

Abstract
A Zn(Cys)4 center has been found in the C-terminal region of the crystal structure of the anaerobic class III ribonucleotide reductase (RNR) from bacteriophage T4. The metal center is structurally related to the zinc ribbon motif and to rubredoxin and rubrerythrin. Mutant enzymes of the homologous RNR from Escherichia coli, in which the coordinating cysteines, conserved in almost all known class III RNR sequences, have been mutated into alanines, are shown to be inactive as the result of their inability to generate the catalytically essential glycyl radical. The possible roles of the metal center are discussed in relationship to the currently proposed reaction mechanism for generation of the glycyl radical in class III RNRs.
Ribonucleotide reductases (RNR) are critical enzymes for the de novo synthesis of DNA in all organisms (1). They catalyze the reduction of the C2′—OH bond in ribonucleotides, thereby transforming them to deoxyribonucleotides. Three classes of RNRs have been identified so far that use similar radical chemistry initiated by a diverse range of metallocofactors (2). Class III RNRs are used by facultative anaerobic bacteria such as Escherichia coli and phages such as bacteriophage T4 for their anaerobic growth (3). A class III RNR is an oxygen-sensitive homodimer α2, encoded by the nrdD gene, that contains, in its active form, a catalytically essential glycyl radical located on a conserved glycine residue of the C-terminal region of the polypeptide (Gly-681 and Gly-580 in E. coli and T4, respectively).
The crystal structure of the oxygen-stable Gly-580 → Ala mutant class III RNR (nrdD) from bacteriophage T4 has shown that the glycine site is located at the tip of a loop near the C terminus of the polypeptide, ≈5 Å away from an essential cysteine residue (Cys-290 in T4) in the substrate-binding site (4, 5). The radical is thus proposed to react with this cysteine to generate an intermediate thiyl radical that serves in turn for abstraction of the H3′ atom of the ribonucleotide substrate (3, 6). The substrate radical is then reduced to the deoxy form by three reducing equivalents provided by formate and the glycine residue, which is converted back to its radical form for another cycle. No crystal structure of the E. coli enzyme is available yet, but the sequence has 57% identity to that of the RNR from T4. Coupled with the complete conservation of all residues hitherto shown to be critical for reactivity, this similarity allows us to infer mechanistic interpretations for one of the enzymes from structural and functional studies on the other.
The activation of protein α, i.e., the introduction of the glycyl radical, is the subject of intense studies in our laboratories (7, 8). In the enzyme from E. coli, the reaction is mediated by the concerted action of three components: (i) S-adenosylmethionine (AdoMet), which is reduced and cleaved into methionine and a putative 5′-deoxyadenosyl radical (Ado°); Ado° then reacts with the glycine residue to generate the glycyl radical by selective hydrogen atom abstraction; (ii) a system for AdoMet reduction consisting of NADPH, flavodoxin oxidoreductase (fpr) and flavodoxin (fldx), which can be substituted for in vitro by strong chemical reductants such as sodium dithionite or photoreduced 5-deazaflavin; (iii) a 17.5-kDa protein called β, or “activase,” which contains an oxygen-sensitive [4Fe-4S]2+/1+ center catalyzing electron transfer from fldx to AdoMet (9, 10). The final glycyl radical is on protein α, but the machinery initiating the radical chemistry is on protein β. However, the activase has not yet been structurally characterized, and no information is available regarding communication between α and β, in particular how radicals are transferred from β to α.
After purification, protein α from E. coli is in the so-called “oxidized” form, which contains a critical disulfide bridge and is inactive. This bridge can be reduced and the protein converted into an active “reduced” form by anaerobic incubation with DTT or with the NADPH:thioredoxin reductase:thioredoxin system (11). The exact location of this disulfide bridge is still unknown and has been proposed to reside in one of two CysX2Cys sequences, one completely conserved and one almost completely conserved (Fig. 1), at the C-terminal end of protein α (11). Mutation of any of these cysteines (Cys-543, Cys-546, Cys-561, or Cys-564) to serine in RNR from bacteriophage T4 eliminated production of the glycyl radical, as indirectly shown from the absence of cleavage of the α polypeptide at the radical site during exposure to air (5). In the initial 3D structure of the enzyme from bacteriophage T4, these cysteines were shown to belong to a portion of the C-terminal region of the polypeptide, between the end of the last β-strand of the α−β barrel and the loop containing the glycyl radical residue (residues 543–570) that was disordered in the crystal (4).
Fig 1.

Sequence alignment of the C-terminal region of the NrdD polypeptide containing the four almost completely conserved cysteine residues. Totally conserved residues are shown in black boxes. The secondary structure as observed in the crystal structure of NrdD from bacteriophage T4 is shown on the top line. T = turn. Figure generated by using ESPRIPT (43). Key to sequence names: Bp_T4, bacteriophage T4; S_mut, Streptococcus mutans; S_pyo, Streptococcus pyogenes; L_lac, Lactococcus lactis; E_fae, Enterococcus faecalis; S_auC, Staphyloccocus aureus COL/NCTC; V_cho, Vibrio cholerae; H_inf, Haemophilus influenzae; S_put, Shewanella putrefaciens; Y_pes, Yersinia pestis; E_col, Escherichia coli; S_typ, Salmonella typhimurium; C_ace, Clostridium acetobutylicum; P_aby, Pyroccocus abyssi; P_hor, Pyrococcus horikoshii; M_jan, Methanococcus jannaschii; M_the, Methanobacterium thermoautotrophicum; A_eut, Alcaligenes eutropha (Ralstonia eutropha); P_aer, Pseudomonas aeruginosa; C_dip, Clostridium difficile.
In this paper we further investigate the role of the equivalent cysteine residues of the C terminus of E. coli protein α with the help of original site-directed mutants, and confirm that they are critical for glycyl radical formation (5). The E. coli system has the advantage over the bacteriophage one in that it allows pure preparations of the β activase and thus more accurate characterization of the radical generation reaction. Furthermore, by using the T4 enzyme, we present the 3D structure of the previously disordered region and show that it contains a M(Cys)4 metal center with structural homology to rubredoxin and the zinc ribbon family. We discuss the implications of this center for interactions with the activase and for the mechanism of generation of the glycyl radical.
Materials and Methods
Materials.
E. coli BL21(DE3) was from Novagen; JM109(DE3) and DH5α were from Promega. Enzymes for DNA manipulation were from Fermentas MBI, New England Biolabs. Synthetic oligonucleotides and bacterial growth medium were obtained from Invitrogen.
Site-Directed Mutagenesis.
To substitute cysteine residues in protein α with alanine, the QuikChange mutagenesis protocol (Stratagene) was used with appropriate primers (Table 1) according to the manufacturer. Mutagenesis was carried out on plasmid pRSS, carrying the nrdD gene as a template (12). Mutations were confirmed by DNA sequencing.
Table 1.
Primers for site-directed mutagenesis of the nrdD gene
| Plasmids | Primers |
|---|---|
| pRSSC644A | CCG ATT GAT GAG GCC TAC GAG |
| CTC GTA GGC CTC ATC AAT CGG | |
| pRSSC647A | C TAC GAG GCT GGC TTT AC |
| GT AAA GCC AGC CTC GTA G | |
| pRSSC662A | C TTC ACT GCC CCG AAA TG |
| CA TTT CGG GGC AGT GAA G | |
| pRSSC665A | CCG AAA GCT GGT AAC CAT GAC |
| GTC ATG GTT ACC AGC TTT CGG |
Plasmids encoding mutated protein α and corresponding primers for site-directed mutagenesis. The primers (upper) and their complements (lower) are broken into codons, and the changed bases are in bold.
Expression and Purification Methods.
Wild-type and mutant Cys-to-Ala protein α were obtained from anaerobic cultures of recombinant E. coli cells JM109(DE3) transformed with the appropriate plasmid: pRSS or pRSSC644A, pRSSC647A, pRSSC662A, pRSSC665A. A published purification procedure was used, with an additional gel filtration step using a Superdex 200 column equilibrated with 50 mM Tris⋅HCl, 0.1 M KCl (pH 8) (13). Protein β (apo and reconstituted forms) was obtained as described (14).
Assays.
Ribonucleotide reduction was routinely measured as the anaerobic reduction of CTP to dCTP as described (11). Glycyl radical formation during anaerobic incubation of a mixture of proteins α and β (50–200 μM), in the presence of 3 mM AdoMet, 10 mM DTT, 2 mM NADPH, 0.3 μg/ml fldx, 0.1 μg/ml flavodoxin reductase in 40 mM Tris⋅Cl, 40 mM KCl, 0.5 mM EDTA (pH 8), was monitored by EPR spectroscopy as described (7). AdoMet reductive cleavage was assayed from the amount of methionine formed in the above reaction mixture after a 1-h incubation (7, 8).
Protein concentration was determined according to Bradford, with BSA as a standard (15).
Spectroscopy.
The cluster of protein β (50–200 μM) was analyzed by EPR spectroscopy after anaerobic reduction with 2 mM sodium dithionite and desalting by NAP-10 chromatography inside a glove box. X-band EPR spectra were recorded on a Bruker Instruments (Billerica, MA) ESP 300D spectrometer equipped with an Oxford Instruments ESR 900 flow cryostat (4.2–300 K). Circular dichroism was performed by using a Jasco (Easton, MD) J-810 spectropolarimeter at 20°C.
Binding Experiments.
Protein α (WT or mutants; 10 nmol) was incubated inside a glove box with reconstituted protein β (3.7–4.1 Fe per monomer; 8 nmol) in 100 mM Tris⋅Cl, 50 mM KCl (pH 8.0) (1 ml). Thereafter, the light brown solutions were loaded at 0.2 ml⋅min−1 on top of a 1-ml dATP-Sepharose affinity column (16). The column was washed with 6 ml buffer in 1.5-ml fractions, and the bound proteins were released by admission of ATP (1 mM) in buffer. Aliquots (containing 2- to 3-μg proteins) of each fraction were analyzed by SDS/PAGE (15%) after Coomassie blue staining and destaining. The α and β proteins were quantitated by densitometry.
Crystal Structure Determination.
The G580A mutant of the α subunit of the anaerobic RNR from bacteriophage T4 was crystallized as described (4). A single crystal was soaked in a solution containing 4 mM dGTP, 4 mM ATP, 45% PEG 400, and 0.1 M Tris (pH 7.5) for 48 h, then flash-cooled in liquid N2. Data were collected at beam line I711 of the MAX-Lab synchrotron (Lund, Sweden), on a Mar345 image plate detector, at a temperature of 100 K. Details of the data set are presented in Table 2. A structure derived from this crystal was presented in an earlier article (17); however, the structure of the metal center has not previously been reported. Data were integrated and scaled by using HKL (18) and reduced by using CCP4 (19). Analyses of anomalous signal were done by using scala. Structures were refined by using cns (20) and refmac (21) with maximum likelihood target functions.
Table 2.
Data collection and refinement
| Data set | Synchrotron | Wavelength, Å | Diffraction range, Å | Rmerge, % | % complete | 〈m〉 | Rmodel (F), % | Rfree (F), % |
|---|---|---|---|---|---|---|---|---|
| dGTP/ATP/ formate/KCl | I711 MAX-Lab | 1.043 | 20–2.45 | 7.6 | 95 | 4.5 | 20.7 | 24.3 |
| Fe edge | BW7A Hamburg | 1.743 | 40–4.4 | 9.4/3.1* | 100 | 8.8 | — | — |
| Zn edge | I711 MAX-Lab | 1.254 | 23–3.5 | 8.6/3.5* | 99 | 6.4 | 26.9 | 30.8 |
Rmerge (I) = ∑j,k|Ij,k− 〈I〉j|/∑j,kIj,k, where Ij,k are the k individual observations of each reflection j and 〈I〉j is the value after weighted averaging. Rmodel(F) = ∑hkl∥ Fo| − |Fc∥/∑hkl|Fc|, where |Fo| and |Fc| are the observed and calculated structure factors, respectively.
Second value is the R factor for Friedel mates; 〈m〉 = multiplicity of observation.
Another protein α mutant, N311A, was crystallized and flash-cooled under essentially identical conditions. Fluorescence scans of the Fe and Zn K edges were carried out at beamline BW7A of the EMBL outstation in Hamburg, Germany, and a data set to 4.4-Å resolution was collected from this crystal at an x-ray energy slightly above the K absorption edge for Fe. A data set to 3.5-Å resolution slightly above the Zn K edge was later collected from the same crystal at station I711 of MAX-Lab. A marccd detector was used in both cases. The structure of native protein α was refined against these data to produce phases for an anomalous difference Fourier. For the Zn edge data, rigid body refinement was followed by tightly restrained atomic refinement and grouped B-factor refinement. For the Fe edge data, only rigid body refinement was done. Structure factors were calculated by using sfall, and difference Fouriers were calculated by using fft.
Structural homology searches on the Protein Data Bank were carried out by using the DALI (22) and TOP (23) servers. The coordinates of residues 538–582 were submitted to the servers.
Results
Site-Directed Mutagenesis and Purification of Mutated α Proteins from E. coli.
The different mutated plasmids were prepared from plasmid pRSS as a template by using a PCR-based method. The mutated enzymes, in which one of the four cysteines Cys-644, Cys-647, Cys-662, and Cys-665 was changed into an alanine, were overexpressed in E. coli JM109(DE3) under conditions similar to the wild type. In all cases, SDS/PAGE analysis of both whole cells and soluble extracts showed a good level of expression of the proteins, comparable to that of the wild type.
All of the mutants were purified aerobically according to standard procedures developed for the wild-type enzyme except for slight modifications described in Materials and Methods that resulted in preparations of improved purity (13). All of the mutants were comparable to the wild-type protein in terms of stability, solubility, and chromatographic behavior. Furthermore, the circular dichroism spectrum was not significantly affected by the mutations (data not shown).
Interaction of α Proteins with the β Protein.
The wild-type α protein forms a rather tight complex with protein β, the activase, and this interaction is required for the introduction of the glycyl radical into protein α (24). Whether the mutant proteins also make a complex with protein β can be probed by using two assays. The first one is based on EPR spectroscopy. As a matter of fact, both the shape and the g-tensor of the EPR spectrum of the reduced S = 1/2 [4Fe-4S]+ cluster of protein β (Fig. 2A), as well as its microwave power saturation properties, are significantly modified by binding to protein α. As shown in Fig. 2, the EPR signal, initially rhombic (g = 2.03, 1.92, 1.86; Fig. 2A), becomes axial (g = 2.02, 1.93; Fig. 2B), and the P1/2 value shifts from 0.6 mW to ≈60 μW at 10 K (data not shown). A similar effect was obtained with the Cys-to-Ala protein α variants (Fig. 2C).
Fig 2.

X-band EPR spectra of reduced protein β (90 μM), alone (A) or complexed with wild-type (B) or C665A mutant (C) protein α. Recording conditions: temperature of 10 K, microwave power of 1 mW, and modulation of 1 mT.
Complex formation can also be shown by experiments in which stoichiometric amounts of α and β are loaded onto a dATP-Sepharose affinity column. Such a column selectively binds protein α, but protein β is also retained as a consequence of tight binding to protein α. Protein α is eluted with an ATP-containing buffer, and complex formation is thus revealed from the presence of protein β in this fraction. When this experiment was carried out with solutions containing a 1:0.8 mixture of mutant α protein and wild-type protein β, elution with ATP resulted in a fraction containing both proteins, as demonstrated by SDS/PAGE (data not shown).
Enzymatic Activities of the Mutant Proteins.
The ability of each mutant to catalyze the reduction of CTP by formate in the presence of the allosteric effector ATP was assayed anaerobically after a 1-h activation in the presence of a stoichiometric amount of protein β, AdoMet, DTT, and a source of electrons (NADPH, flavodoxin reductase, fldx), as described (11). As shown in Table 3, three mutants, Cys-644 → Ala, Cys-662 → Ala, and Cys-665 → Ala, were devoid of significant enzyme activity whereas the Cys-647 → Ala mutant displayed 2% of activity with respect to the wild type. However, this value is within the error of the enzyme assay. Thus, the results confirm the importance of the four cysteines for RNR activity.
Table 3.
Biochemical properties of WT and mutant protein α
| Proteins | CTP reductase activity, nmol/min/mg* | EPR Gly radical† | Methionine, mol/mol α† |
|---|---|---|---|
| WT | 280 | + | 0.95 |
| C644A | <1 | ND | 0.03 |
| C647A | 5.5 | ND | 0.07 |
| C662A | <1 | ND | 0.04 |
| C665A | <1 | ND | 0.10 |
ND, not detectable.
Two to 4 μg of each protein was assayed for CTP reduction under standard conditions.
Under standard activation conditions and after a 1-h reaction, the same enzyme solution was used both for glycyl radical determination by EPR spectroscopy and for methionine quantification.
To obtain further insights into the function of these cysteines, the ability of the mutants to generate the glycyl radical during the activation step was investigated. Because the radical displays a characteristic doublet EPR signal at g = 2.003 − (3) (25), the reaction of each mutant with AdoMet and the NADPH/flavodoxin reductase/fldx system in the presence of DTT and the activase under anaerobic conditions was monitored by EPR spectroscopy. After 1 h of reaction, no EPR signal could be detected for any of the mutant proteins. The lack of CTP reductase activity of the mutant proteins can thus be simply explained by their inability to generate the catalytically essential glycyl radical.
An aliquot of each of the above reaction mixtures was also analyzed for its methionine content as a probe for AdoMet reductive cleavage, a prerequisite for activation. As shown in Table 2, the amount of methionine produced during activation was greatly decreased (to 3–10%) in the case of the Cys-to-Ala mutants when compared with the wild-type protein.
Cys-543, Cys-546, Cys-561, and Cys-564 in Protein α from Bacteriophage T4 Provide a Metal-Binding Site.
The overall structure of protein α from bacteriophage T4 suggested that the binding site of protein β would be on the face of the molecule where the glycyl radical loop enters the β-barrel (4). It is thus interesting that the four previously disordered C-terminal cysteine residues of protein α form a metal-binding site in this region. Fig. 3b shows electron density for this center, and Fig. 5a shows its overall structure. The structure is well refined, with an Rfree of 24.3%. It is derived from a crystal soaked with dGTP and ATP to study the allosteric regulation, diffracting to 2.45 Å. The metal domain has also been observed in varying degrees of order in other NrdD structures (PDB codes and ).
Fig 3.

Structure of the metal-binding domain (MBD) in NrdD and its position in the overall structure. (a) A dimer of NrdD, showing the position of the MBD in relation to the glycyl radical. The monomers are drawn in light and dark gray, respectively. The distance from the metal to the Cα atom of Gly-580 is 28 Å, and the distance between the two metal ions in the dimer is 70 Å. Figs. 3a and 5 were made with MOLSCRIPT (44), Figs. 3b and 4c were made with BOBSCRIPT (45). (b) Electron density for the MBD at 2.45-Å resolution. This is a 2 |Fo| − |Fc| map, with phases from the refined model, contoured at 0.9 σ. An all-atom model for residues 541–574 of NrdD and the metal atom is superimposed.
Fig 5.

Comparison of the fold of the MBD with rubrerythrin and the Zn ribbon motif from transcription factor IID. The coordinating cysteines are labeled. (a) The MBD from NrdD, residues 541–574. (b) Rubrerythrin, C-terminal residues 144–191 (31). (c) Transcription factor IIB N-terminal domain, residues 1–50 from the first model in the PDB file of the NMR structure (26).
The M(Cys)4 domain is on the outer edge of the α component, on a flexible arm. The average main-chain B-factor between residues 541 and 574 is 78 Å2, compared with a value of 43 Å2 for the whole structure. The distance from the metal atom to the Cα atom of the mutated glycyl radical residue Ala-580 is 25 Å. Cys-543, Cys-546, Cys-561, and Cys-564 coordinate the metal ion tetrahedrally. The coordination distances to the Sγ atoms of the four cysteines are 2.1, 2.2, 2.5, and 2.6 Å. These discrepancies from ideal values are most likely due to the high mobility of this region and consequent uncertainty in the atomic coordinates.
The metal-binding motif begins at the C-terminal end of the last β-strand (βJ) of the 10-stranded α−β barrel of the RNR superfamily. It ends in the first strand of the C-terminal hairpin protruding back into the barrel and housing the glycyl radical site. The two CysX2Cys motifs are found in loops. The region between the two motifs is a short hairpin with two β-strands. Thus, the metal-binding site is constructed with a minimum of extra residues.
The high resolution x-ray experiment did not unambiguously identify the metal ion. Fluorescence scans of the Fe and Zn K edges are shown in Fig. 4, and optimized anomalous data sets collected near these edges are summarized in Table 2. The Zn edge scan clearly shows the presence of Zn (Fig. 4a) whereas the Fe edge scan (Fig. 4b) shows very little Fe in the crystal. The quality of the available crystals allowed collection of data to only 3.5 Å at the Zn edge. Nevertheless the first peak in the Zn anomalous difference Fourier, at 10.7 σ, is in close proximity to the metal ion modeled in the high resolution structure. The quality of the electron density for this domain at 3.5-Å resolution does not allow the surrounding residues to be unambiguously placed, so the anomalous peak is off-center with regard to the presumed position of the Zn atom (Fig. 4c). However, the peak corresponds exactly to a 6.1-σ peak in the |Fo| − |Fc| difference Fourier, which appears when the metal ion is omitted from phase calculation. As a control, the next two highest peaks in the Zn anomalous difference Fourier, at 5.1 and 4.9 σ, correspond to the Sγ atoms of the well ordered Cys-449 and Cys-80, because S has some residual anomalous signal at 1.254 Å. Less intense peaks are noise. A similar anomalous difference Fourier at 4.4-Å resolution calculated with data collected from the same crystal at the Fe edge (not shown) contains a peak at a similar position but only at the same level as the peaks from ordered sulfurs. Taken together, these results identify the metal bound by the four cysteines as mainly Zn, with trace amounts of Fe.
Fig 4.

Identity of the metal as shown by absorption edge scans and anomalous difference Fouriers. (a) X-ray fluorescence scan of the K absorption edge of Zn. The fluorescence is given in arbitrary units. In addition, the energy scale is not exactly calibrated due to small errors in the calibration of the synchrotron station. (b) As in a but for the K edge of Fe. (c) Anomalous difference Fourier (thin gray lines) and |Fo| − |Fc| difference Fourier (thick black lines) for Zn. The anomalous map is contoured at 5 σ, the |Fo| − |Fc| map at 4 σ. For the latter map, the Zn atom was omitted from the coordinates, which were then given random displacements of rms 0.1 Å and refined by using REFMAC.
Structural Homology to Other Metallodomains.
Structural homology searches were run by using the DALI and TOP servers. The only statistically significant homology reported by the DALI server was to the Zn-binding domain of valyl tRNA synthetase. However, TOP, which additionally allows permutation of secondary structure elements, reported statistically significant structural homology to several cysteine-containing metal centers, mostly coordinating Zn. The closest structural neighbor reported by TOP was an NMR structure of the N-terminal domain of transcription factor IIB (refs. 26; PDB code ; Fig. 5c), containing a zinc ribbon motif, which is a rubredoxin-like motif consisting of three β-strands connected by loops that coordinate a Zn ion through four Cys residues. Twenty-two residues could be superimposed with an rms deviation in Cα positions of 0.83 Å, including the four Cys ligands. Zinc ribbons have also been found in transcription factor IIS (27), the single-stranded DNA binding protein RPA (28), in a variant form in ribosomal protein L36 (29), and in the RNA-dependent protein deacetylase SIR2-Af1 (30). The next nearest structural neighbors were the rubredoxin-like four-cysteine centers of Fe and Zn rubrerythrins (31, 32; PDB codes and ; Fig. 5b). Twenty-one Cα atoms in Fe rubrerythrin matched with an rms deviation of 0.77 Å, including the Cys ligands. However, the secondary structure elements are permuted with respect to NrdD, and the two CysX2Cys motifs are swapped in the primary structure (Fig. 5b).
Discussion
The crystal structure of protein α of the anaerobic RNR from bacteriophage T4 shows the presence of a M(Cys)4 metal center in the C terminus. Mutants of the highly homologous enzyme from E. coli confirm the importance of the four coordinating cysteines, present in one fully conserved (Cys-662, Cys-665) and one less conserved (Cys-644, Cys-647) CysX2Cys sequence motif of protein α (Fig. 1). Mutation of these cysteines to alanine results in an enzyme that still forms a complex with the activase but cannot acquire the catalytically essential glycyl radical from the radical-generating machinery, as shown directly by EPR spectroscopy, and thus cannot be activated for ribonucleotide reduction.
How can these cysteines in protein α participate in the formation of the glycyl radical? In its simplest form, the mechanism for that reaction is (33):
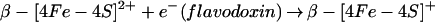 |
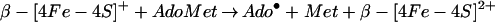 |
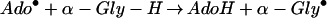 |
We can exclude mechanisms in which the cysteines act as radical transfer sites from the 5′-deoxyadenosyl radical and the glycyl radical (Eq. 3), forming intermediate thiyl radicals. We have indeed shown, by using protein α preparations labeled with deuterated glycines, that deuterium is incorporated into 5′-deoxyadenosine, which proves that the 5′-deoxyadenosyl radical directly abstracts the hydrogen atom of the glycine residue (unpublished results). This behavior has also been demonstrated for another glycyl radical enzyme, pyruvate-formate lyase (34).
The discovery that these four cysteines provide a metal-binding site suggests new mechanistic hypotheses. To our knowledge, there have been no previous observations of metal binding in the large component of a RNR. The structural homology to the Zn ribbon family and to rubrerythrins clearly shows that the metal binding is not artefactual. However, because the Zn ribbon is also structurally similar to rubredoxins [a well characterized class of non-heme iron proteins (35)] and because iron sites in rubredoxins can be occupied by Zn, in particular during heterologous expression in E. coli (36), the architecture of the metal-binding site does not in itself allow us to definitively say whether the center has a redox or a structural role (37). Analysis of metals in the crystal revealed the presence of both Fe and Zn, although Fe was present only in very small amounts. This finding raises the question whether the true active center contains Fe or Zn.
If the large subunit of a class III RNR contained a Zn(Cys)4 center, the simplest conceivable role for it would be a structural one: it could in some way organize the C-terminal end of protein α and thus control the accessibility/reactivity of the glycine residue, thus optimizing reaction 3. Mutation of the cysteines would result in proteins in which oxidation of the glycine residue would thus be much less efficient. This control in turn would explain why so little methionine from AdoMet is produced during activation of these mutants (Table 3). Indeed, reduction of AdoMet by fldx to methionine and the 5′-deoxyadenosyl radical (Eqs. 1 and 2) is highly thermodynamically unfavorable and needs to be coupled with glycyl radical formation (Eq. 3) (7). This coupling was previously nicely shown from experiments using a mutant α protein in which the glycine residue was changed to an alanine (7). An alternative structural role could be in promoting AdoMet binding by completing the AdoMet-binding site, which presumably lies principally on protein β because activation of AdoMet is achievable by using protein β alone (8).
Such a structural function could also be fulfilled by an iron center, if we allow for the possibility that the Zn atom may be an artifact of purification and that the true metal is Fe. If this were the case, additional roles for this center would be conceivable, for example, in electron transfer from fldx during the initial reduction of the [4Fe-4S]2+ cluster (Eq. 1). However, this particular hypothesis is unlikely because the redox potentials for Fe3+/2+ in rubredoxins range from approximately −100 to +50 mV (38) whereas the redox potential of the [4Fe-4S]2+/+ cluster in the activase is very negative, −550 mV with respect to NHE (7). We also might consider as a possibility that, in concert with the structural role invoked above, the CysX2Cys motifs use their chelating capacity to convert the cluster into a more reactive [3Fe-4S] one as suggested before (8).
In fact, the requirement for such a rubredoxin-like metal center is not a general property of glycyl-radical enzymes. Rather, it distinguishes class III RNR from all of the other enzymes of that family because the twin proximal CysX2Cys motifs are not present in the large subunits of any of the other characterized proteins of that class: pyruvate-formate lyase (39), p-hydroxyphenylacetate decarboxylase (40), benzylsuccinate synthase (41), or 2-keto acid-formate lyase (42). Additionally, the twin CysX2Cys motif seems to be present at the C terminus of several class II RNRs, which use adenosylcobalamin as a radical source. Assuming that this motif builds a similar metal center to the one seen here, it is hard to imagine that it could have a redox role in the class II RNRs. Altogether, these observations rather suggest a structural role for the metal center. The results presented here open unanticipated directions for future research aimed at understanding the fascinating chemistry of class III RNRs. Further experiments should first focus on the identification of the physiological metal ion in the rubredoxin-like center and of its function.
Acknowledgments
We thank the staff at MAX-Lab (Y. Cerenius) and European Molecular Biology Laboratory (EMBL) Hamburg (C. Enroth) for help with data collection, and V. Forge (Commissariat à l'Energie Atomique Grenoble) for CD experiments. Research was funded by grants from the Swedish Research Council and Crafoords Stiftelse (to D.T.L.). We acknowledge financial support from the European Community–Access to Research Infrastructure Action of the Improving Human Potential Program (to the EMBL Hamburg Outstation, Contract HPRI-CT-1999-00017).
Abbreviations
RNR, ribonucleotide reductase
AdoMet, S-adenosylmethionine
fldx, flavodoxin
MBD, metal-binding domain
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The further refined coordinates of the dGTP complex have been deposited in the Protein Data Bank, www.rcsb.org, with PDB ID code replacing entry .
References
- 1.Jordan A. & Reichard, P. (1998) Annu. Rev. Biochem. 67, 71-98. [DOI] [PubMed] [Google Scholar]
- 2.Sjöberg B.-M. (1997) Struct. Bonding 88, 139-173. [Google Scholar]
- 3.Fontecave M., Mulliez, E. & Logan, D. T. (2002) Prog. Nucleic Acid Res. Mol. Biol. 72, 95-127. [DOI] [PubMed] [Google Scholar]
- 4.Logan D. T., Andersson, J., Sjöberg, B.-M. & Nordlund, P. (1999) Science 283, 1499-1504. [DOI] [PubMed] [Google Scholar]
- 5.Andersson J., Westman, M., Sahlin, M. & Sjöberg, B.-M. (2000) J. Biol. Chem. 275, 19449-19455. [DOI] [PubMed] [Google Scholar]
- 6.Fontecave M. & Mulliez, E. (1999) in Chemistry and Biochemistry of B12, ed. Banerjee, R. (Wiley, New York), pp. 731–756.
- 7.Mulliez E., Padovani, D., Atta, M., Alcouffe, C. & Fontecave, M. (2001) Biochemistry 40, 3730-3736. [DOI] [PubMed] [Google Scholar]
- 8.Padovani D., Thomas, F., Trautwein, A. X., Mulliez, E. & Fontecave, M. (2001) Biochemistry 40, 6713-6719. [DOI] [PubMed] [Google Scholar]
- 9.Tamarit J., Gerez, C., Meier, C., Mulliez, E., Trautwein, A. X. & Fontecave, M. (2000) J. Biol. Chem. 275, 15669-15675. [DOI] [PubMed] [Google Scholar]
- 10.Ollagnier S., Meier, C., Mulliez, E., Gaillard, J., Schuenemann, V., Trautwein, A. X., Mattioli, T., Lutz, M. & Fontecave, M. (1999) J. Am. Chem. Soc. 121, 6344-6350. [Google Scholar]
- 11.Padovani D., Mulliez, E. & Fontecave, M. (2001) J. Biol. Chem. 276, 9587-9590. [DOI] [PubMed] [Google Scholar]
- 12.Sun X., Eliasson, R., Pontis, E., Andersson, J., Buist, G., Sjöberg, B.-M. & Reichard, P. (1995) J. Biol. Chem. 270, 2443-2446. [DOI] [PubMed] [Google Scholar]
- 13.Young P., Andersson, J., Sahlin, M. & Sjöberg, B.-M. (1996) J. Biol. Chem. 271, 20770-20775. [DOI] [PubMed] [Google Scholar]
- 14.Tamarit J., Mulliez, E., Meier, C., Trautwein, A. X. & Fontecave, M. (1999) J. Biol. Chem. 274, 31291-31296. [DOI] [PubMed] [Google Scholar]
- 15.Bradford M. M. (1976) Anal. Biochem. 72, 248-254. [DOI] [PubMed] [Google Scholar]
- 16.Eliasson R., Pontis, E., Fontecave, M., Gerez, C., Harder, J., Jörnvall, H., Krook, M. & Reichard, P. (1992) J. Biol. Chem. 267, 25541-25547. [PubMed] [Google Scholar]
- 17.Larsson K.-M., Andersson, J., Sjöberg, B.-M., Nordlund, P. & Logan, D. T. (2001) Structure 9, 739-750. [DOI] [PubMed] [Google Scholar]
- 18.Otwinowski Z. & Minor, W. (1997) in Methods in Enzymology, ed. Sweet, R. M. (Academic, New York), Vol. 276, pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project No. 4 (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
- 20.Brünger A. & Warren, G. L. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 21.Murshudov G. N., Vagin, A. A. & Dodson, E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240-255. [DOI] [PubMed] [Google Scholar]
- 22.Holm L. & Sander, C. (1995) Trends Biochem. Sci. 20, 478-480. [DOI] [PubMed] [Google Scholar]
- 23.Lu G. (2000) J. Appl. Crystallogr. 33, 176-183. [Google Scholar]
- 24.Ollagnier S., Mulliez, E., Gaillard, J., Eliasson, R., Fontecave, M. & Reichard, P. (1996) J. Biol. Chem. 271, 9410-9416. [DOI] [PubMed] [Google Scholar]
- 25.Sun X., Ollagnier, S., Schmidt, P. P., Atta, M., Mulliez, E., Lepape, L., Eliasson, R., Gräslund, A., Fontecave, M., Reichard, P. & Sjöberg, B.-M. (1996) J. Biol. Chem. 271, 6827-6831. [PubMed] [Google Scholar]
- 26.Zhu W., Zeng, Q., Colangelo, C. M., Lewis, M., Summers, M. F. & Scott, R. A. (1996) Nat. Struct. Biol. 3, 122-124. [DOI] [PubMed] [Google Scholar]
- 27.Qian X., Jeon, C., Yoon, H., Agarwal, K. & Weiss, M. A. (1993) Nature 365, 277-279. [DOI] [PubMed] [Google Scholar]
- 28.Bochkareva E., Korolev, S., Lees-Miller, S. P. & Bochkarev, A. (2002) EMBO J. 21, 1855-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Härd T., Rak, A., Allard, P., Kloo, L. & Garber, M. (2000) J. Mol. Biol. 296, 169-180. [DOI] [PubMed] [Google Scholar]
- 30.Min J., Landry, J., Sternglanz, R. & Xu, R. M. (2001) Cell 105, 269-279. [DOI] [PubMed] [Google Scholar]
- 31.deMaré F., Kurtz, D. M., Jr. & Nordlund, P. (1996) Nat. Struct. Biol. 3, 539-546. [DOI] [PubMed] [Google Scholar]
- 32.Sieker L. C., Holmes, M., Le Trong, I., Turley, S., Santarsiero, B. D., Liu, M. Y., LeGall, J. & Stenkamp, R. E. (1999) Nat. Struct. Biol. 6, 308-309. [DOI] [PubMed] [Google Scholar]
- 33.Fontecave M., Mulliez, E. & Ollagnier-de-Choudens, S. (2001) Curr. Opin. Chem. Biol. 5, 506-511. [DOI] [PubMed] [Google Scholar]
- 34.Frey M., Rothe, M., Wagner, A. F. V. & Knappe, J. (1994) J. Biol. Chem. 269, 12432-12437. [PubMed] [Google Scholar]
- 35.Meyer J. & Moulis, J.-M. (2001) in Handbook of Metalloproteins, eds. Messerschmidt, A., Huber, R., Poulos, T. & Weighardt, K. (Wiley, New York), pp. 505–517.
- 36.Pétillot V., Forest, E., Mathieu, I., Meyer, J. & Moulis, J.-M. (1993) Biochem. J. 296, 657-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dauter Z., Wilson, K. S., Sieker, L. C., Moulis, J.-M. & Meyer, J. (1996) Proc. Natl. Acad. Sci. USA. 93, 8836-8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eidsness M. K., Burden, A. E., Richie, K. A., Kurtz, D. M., Scott, R. A., Smith, E. T., Ichiye, T., Beard, B., Min, T. P. & Kang, C. H. (1999) Biochemistry 38, 14803-14809. [DOI] [PubMed] [Google Scholar]
- 39.Rödel W., Plager, W., Frank, R. & Knappe, J. (1988) Eur. J. Biochem. 177, 153-158. [DOI] [PubMed] [Google Scholar]
- 40.Thorsten S. & Andre, P. I. (2001) Eur. J. Biochem. 268, 1363-1372. [DOI] [PubMed] [Google Scholar]
- 41.Leuthner B., Leitwein, C., Schultz, H., Horth, P., Haehnel, W., Schiltz, E., Schagger, H. & Heider, J. (1998) Mol. Microbiol. 28, 615-618. [DOI] [PubMed] [Google Scholar]
- 42.Sawers G. (1999) FEMS Microbiol. Rev. 22, 543-551. [Google Scholar]
- 43.Gouet P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999) Bioinformatics 15, 305-308. [DOI] [PubMed] [Google Scholar]
- 44.Kraulis P. J. (1991) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 45.Esnouf R. M. (1999) Acta Crystallogr. D 55, 938-940. [DOI] [PubMed] [Google Scholar]