoxide via the catalytic electron transport chain: NADPH → spinach ferredoxin:NADP+ oxidoreductase → rubredoxin → 2Fe-SOR → superoxide (30). In these experiments a precalibrated flux of superoxide was generated by using the xanthine/xanthine oxidase catalyzed reduction of dioxygen, and the expected stoichiometry of 1 mol NADPH oxidized:2 mol superoxide reduced for reduction to hydrogen peroxide was confirmed, after correcting for the background NADPH consumption rate in the absence of SOR. A high level of catalase activity was added to ensure that the NADPH consumption is not caused by its reduction of hydrogen peroxide. Fig. 6 shows that the enhancement of the NADPH consumption rate by C13S 2Fe-SOR in this assay is similar to that of the wild-type protein. The traces for wild-type and C13S 2Fe-SOR correspond to NADPH consumption rates of 8.3 and 7.9 μM NADPH per min, respectively. Subtracting the background (no SOR) consumption rate of 1.7 μM NADPH per min gives corrected consumption rates, 6.6 and 6.2 μM NADPH per min for wild-type and C13S 2Fe-SORs, which are approximately half of the precalibrated superoxide flux, 12 μM superoxide per min, i.e., the stoichiometry expected for reaction 1. The N-terminal 2Fe-SOR showed a background-corrected NADH consumption rate of 1.2 μM NADPH per min. No NADPH consumption above background was observed if any of the components were omitted from the reaction mixture.
The Role of the [Fe(SCys)4] Site in 2Fe-SOR.
The results described above demonstrate that the recombinant D. vulgaris C13S 2Fe-SOR variant contains approximately one iron per monomer, contains neither [Fe(SCys)4] nor [Fe(SCys)3(OSer)] sites, but retains a [Fe(NHis)4(SCys)] site with spectroscopic and redox properties that are nearly indistinguishable from those of the wild-type protein. The pulse radiolysis kinetics and SOR assays on C13S 2Fe-SOR show that destruction of the [Fe(SCys)4] site neither affects superoxide reactivity nor unmasks latent SOD activity of the [Fe(NHis)4(SCys)] site in 2Fe-SOR. The growth complementation results show that the native [Fe(SCys)4] site is not necessary for D. vulgaris 2Fe-SOR to catalyze reaction 1 at a rate sufficient to reduce superoxide to nonlethal levels in E. coli; the [Fe(NHis)4(SCys)] site in C13S 2Fe-SOR is apparently sufficient for this function even though nonnative electron donors must supply the reducing equivalents. The SOR assay described in this work used D. vulgaris rubredoxin, a small electron transfer protein, as proximal electron donor to the 2Fe-SOR. C13S 2Fe-SOR was as active as the wild-type protein in this assay, indicating that rubredoxin efficiently donates electrons directly to the [Fe(NHis)4(SCys)] site. The native source(s) of reducing equivalents for SORs' catalysis of reaction 1 is(are) currently unknown, although both genetic and biochemical evidence suggest rubredoxin as the prime candidate for proximal electron donor to D. vulgaris 2Fe-SOR (30, 47). In vitro experiments have shown that D. vulgaris rubredoxin catalyzes reduction of both the [Fe(SCys)4] and [Fe(NHis)4(SCys)] sites of D. vulgaris 2Fe-SOR, but that reduction of the latter site is at least 4-fold faster than the former (30). The results presented here also indicate that the [Fe(SCys)4] site does not “tune” the redox properties of the [Fe(NHis)4(SCys)] site in 2Fe-SOR and argues against the recent proposal that a conserved tyrosine residue participates in a superexchange electron transfer pathway between the [Fe(SCys)4] and [Fe(NHis)4(SCys)] sites (38). A role for this tyrosine in facilitating electron transfer between rubredoxin (or other exogenous electron donors) and the [Fe(NHis)4(SCys)] site of 2Fe-SOR remains a possibility.
Because the [Fe(SCys)4] site apparently does not participate in the superoxide reductase activity of 2Fe-SOR, alternative functions must be considered. The stability and full occupancy of the [Fe(SCys)4] sites in 2Fe-SORs argues against an iron uptake/delivery function. The lack of apparent interactions with the [Fe(NHis)4(SCys)] site does not favor an intramolecular redox signaling function for the [Fe(SCys)4] site. Although the isolated C13S 2Fe-SOR is somewhat less stable than the wild-type protein to long-term storage, a purely structural role for the [Fe(SCys)4] site seems inconsistent with the wild-type-like homodimer and activities of the C13S variant. The properties of a 1Fe-SOR from Treponema pallidum further argue against a structural role for the [Fe(SCys)4] site. The T. pallidum SOR contains an N-terminal domain with significant amino acid sequence homology to that of D. vulgaris 2Fe-SOR, but the T. pallidum SOR lacks three of the four cysteine residues that supply ligands to the [Fe(SCys)4] site in 2Fe-SORs and contains no metal or other cofactors in its N-terminal domain (5, 34). In fact, the N- and C-terminal domains of D. vulgaris 2Fe-SOR have even higher sequence homologies to two distinct proteins from Desulfovibrio gigas: the N-terminal domain is homologous to the small [Fe(SCys)4]-containing protein, desulforedoxin, and the C-terminal domain to a 1Fe-SOR called neelaredoxin (27, 28, 47). Notably, the D. gigas desulforedoxin and neelaredoxin have never been demonstrated to be redox partners. The possibility of an as yet undiscovered physiological redox partner for the [Fe(SCys)4] site in 2Fe-SORs cannot, however, be ruled out. Another possible role for the [Fe(SCys)4] site, for which we know of no precedent, is catalytic one-electron reduction of dioxygen to generate superoxide, which, if rapidly scavenged by the ferrous [Fe(NHis)4(SCys)] site, would circumvent the unfavorable redox potential difference between the [Fe(SCys)4] site and the O2/O couple. The 2Fe-SOR could, thus, lower not only the intracellular superoxide but also the dioxygen concentration.
couple. The 2Fe-SOR could, thus, lower not only the intracellular superoxide but also the dioxygen concentration.
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of Frank Rusnak. We thank Dr. Eric Coulter and Radu Silaghi-Dumitrescu for experimental assistance and helpful discussions. This work was supported by National Institutes of Health Grant GM 40388 (to D.M.K.). Pulse radiolysis studies were carried out at the Center for Radiation Chemical Research, Brookhaven National Laboratory (D.E.C.), which is supported under Contract DE-AC02-98CH10886 with the U.S. Department of Energy and supported by its Division of Chemical Sciences, Office of Basic Energy Sciences.
Abbreviations
- SOR
superoxide reductase
- SOD
superoxide dismutase
- 2Fe-SOR
two-iron SOR
- NHE
normal hydrogen electrode
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
The absorption spectrum of the ferric [Fe(SCys)4] site in a genetically engineered protein fragment corresponding to the N-terminal [Fe(SCys)4]-containing domain of D. vulgaris 2Fe-SOR (residues 1–38) is provided as supporting information. The metal content and spectroscopic properties of this protein, which we call N-terminal 2Fe-SOR, were found to be very similar to those previously described for the same recombinant protein fragment by Ascenso et al. (27).
During isolation and purification, a small quantity of C13S 2Fe-SOR was eluted as a separate fraction that, based on its absorption spectrum, may have contained a small portion of ferric [Fe(SCys)3(OSer)] sites. However, the visible absorption attributed to these sites bleached irreversibly after several minutes at room temperature, whereas a much more intense blue color caused by the ferric [Fe(NHis)4(SCys)] sites could be regenerated by addition of oxidants.
References
- 1.Pianzzola M J, Soubes M, Touati D. J Bacteriol. 1996;178:6736–6742. doi: 10.1128/jb.178.23.6736-6742.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liochev S I, Fridovich I. J Biol Chem. 1997;272:25573–25575. doi: 10.1074/jbc.272.41.25573. [DOI] [PubMed] [Google Scholar]
- 3.Jenney F E, Jr, Verhagen M F J M, Cui X, Adams M W W. Science. 1999;286:306–309. doi: 10.1126/science.286.5438.306. [DOI] [PubMed] [Google Scholar]
- 4.Lombard M, Fontecave M, Touati D, Niviere V. J Biol Chem. 2000;275:115–121. doi: 10.1074/jbc.275.1.115. [DOI] [PubMed] [Google Scholar]
- 5.Lombard M, Touati D, Fontecave M, Niviere V. J Biol Chem. 2000;275:27021–27026. doi: 10.1074/jbc.M004201200. [DOI] [PubMed] [Google Scholar]
- 6.Kurtz D M, Jr, Coulter E D. Inorg Chem. 2001;14:407–435. [Google Scholar]
- 7.Lumppio H L, Shenvi N V, Summers A O, Voordouw G, Kurtz D M., Jr J Bacteriol. 2001;183:101–108. doi: 10.1128/JB.183.1.101-108.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fournier M, Zhang Y, Wildschut J D, Dolla A, Voordouw J K, Schriemer D C, Voordouw G. J Bacteriol. 2003;185:71–79. doi: 10.1128/JB.185.1.71-79.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurtz D M, Jr, Coulter E D. J Biol Inorg Chem. 2002;7:653–658. doi: 10.1007/s00775-002-0360-4. [DOI] [PubMed] [Google Scholar]
- 10.Adams M W W, Jenney F E, Jr, Clay M D, Johnson M K. J Biol Inorg Chem. 2002;7:647–652. doi: 10.1007/s00775-002-0359-x. [DOI] [PubMed] [Google Scholar]
- 11.Imlay J A. J Biol Inorg Chem. 2002;7:659–663. doi: 10.1007/s00775-002-0361-3. [DOI] [PubMed] [Google Scholar]
- 12.Auchère F, Rusnak F. J Biol Inorg Chem. 2002;7:664–667. doi: 10.1007/s00775-002-0362-2. [DOI] [PubMed] [Google Scholar]
- 13.Abreu I A, Xavier A V, LeGall J, Cabelli D E, Teixeira M. J Biol Inorg Chem. 2002;7:668–674. doi: 10.1007/s00775-002-0363-1. [DOI] [PubMed] [Google Scholar]
- 14.Shearer J, Nehring J, Lovell S, Kaminsky W, Kovacs J A. Inorg Chem. 2001;40:5483–5484. doi: 10.1021/ic010221l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shearer J, Scarrow R C, Kovacs J A. J Am Chem Soc. 2002;124:11709–11717. doi: 10.1021/ja012722b. [DOI] [PubMed] [Google Scholar]
- 16.Mathe C, Mattioli T A, Horner O, Lombard M, Latour J-M, Fontecave M, Niviere V. J Am Chem Soc. 2002;124:4966–4967. doi: 10.1021/ja025707v. [DOI] [PubMed] [Google Scholar]
- 17.Coulter E D, Emerson J P, Kurtz D M, Jr, Cabelli D E. J Am Chem Soc. 2000;122:11555–11556. [Google Scholar]
- 18.Niviere V, Lombard M, Fontecave M, Houee-Levin C. FEBS Lett. 2001;497:171–173. doi: 10.1016/s0014-5793(01)02468-1. [DOI] [PubMed] [Google Scholar]
- 19.Lombard M, Houee-Levin C, Touati D, Fontecave M, Niviere V. Biochemistry. 2001;40:5032–5040. doi: 10.1021/bi0023908. [DOI] [PubMed] [Google Scholar]
- 20.Emerson J P, Coulter E D, Cabelli D E, Phillips R S, Kurtz D M., Jr Biochemistry. 2002;41:4348–4357. doi: 10.1021/bi0119159. [DOI] [PubMed] [Google Scholar]
- 21.Yeh A P, Hu Y, Jenney F E, Jr, Adams M W, Rees D C. Biochemistry. 2000;39:2499–2508. doi: 10.1021/bi992428k. [DOI] [PubMed] [Google Scholar]
- 22.Abreu I A, Saraiva L M, Soares C M, Teixeira M, Cabelli D E. J Biol Chem. 2001;276:38995–39001. doi: 10.1074/jbc.M103232200. [DOI] [PubMed] [Google Scholar]
- 23.Verhagen M F J M, Voorhorst W G, Kolkman J A, Wolbert R B, Hagen W R. FEBS Lett. 1993;336:13–18. doi: 10.1016/0014-5793(93)81599-u. [DOI] [PubMed] [Google Scholar]
- 24.Coehlo A V, Matias P, Fülop V, Thomson A, Gonzalez A, Carrondo M A. J Biol Inorg Chem. 1997;2:680–689. [Google Scholar]
- 25.Bernstein H J. Trends Biochem Sci. 2000;25:453–455. doi: 10.1016/s0968-0004(00)01606-6. [DOI] [PubMed] [Google Scholar]
- 26.Page C C, Moser C C, Chen X, Dutton P L. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 27.Ascenso C, Rusnak F, Cabrito I, Lima M J, Naylor S, Moura I, Moura J J G. J Biol Inorg Chem. 2000;5:720–729. doi: 10.1007/s007750000161. [DOI] [PubMed] [Google Scholar]
- 28.Silva G, LeGall J, Xavier A V, Teixeira M, Rodrigues-Pousada C. J Bacteriol. 2001;183:4413–4420. doi: 10.1128/JB.183.4.4413-4420.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ausubel F A, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley Interscience; 1990. [Google Scholar]
- 30.Coulter E D, Kurtz D M., Jr Arch Biochem Biophys. 2001;394:76–86. doi: 10.1006/abbi.2001.2531. [DOI] [PubMed] [Google Scholar]
- 31.Carlioz A, Touati D. EMBO J. 1986;5:623–630. doi: 10.1002/j.1460-2075.1986.tb04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tavares P, Ravi N, Moura J J G, LeGall J, Huang Y-H, Crouse B R, Johnson M K, Huynh B H, Moura I. J Biol Chem. 1994;269:10504–10510. [PubMed] [Google Scholar]
- 33.Abreu I A, Saraiva L M, Carita J, Huber H, Stetter K O, Cabelli D, Teixeira M. Mol Microbiol. 2000;38:322–334. doi: 10.1046/j.1365-2958.2000.02121.x. [DOI] [PubMed] [Google Scholar]
- 34.Jovanovic T, Ascenso C, Hazlett K R, Sikkink R, Krebs C, Litwiller R, Benson L M, Moura I, Moura J J, Radolf J D, et al. J Biol Chem. 2000;275:28439–28448. doi: 10.1074/jbc.M003314200. [DOI] [PubMed] [Google Scholar]
- 35.Apiyo D, Jones K, Guidry J, Wittung-Stafshede P. Biochemistry. 2001;40:4940–4948. doi: 10.1021/bi002653y. [DOI] [PubMed] [Google Scholar]
- 36.Chen L, Sharma P, Le Gall J, Mariano A M, Teixeira M, Xavier A. Eur J Biochem. 1994;226:613–618. doi: 10.1111/j.1432-1033.1994.tb20087.x. [DOI] [PubMed] [Google Scholar]
- 37.Clay M D, Jenney F E, Jr, Hagedoorn P L, George G N, Adams M W W, Johnson M K. J Am Chem Soc. 2002;124:788–805. doi: 10.1021/ja016889g. [DOI] [PubMed] [Google Scholar]
- 38.Clay M D, Jenney F E, Jr, Noh H J, Hagedoorn P L, Adams M W, Johnson M K. Biochemistry. 2002;41:9833–9841. doi: 10.1021/bi025833b. [DOI] [PubMed] [Google Scholar]
- 39.Xiao Z, Lavery M J, Ayhan M, Scrofani S D B, Wilce M C J, Guss J M, Tregloan P A, George G N, Wedd A G. J Am Chem Soc. 1997;120:4135–4150. [Google Scholar]
- 40.Coulter E D, Shenvi N V, Beharry Z, Smith J J, Prickril B C, Kurtz D M., Jr Inorg Chim Acta. 2000;297:231–234. [Google Scholar]
- 41.Berthomieu C, Dupeyrat F, Fontecave M, Vermeglio A, Niviere V. Biochemistry. 2002;41:10360–10368. doi: 10.1021/bi020344x. [DOI] [PubMed] [Google Scholar]
- 42.Touati D. Arch Biochem Biophys. 2000;373:1–6. doi: 10.1006/abbi.1999.1518. [DOI] [PubMed] [Google Scholar]
- 43.McCord J M, Fridovich I. J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 44.Bielski B H J, Cabelli D E, Arudi R L. J Phys Chem Ref Data. 1985;14:1041–1100. [Google Scholar]
- 45.Goto J J, Gralla E B, Valentine J S, Cabelli D E. J Biol Chem. 1998;273:30104–30109. doi: 10.1074/jbc.273.46.30104. [DOI] [PubMed] [Google Scholar]
- 46.Fisher C L, Cabelli D E, Tainer J A, Hallewell R A, Getzoff E D. Proteins Struct Funct Genet. 1994;19:24–34. doi: 10.1002/prot.340190105. [DOI] [PubMed] [Google Scholar]
- 47.Brumlik M J, Voordouw G. J Bacteriol. 1989;171:4996–5004. doi: 10.1128/jb.171.9.4996-5004.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.