

Abstract
Although analysis strategies exist for probing a diverse array of molecular properties, most of these approaches are not amenable to the study of reaction intermediates and other transient species. Separations in particular can provide detailed information on attributes not readily measured by spectroscopy but typically are performed over time scales much longer than the life span of highly unstable compounds. Here we report the development of an electrophoretic strategy that dramatically extends the practical speed limit for fractionations and demonstrate its utility in examining transient hydroxyindole photoproducts. Fluorescent reaction intermediates are optically generated in femtoliter volumes within a flowing reagent stream and are differentially transported at velocities as large as 1.3 m⋅s−1, thereby minimizing band variance and allowing multicomponent reaction mixtures to be resolved over separation paths as short as 9 μm. Analyte migration times and band variances do not deviate significantly from basic theory for separations performed with fields that exceed 0.1 MV⋅cm−1, indicating that effects from Joule heating are minor. We demonstrate the feasibility of achieving baseline resolution of a binary mixture in <10 μs, nearly 100-fold faster than previously possible. Application of this approach to the study of a range of short-lived molecules should be feasible.
Although of tremendous value, time-resolved spectroscopy ultimately is limited in its ability to probe transient solution-phase molecules. Accurate interpretation of spectroscopic data can be problematic when spectral features are broad or reaction environments contain multiple chemical species. Measurements of molecular transport behavior in defined fields or chromatographic flow environments can offer insights into properties not readily probed by spectroscopy alone and in many cases can be used to derive information on a large number of components within complex sample mixtures. Unfortunately, because such separation procedures typically require minutes or longer to complete, they have not been suitable for studies of highly unstable molecules.
The speed of a separation is limited by the time required to transport a species of interest over a distance sufficiently long to isolate it from other detectable components. In chromatographic separations, the velocity of an analyte typically is restricted by its transfer rates between phases, with plate heights becoming prohibitively large under high flow-rate conditions. In contrast, capillary electrophoresis (CE) is not constrained by partitioning or adsorption-desorption kinetics and, consequently, has been adapted for analyses on low- and subsecond time scales (1–6). In CE, the migration time (t) of an analyte scales as the separation distance (L) and inversely with the applied field (E),
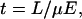 |
1 |
where μ is the sum of the electrophoretic and electroosmotic mobilities. Under ideal circumstances in which diffusion is the sole determinant of spatial bandwidths, Jorgenson and Lukacs (7) showed that the number of theoretical plates (N) produced in a separation depends only on the voltage (V) applied across the separation region, regardless of separation distance,
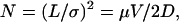 |
2 |
where σ is the rms bandwidth, and D is the analyte diffusion coefficient. From Eq. 2, it appears feasible to achieve faster separations simply by applying a given voltage across ever shorter separation channels. Ultimately, however, Joule heating places a practical limit on the strength of the electric field that can be used, and variances caused by the injection-plug length and spatial extent of the detection region degrade separation efficiency (7, 8).
Although electric-field limitations are important, effects from Joule heating can be minimized by using extremely narrow-bore channels (i.e., ≤10-μm i.d.) and low-conductivity buffer systems. Accordingly, the greatest challenge for high-speed CE has been reducing injection-plug lengths to dimensions suitable for ultrashort separation distances. A microfluidics-based strategy, in which samples are electroosmotically introduced into a separation region within a network of channels, has been used with very large electric fields (≈50 kV⋅cm−1) to fractionate dichlorofluorescein and rhodamine in ≈1 ms, the fastest separation published to date (3). The speed and efficiency of this approach, however, ultimately are limited by the time required to establish on-chip fields that are necessary for sample introduction and subsequent fractionation.
Optical gating can be used as an alternative to microfluidics-based sampling to modulate the composition of flowing analyte streams more rapidly (1). This approach relies on a high-intensity laser to photodestroy fluorescent analytes efficiently as they enter a separation region. When a separation is to be performed the laser beam is blocked, thereby allowing a small plug of fluorescent material to pass unaltered into the separation region for electrophoretic analysis. Because the separation voltage is applied to the capillary at all times, capacitive charging times do not limit how narrow a sample plug can be; rather, plug sizes are determined by the dimensions of the laser focus, the time required for optical switching, and the velocity at which samples migrate through the gating site. Convolution of these factors has the potential to yield sample plugs <1-μm long even under conditions of extremely high-velocity solution flow.
Optically gated CE was devised originally as a means to sequentially sample from a chromatographic effluent (1), not to probe transient molecular states. Molecules undergoing photoalteration were rendered undetectable, allowing fluorescent peaks to be detected on a low background. Studies on multiphoton-excited photoreactions of biological molecules (9–11), in which thermally labile fluorescent photoproducts are generated from nonfluorescent reactants, suggested that optical gating could be a useful means for producing plugs of evolving chemical mixtures for electrophoretic analysis. Note that in this case optical gating creates rather than destroys fluorescent molecules. We have demonstrated that by using high numerical aperture optics to minimize the dimensions of the optical gate and probe sites, optically gated CE separations can fractionate cationic from anionic hydroxyindole photoproducts across migration paths of 9–12 μm, the shortest separation distances reported to date (5). Although this approach was capable of analyzing mixtures on low-millisecond time scales, the speed and efficiency of such separations were limited by the magnitude of the electric field that could be applied to the separation region (≈2,000 V⋅cm−1).
Here we report the use of a geometric capillary modification to amplify electric fields experienced at optically gated separation regions, an approach that yields large reductions in separation times and correspondingly large improvements in separation efficiency. In these experiments, mixtures of 5-hydroxyindoles are introduced continuously by using extremely high electric fields into a region of fused silica capillary that has been pulled to an hourglass shape. As reactant molecules enter the most constrained region of the channel, plugs of visible-emitting multiphoton photoproducts are created in a gated laser focus. These plugs are electrophoretically fractionated into component bands and are detected via two-photon excited fluorescence ≈10 μm downstream at a probe focus (Fig. 1a). Baseline resolution of a binary mixture of positive and neutral transient photoproducts is accomplished in times as short as 10 μs, nearly 100-fold faster than previously reported for any fractionation technique.
Figure 1.

(a) Schematic of the photoreaction and probe regions in optically gated microsecond separations. Femtoliter reaction plugs are created in flowing reagent streams by a gate focus that is switched to high intensity for ≈1–2 μs; transient reaction products migrate according to charge-to-drag ratios, providing the capacity to analyze reaction mixtures within microseconds. As depicted, photoproduct diffusion is minor on the time scales of these separations. (b) A typical hourglass structure created in the central portion of a short fused-silica capillary (29-μm i.d. and 320-μm o.d. in unpulled regions). By reducing the cross-sectional area by a factor of ≈30–40, fields in excess of 0.1 MV⋅cm−1 can be generated in short capillary stretches by using an applied potential of 20 kV. (Scale bar, 250 μm.) The boxed inset shows the central (≈60-μm) region of the hourglass where separations are performed. (c) Schematic of the electrophoresis assembly. Index-matching glycerin is used to fill microscopic gaps between the capillary and the underlying coverslip. The microscope objective (obj) focuses two separate laser beams, a microsecond-gated photoreaction beam and a continuous probe beam, to positions in the capillary separated by ≈10 μm.
Methods
A 76-MHz Coherent Mira 900-F femtosecond mode-locked Ti/S laser (≈750 nm) pumped by a 10-W Coherent Verdi frequency-doubled (532-nm) neodymium/vanadate laser is used in all studies. Output from the Mira is split into separate gate and probe beams by using a half-wave plate and a polarizing beamsplitter. The intensity of the gate beam is modified by a second half-wave plate/polarizer pair and is electrooptically modulated by using a Pockel's cell (350–50, Conoptics, Danbury, CT) driven by a high-speed delay generator (DG-535, Stanford Research, Sunnyvale, CA). The Pockel's cell, which has rise and fall times of <1 μs, is modulated to produce ≈1- to 2-μs gate periods (≈75–150 laser pulses per gate, ≈3 nJ per pulse) at a repetition rate of 500 Hz. The unmodulated probe beam typically is operated at an average power of ≈50 mW (≈0.4 nJ per pulse), a level sufficient to probe reaction intermediates while minimizing additional photoreaction.† The two beams are recombined slightly off axis from each other by using a polarizing beamsplitter, passed through a long-pass dichroic mirror (630DCXRU, Chroma Technology, Brattleboro, VT), and reflected into a 1.3-numerical aperture ×100 oil-immersion microscope objective (Fluar, Zeiss) with a 90/10 beamsplitter (i.e., 90% nominal reflectance) situated directly below the objective. Because the modulated and unmodulated beams are not exactly colinear, the two beam waists are formed at different positions in the focal plane. Mirror-tilt adjustments are used to align and separate the focal spots. By using this optical system, multiphoton focal volumes <1 μm3 can be produced (10).
Visible fluorescence from hydroxyindole photoproducts is epi-collected by using the ×100 objective, directed back along the laser-propagation path by the 90/10 beamsplitter, and reflected toward a bialkali photomultiplier tube (HC-125, Hamamatsu, Middlesex, NJ) by the dichroic mirror. Residual laser scatter is rejected from the light-collection pathway by using a 5-cm saturated aqueous CuSO4 filter and three 3-mm-thick BG 39 filters. Signal from the photomultiplier is acquired in a photon-counting mode by using a multichannel scaler (SR430, Stanford Research), which allows data to be summed on the fly from sequential electrophoretic runs. For some studies, a second detection channel is added for measurement of UV fluorescence. In this case, a UV-reflecting long-pass dichroic mirror (375DCLP, Chroma Technology) is inserted between the 90/10 beamsplitter and the visible-reflecting dichroic mirror. UV fluorescence reflected from the beam path at this position is passed through two UV-transmitting filters (Barr Associates, Westford, MA) and is measured by using a second photomultiplier and an SR430 multichannel scaler.
Fused-silica capillaries (Polymicro Technologies, Phoenix) are pulled to hourglass shapes (Fig. 1b) by using a micropipet puller (P-2000, Sutter Instruments, Novato, CA). Various pull sequences can be used to create a range of hourglass dimensions. For the studies reported here, a procedure is used that results in capillary waist diameters ≈5- to 7-fold smaller than in the unpulled capillary regions, with the total hourglass extending over a 1- to 2-mm-long region located centrally on a capillary that is ≈6 cm long. Throughout the hourglass, the i.d./o.d. ratio remains similar, and the most narrow capillary region typically extends over a distance of 50–100 μm (Fig. 1b, boxed inset).
A modified inverted optical microscope (Axiovert 135, Zeiss) serves as a stable platform for the capillary and objective and provides a means to image the channel for alignment purposes. Pulled capillaries are secured under a milled Plexiglas block such that the hourglass region coincides with a drilled access hole (Fig. 1c). A borosilicate glass coverslip (no. 0) is mounted directly beneath the capillary and sealed against the block to form a well surrounding the pulled region of the capillary. Refractive index-matching fluid (glycerin) is placed in the well to fill the narrow gap between the capillary and the underlying coverslip, an approach that minimizes optical aberrations caused by the curved surface of the capillary.
The sample is transported electrokinetically between inlet and outlet vials, which are sealed with septa to confine high-potential solutions. Platinum electrode wires and small-diameter plastic tubing (used for deoxygenation of phosphate-buffered solutions) are fixed in place at the base of each vial by using epoxy. The entire assembly, including buffer reservoirs and insulating Plexiglas sheets, is bolted to a three-axis translation stage (562, Newport, Fountain Valley, CA). Motorized actuators (860A, Soma, Irvine, CA) are used for final alignment of the capillary with the laser foci. Separation voltages are applied by using two dual-polarity high-voltage power supplies (CZE1000, Spellman, Hauppage, NY) connected to the same ground and operating at opposite polarities.
All reagents are used as received from Aldrich. 5-Hydroxytryptamine [serotonin (5HT)] creatinine sulfate and 5-hydroxytryptophan (5HTrp) sample solutions are prepared fresh daily in electrophoresis buffer (5 mM sodium Hepes, pH 7.1 or 5 mM sodium phosphate, pH 7.0) and filtered by using 0.20-μm syringe filters (Corning, Corning, NY). Buffer concentrations are minimized to limit current in high-field separations and, in the case of Hepes, to minimize production of hydroxyindole photopolymers. Phosphate-buffered samples are deoxygenated for ≈15 min by slowly bubbling argon gas through the inlet vial (9). For velocity-scaling studies, capillaries and buffer reservoirs are filled with solution and equilibrated by application of high voltage for ≈30 min before use.
Results and Discussion
The capability of this system to analyze transient photochemical products was evaluated by using different separation fields and data-acquisition rates (Fig. 2). Here a potential of either 10 or 20 kV was applied across a 6.1-cm-long capillary (original i.d. and o.d., 29 and 320 μm, respectively). The total pull region on this capillary extended for ≈1.5 mm, reaching a minimum channel diameter of ≈5 μm at the hourglass waist. Analysis with a 20-kV potential achieves extremely rapid baseline resolution of 5HT and 5HTrp transient photoproducts, with the two species migrating through the 10-μm separation distance in 14.0 and 19.5 μs, respectively. The bottom two traces in Fig. 2 show separate 20-kV analyses performed under identical conditions with the exception of data-acquisition rate. The analysis with a higher signal-to-noise (open circles) was acquired by using 640-ns data bins, a size that results in moderate undersampling (five to six bins per peak). By decreasing the data-bin size to 320 ns (black circles), somewhat better peak definition is achieved (≥10 bins per peak). The top trace in Fig. 2 shows an analysis of the hydroxyindole mixture acquired with a 10-kV separation potential. Here the time axis is compressed 2-fold (i.e., the trace extends from 0 to 70 μs) to enable a direct visual comparison with the 20-kV separations. Analyte velocities scale as expected with separation potential, and the spatial widths of bands are similar at the two field settings.
Figure 2.

Electrophoretic characterization of transient photochemical products created from mixtures of the 5-hydroxyindoles 5HT and 5HTrp (both 250 μM) by using a separation distance of 10 μm. The bottom two traces are analyses of a reaction mixture produced by using a field of 0.12 MV⋅cm−1 (20 kV). Baseline resolution of the two fluorescent products is achieved in ≈19.5 μs. Photon-counting bin sizes were 640 ns (open circles) and 320 ns (black circles). The top trace (gray circles) demonstrates analysis with 0.06 MV⋅cm−1 (10 kV) and 640-ns data bins and has been compressed 2-fold in the horizontal dimension to facilitate comparison to the faster, higher-field separations. The nominal gating period for each analysis was 2 μs; ≈50,500 and 65,500 repeat electrophoretic events were summed to generate the 10- and 20-kV traces, respectively. The y axis values represent actual counts for the 20-kV (640-ns) trace, with the other analyses displaced vertically for ease of visual comparison.
The duration of the gate period, nominally 2 μs for all three separations, was evaluated by focusing the Ti/S beam into a fluorescein solution with relatively low laser powers (≈5 mW). Two-photon excited emission from this solution (Fig. 2 Inset) shows a somewhat longer time to reach 90% plateau (≈0.7 μs) than to return 90% to baseline (≈0.5 μs) and an overall rms duration (σgate) of ≈0.57 μs. Provided that transition saturation does not occur, the higher intensity dependence of hydroxyindole photoreaction should cause somewhat steeper rise and fall times than those associated with this fluorescence transient.
For an unpulled 6-cm-long capillary, application of a potential between inlet and outlet reservoirs would establish an axially uniform field of only ≈0.16 V⋅cm−1 for every volt applied. Because the mobilities of 5HTrp and its transient photoproduct are indistinguishable on microsecond time scales,‡ knowledge of 5HTrp mobility (determined by using conventional CE separations to be 4.4 × 10−4 cm2⋅V−1⋅s−1) provides a means to estimate fields within hourglass structures. For the separations shown in Fig. 2, a field of nearly 6 V⋅cm−1 was generated per volt applied to the capillary, an enhancement of ≈35-fold relative to an unpulled channel. Such fields are similar to what is predicted by using a simple model based on Ohm's law and the geometry of the hourglass region.
A detailed examination of analyte velocities and bandwidths was performed by using separation fields ranging up to 0.12 MV⋅cm−1 (applied potentials of 5–20 kV). Migration velocities for photoproducts scale linearly with potential (Fig. 3a), reaching values of 0.72 m⋅s−1 (5HT product) and 0.52 m⋅s−1 (5HTrp product) at 20 kV. Linear least-square fits show excellent agreement with the data, indicating that heating does not alter mobilities measurably in this study.
Figure 3.

(a) Plots of velocity versus applied potential for 5HT and 5HTrp photoproducts. Both plots scale linearly over the range of potentials examined here, demonstrating that Joule heating does not alter mobilities significantly even for fields >0.1 MV⋅cm−1. Duplicate measurements were made at each applied potential value. (b) Band variance (σ2, μm2) for 5HT and 5HTrp as a function of peak migration time with 2-μs nominal gating periods for each analysis. The solid curve represents ideal band variance calculated by using an rms focal dimension (σfocus) of 0.265 μm, an rms gating duration (σgate) of 0.57 μs, and a product diffusion coefficient (D) of 6.5 × 10−6 cm2⋅s−1. The same reagent solutions and capillary were used to acquire data in Figs. 2 and 3.
Spatial variances (σ2) of transient photoproduct peaks were determined for each of the 16 analyses (i.e., duplicates of four different field settings for both compounds) examined in Fig. 3a. Band variances in these studies are largest for species migrating most rapidly (Fig. 3b), a result that is expected even in the absence of Joule-heating effects. In a basic model that does not consider contributions of temperature gradients to band broadening, the overall variance is represented as the sum of terms related to sample introduction [(σintro)2], diffusion during the separation process, and a finite detection site [(σdet)2].
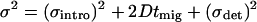 |
3 |
Here tmig is the average migration time of a given analyte. Detection variance can be represented as σ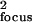 , where σfocus is the rms focal dimension (dependent on focal spot size and the effective intensity-dependence of excitation). Variance from sample introduction depends on both this focal dimension and analyte migration during the gating period (calculated as the product of analyte velocity, v, and σgate, the rms gate duration). The overall spatial variance therefore is
, where σfocus is the rms focal dimension (dependent on focal spot size and the effective intensity-dependence of excitation). Variance from sample introduction depends on both this focal dimension and analyte migration during the gating period (calculated as the product of analyte velocity, v, and σgate, the rms gate duration). The overall spatial variance therefore is
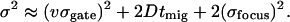 |
4 |
In this equation, velocities and migration times are known. By using the rms gate duration measured for fluorescein (0.57 μs) and a diffusion coefficient (6.5 × 10−6 cm2⋅s−1) that assumes structural similarity between product and parent molecules (12), predictions of variance versus migration time for different σfocus values can be compared with experimental results. The curve in Fig. 3b was produced by using an rms focal dimension of 0.265 μm, a reasonable size for the optical configuration used in these studies.§
The feasibility of using significantly higher fields to electrophoretically characterize transient solutions on shorter time scales is demonstrated in Fig. 4. Here a field estimated at ≈0.15 MV⋅cm−1 was used to fractionate 5HT and 5HTrp photoproducts in a more narrow capillary (originally, 15-μm i.d. and 360-μm o.d.), with both peaks migrating through the 9-μm separation distance in <10 μs.¶ To maintain reasonable separation efficiencies, the nominal gating period was reduced to 1 μs. Because photoproduct signal is diminished when using extremely brief gate periods and for very large migration velocities, it was necessary to maintain the data-bin size at 640 ns to detect peaks above baseline noise; as a result, bands are undersampled (approximately three to five bins per peak). Better signal-to-noise ratios could be achieved for smaller bin sizes by summing a greater number of analyses, but the multichannel scaler used in the current studies is limited to acquisition of ≈65,500 repeat events.
Figure 4.

Electrophoretic resolution of 5HT and 5HTrp photoproducts in 10 μs. Here, a field estimated to be ≈0.15 MV⋅cm−1 (35 kV) was used to fractionate components over a separation distance of 9 μm. The sample solution contained deoxygenated phosphate buffer (5 mM, pH 7.0) and 5HT and 5HTrp (both at 500 μM). Approximately 65,500 repeat electrophoretic events were summed to generate this trace, and the data-bin size was 640 ns. The nominal gate duration was 1 μs, and the probe beam was operated at ≈100 mW.
Assuming that greater signal averaging is performed, it should be feasible to reduce separation times further by using some combination of larger field strengths, lower viscosity solvents, and shorter separation distances; however, unless shorter sample plugs can be generated, reductions in separation efficiency eventually will become prohibitive. The use of a pulse picker or an acoustooptic modulator in place of the current Pockel's cell could reduce gate durations substantially, a necessity if submicrosecond analysis times are to be achieved. The fields required for such improvements in separation time may cause significant radial temperature gradients, and a resultant degradation of separation efficiency, unless smaller-diameter separation channels are used. Ultimately, the speed at which optically gated separations can be performed is limited by the kinetics of the relevant photochemistry, which have not yet been fully explored for the hydroxyindole reactions examined here.
Because of the small spatial extent of the injection plugs and high-velocity transport, >20 million theoretical plates were generated per second for the 20-kV separations shown in Fig. 2, a value that is nearly 100-fold greater than was possible previously (6). Although the resultant theoretical plate numbers are modest (N ≈ 300–500), separation efficiencies are ≈10-fold greater than earlier millisecond separations (3, 5). Moreover, relatively minor modifications of the current instrument and separation-capillary geometry should make it feasible to perform fractionations over much longer distances by using fields similar to those in the current studies. Because the primary causes of variance in the current studies are those associated with the sample-injection length and the detection process, a <2-fold increase in variance is predicted for 5HTrp when the total migration distance is increased from 10 to 100 μm (for E = 0.12 MV⋅cm−1, a 2-μs gate duration, and the same focal dimensions used in the current studies). As a consequence, such a separation is expected to yield a >50-fold increase in theoretical plate number (Nexp > 2 × 104). It thus appears feasible to perform CE separations in <0.25 ms with efficiencies comparable to those typical of high-performance liquid chromatography.
Possibilities exist for applying this high-speed fractionation technique to the study of other transient species. Photoreactions involving various classes of aromatic molecules generate products with new fluorescence properties (13–16), some of which may proceed through intermediates amenable to this analysis strategy. As a more general approach for studying photoreactions that do not normally yield fluorescent intermediates, it may be feasible to conjugate a hydroxyindole tag to a photochemically active species of interest, thus imparting reactants with the same fluorescent photoswitch used in optically gated separations here. Moreover, this approach would not be limited to investigations of photochemistry. By initiating reactions through microsecond photolytic release of reactants from caged precursors (17) or by microsecond mixing of reagents within microfabricated channel networks (18), intermediates in thermal reactions could be probed on submillisecond time scales. Characterization of transient macromolecular conformations also could be achieved in a similar manner. By simultaneously initiating protein folding‖ and phototransformation of a 5HTrp residue (substituted for tryptophan or appended to a terminus), high-speed electrophoresis could be used to clarify chemical and physical properties of folding intermediates that persist for as little as tens of microseconds.
Acknowledgments
We thank E. Okerberg, M. J. Gordon, and M. Gostkowski for discussions and experimental assistance. We gratefully acknowledge support for this work from Robert A. Welch Foundation Grant F-1331, Eli Lilly, and National Science Foundation Grant CHE-9734258.
Abbreviations
- CE
capillary electrophoresis
- 5HT
serotonin
- 5HTrp
5-hydroxytryptophan
Footnotes
A variety of recent technologies now make it possible to initiate protein folding on microsecond or faster time scales, including high-speed mixing (18), photolysis of intramolecular tethers (19, 20), and photoexcited electron transfer (21).
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 3545.
Generation of visible-emitting hydroxyindole photoproducts depends on absorption of three to four near-IR photons, leading (in nonsaturating conditions) to an intensity dependence of I3 to I4 (9–11) Because fluorescence excitation of products occurs via two-photon absorption, this process ideally scales as I2 and can be accomplished by using significantly lower laser powers than are required for efficient photoreaction.
High-intensity optical gating of 5HTrp (a neutral zwitterion at pH 7.1) creates a photobleached “hole” that migrates at the velocity of the unreacted indole and results in a negative-going peak in the baseline level of UV fluorescence. On time scales ranging from ≈40 to 200 μs, 5HTrp comigrates with its fluorescent photoproduct, indicating that the 5HTrp photoproduct is neutral (and the 5HT photoproduct is positively charged). Shorter times were not examined here, because the poor signal-to-noise ratio for hole measurements required the use of long gate durations.
This value represents an average of the gate and probe foci dimensions, which are likely to be somewhat different as a result of the different dependencies on laser intensity of the photoreaction and detection events and the possibility that these processes are saturated to different degrees.
5HTrp mobility was determined in a conventional CE analysis by using 5 mM phosphate separation buffer (containing 500 μM 5HT and 500 μM 5HTrp) and used to estimate the field in this fractionation based on an assumption that analyte mobility is constant.
References
- 1.Monnig C A, Jorgenson J W. Anal Chem. 1991;63:802–807. doi: 10.1021/ac00008a013. [DOI] [PubMed] [Google Scholar]
- 2.Tao L, Thompson J E, Kennedy R T. Anal Chem. 1998;70:4015–4022. doi: 10.1021/ac980324u. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson S C, Culbertson C T, Daler J E, Ramsey J M. Anal Chem. 1998;70:3476–3480. [Google Scholar]
- 4.Lapos J A, Ewing A G. Anal Chem. 2000;72:4598–4602. doi: 10.1021/ac000581a. [DOI] [PubMed] [Google Scholar]
- 5.Gordon M J, Okerberg E, Gostkowski M L, Shear J B. J Am Chem Soc. 2001;123:10780–10781. doi: 10.1021/ja016898p. [DOI] [PubMed] [Google Scholar]
- 6.Kennedy R T, German I, Thompson J E, Witowski S R. Chem Rev (Washington, DC) 1999;99:3081–3131. doi: 10.1021/cr980134j. [DOI] [PubMed] [Google Scholar]
- 7.Jorgenson J W, Lukacs K D. Anal Chem. 1981;53:1298–1302. [Google Scholar]
- 8.Huang H, Coleman W F, Zare R N. J Chromatogr. 1989;480:95–110. [Google Scholar]
- 9.Gostkowski M L, Curey T E, Okerberg E, Kang T, Vanden Bout D A, Shear J B. Anal Chem. 2000;72:3821–3825. doi: 10.1021/ac000278+. [DOI] [PubMed] [Google Scholar]
- 10.Gostkowski M L, Wei J, Shear J B. Anal Biochem. 1998;260:244–250. doi: 10.1006/abio.1998.2723. [DOI] [PubMed] [Google Scholar]
- 11.Shear J B, Xu C, Webb W W. Photochem Photobiol. 1997;65:931–936. doi: 10.1111/j.1751-1097.1997.tb07950.x. [DOI] [PubMed] [Google Scholar]
- 12.Bello M S, Rezzonico R, Righetti P G. Science. 1994;266:773–776. doi: 10.1126/science.266.5186.773. [DOI] [PubMed] [Google Scholar]
- 13.Fairhead H, P. Setlow P. J Bacteriol. 1992;174:2874–2880. doi: 10.1128/jb.174.9.2874-2880.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fujita, H. & Sasaki, M. (1986) Chem. Lett., 545–548.
- 15.Kohen E, Kohen C, Reyftmann J P, Morliere P, Santus R. Biochim Biophys Acta. 1984;805:332–336. doi: 10.1016/0167-4889(84)90015-6. [DOI] [PubMed] [Google Scholar]
- 16.Lores M, Garcia C M, Cela R. J Chromatogr A. 1994;683:31–44. [Google Scholar]
- 17.Grewer C, Jäger J, Carpenter B K, Hess G P. Biochemistry. 2000;39:2063–2070. doi: 10.1021/bi9919652. [DOI] [PubMed] [Google Scholar]
- 18.Knight J B, Vishwanath A, Brody J P, Austin R H. Phys Rev Lett. 1998;80:3863–3866. [Google Scholar]
- 19.Hansen K C, Rock R S, Larsen R W, Chan S I. J Am Chem Soc. 2000;122:11567–11568. [Google Scholar]
- 20.Volk M. Eur J Org Chem. 2001;2001:2605–2621. [Google Scholar]
- 21.Pascher T, Chesick J P, Winkler J R, Gray H B. Science. 1996;271:1558–1560. doi: 10.1126/science.271.5255.1558. [DOI] [PubMed] [Google Scholar]