

Abstract
Cell polarization is required for directed cell migration. We investigated the role of the calcium-dependent protease calpain during neutrophil chemotaxis and found that calpain inhibition induced neutrophil adhesion, polarization, and rapid chemokinesis in the absence of exogenous activators. Resting neutrophils display constitutive calpain activity with μ-calpain being the predominant active isoform. Our findings suggest that constitutive calpain activity in resting neutrophils may function as a negative regulator of protrusion and migration. Specific inhibition of μ-calpain, but not m-calpain, induced neutrophil polarization and chemokinesis. In contrast to IL-8-induced chemokinesis, the chemokinesis induced by calpain inhibition was not reduced in the presence of pertussis toxin, suggesting that calpain functions downstream of G protein-coupled receptors. Further, both calpain inhibition and stimulation with IL-8 and formyl-Met-Leu-Phe (fMLP) induced an increase in Cdc42 and Rac activation. These findings are consistent with the involvement of calpain in chemotaxis pathways. Accordingly, calpain inhibition decreased neutrophil chemotaxis and directional persistence in a gradient of IL-8 and fMLP. Together, these data reveal a previously uncharacterized function for calpain in neutrophils and suggest that localized modulation of calpain activity may regulate neutrophil chemotaxis downstream of G-protein-coupled receptors.
Keywords: migration
Neutrophils, key participants in the innate immune system, are the first responders to inflammatory stimuli such as bacterial infection or tissue injury. This rapid response is achieved by the detection of chemotactic gradients and the polarization and migration of neutrophils toward inflammatory mediators such as formyl-Met-Leu-Phe (fMLP) and IL-8. In the resting state, neutrophils are poorly adherent and exist in a spherical shape. A rapid change in cell morphology occurs in response to inflammatory stimuli such that neutrophils become polarized and migrate toward the inflammatory mediator. The mechanisms that regulate these changes in response to chemoattractants remain poorly understood.
There has been recent progress in understanding how cells migrate directionally in a chemoattractant gradient. Although an even distribution of G protein-coupled chemoattractant receptors (GPCRs) has been found on the surface of cells during chemotaxis (1, 2), recent studies have demonstrated that a gradient of chemoattractant can induce an asymmetry in the localization of phosphatidylinositol phosphate (PIP3) with enhancement at the leading edge of migrating neutrophil-like HL-60 cells (3), implying that GPCR activation leads to localized changes in membrane organization and recruitment of signaling intermediates. Previous studies have demonstrated that calpain activity is regulated by polyphosphoinositides (4), raising the intriguing possibility that calpain is an essential signaling intermediate downstream of chemoattractant-induced membrane lipid reorganization. In this paper, we explored the role of the calcium-dependent cysteine protease calpain in the chemokinesis and chemotaxis of primary neutrophils.
Calpain has been implicated as a critical calcium-dependent regulator of the actin cytoskeleton and cell migration (5–7). Previous studies have demonstrated that calpain plays a central role in fibroblast migration by regulating integrin–cytoskeletal interactions and cell detachment (5). In all cell types examined to date, inhibition of calpain reduces cell migration rates and invasiveness. The mechanisms for these effects include roles in both cell detachment (5, 8) and cell spreading (6, 9, 10). Calpain-deficient fibroblasts (Capn 4−/−) exhibit reduced migration rates, but also reveal a role for calpain in the regulation of cell protrusion (7). Our findings support a role for calpain in regulating neutrophil polarization and directional migration to chemotactic stimuli. In contrast to all other cell types examined to date, calpain inhibition increases the migration of resting neutrophils. We found that inhibition of μ-calpain (calpain I) but not m-calpain (calpain II) promotes neutrophil polarization, spreading, and random migration. However, the random migration induced by calpain inhibition is associated with a decreased capacity for directional migration toward chemotactic stimuli.
Materials and Methods
Reagents and Cell Isolation.
The calpain inhibitors, N-acetyl-Leu-Leu-Nle-CHO (ALLN) and N-acetyl-Leu-Leu-methioninal (ALLM), were resuspended at 10 mg/ml in 100% EtOH, and stocks were used within 48 h. PD150606 was resuspended in anhydrous DMSO and used within 30 days. Calpastatin peptide was used at 79 μM. The cell-permeable cathepsin inhibitor I was resuspended in 100% EtOH and used at 200 μM (11). The proteosome inhibitor, clasto-lactacystin β-lactone (12), was used at 5 μM. Purified porcine m- and μ-calpain and all above reagents were obtained from Calbiochem. Z-Leu-NVa-CONH-CH2-2-pyridyl (compound 73) and Z-Leu-Abu-CONH-(CH2)3-4-morpholinyl (compound 42), specific inhibitors of μ-calpain and m-calpain, respectively, were the kind gift of J. Powers (Georgia Institute of Technology, Atlanta) and used at 75 μM (13). Pertussis toxin (PTx) was obtained from List Biological Laboratories (Campbell, CA). Fibrinogen (Fbg), IL-8, and fMLP were from Sigma. The calpain indicator CMAC,t-BOC-LM (BOC; Molecular Probes) was used at 10 μM. Primary human neutrophils were obtained from healthy donors with informed consent as described (14).
HIV-TAT Constructs.
The pOPI3CID plasmid containing the calpastatin inhibitory domain (CID) was the generous gift of N. Forsberg (Oregon State University, Corvallis) (15). PCR primers (5′-GACACTTCCAACCAAGCTTGCGGCCGCCATG-3′ and 5′-TACTCGGAATTCTATCACACGCCGGTCTTCTTC-3′) containing EcoRI and NcoI sites were used to generate the CID sequence for insertion into the pTAT-hemagglutinin (HA) vector. The pTAT-HA and pTAT-GFP plasmids were the kind gift of S. Dowdy (Howard Hughes Medical Institute, St. Louis) (16, 17). Native purification of the fusion proteins was performed by using previously described methods (17).
Casein Gel.
m- and μ-calpain activities were detected by casein zymography using described methods (7, 18). Briefly, cells were collected and lysed (50 mM Hepes, pH 7.6/150 mM NaCl/1% Triton X-100/5 mM EDTA/10 mM 2-mercaptoethanol (2-ME)/4 μM PMSF/0.05 trypsin inhibitory units (TIU)/ml aprotinin/2 mM phenantroline/2 mM vanadate/phosphatase inhibitor mixture; Sigma). The samples were run on a nondenaturing polyacrylamide gel containing casein. Gel was incubated overnight in 25 mM Tris⋅HCL (pH 7.6) with 5 mM CaCl2 and 10 mM 2-ME, fixed, and stained with Coomassie brilliant blue.
Video Microscopy.
Neutrophils were pretreated for 20 min at 37°C in HyQ-CCM1 (CCM1; Hyclone) containing inhibitor or vehicle control. Samples were plated onto non-tissue culture-treated dishes coated with 2.5 μg/ml Fbg at 37°C for 30 min. IL-8 or fMLP were included for the last 5 min where indicated. Dishes were placed on a heated microscope stage with 10% CO2. Cells were imaged on an Olympus IX-70 inverted microscope (Olympus America, Melville, NY) using a ×20 objective. Images were collected by using a Hitachi (Tokyo) Denshi KP-M1U video camera and captured into scion image v1.62c every 15 s for 20 min by using an LG3 frame grabber (Scion, Frederick, MD).
Calpain Activity Assay.
To visualize calpain activity, neutrophils were preloaded for 20 min in CCM-1 containing treatment as above and 10 μM CMAC,t-BOC-LM at 37°C. Fluorescence was quantified by using a FACSVantage SE (Becton Dickinson).
Chemotaxis Assay.
Neutrophils were pretreated and plated as above. Chemoattractants were loaded into an Eppendorf Femptotip. A chemoattractant gradient was formed by slow release of the chemoattractant from the tip into the medium by using an Eppendorf FemptoJet microinjection system as described (3). Cells were viewed on a Nikon Eclipse TE300 inverted fluorescent microscope. Images were acquired with a Hamamatsu (Hamamatsu Photonics, Hamamatsu City, Japan) cooled charge-coupled device video camera using either a ×20 phase or a ×40 differential interference contrast microscopy objective and captured into metamorph v5.0 (Universal Imaging, Downingtown, PA).
Cell Tracking.
Cell centroid positions were marked in scion image, and cell movement was determined as a function of time in Microsoft excel v8.0. The mean square displacement 〈d2(t)〉 was calculated as a function of time for each cell in the field. Means of at least three separate experiments with at least 36 cells per treatment were tracked (average number of cells tracked was 55). These data were then analyzed by using a persistent random walk model to calculate cell speed and directional persistence (19, 20).
Directional Migration.
Neutrophil migration by transwell was determined by using standard methods (21). In brief, 3 μm pore transwell filters (Costar) were coated with 2.5 μg/ml Fbg. Neutrophils were pretreated for 20 min in CCM-1 containing inhibitor or vehicle control as described. CCM-1 alone or CCM-1 containing 1.25 nM IL-8 was placed in the bottom chamber, and 4 × 105 neutrophils were placed in the top chamber and incubated at 37°C and 10% CO2 for 2 h. EDTA was then added to the top and bottom chambers. The number of neutrophils that migrated across the filter was determined and expressed relative to control.
Rho GTPase Activity Assays.
Cdc42 and Rac activity assays were performed as described (14). Neutrophils were incubated in CCM-1 containing vehicle or 50 μg/ml ALLN for 15 min. Cells were plated on 2.5 μg/ml Fbg for 10 min, and IL-8 or fMLP was added during the last 5 min when appropriate. The cells were lysed, and 300 μg of lysate was affinity precipitated with a PAK binding domain-GST fusion protein that binds to the active, GTP-bound form of Rac and Cdc42. The affinity-precipitated products were run on an SDS/PAGE gel, transferred to nitrocellulose, and blotted for Cdc42 or Rac.
Statistical Analyses.
In all cases, a two-tailed, paired Student's t test was used to compare the treated sample to its control. A value of P ≤ 0.05 was taken as significant.
Results
Calpain Inhibition Modulates Neutrophil Adhesion and Polarization.
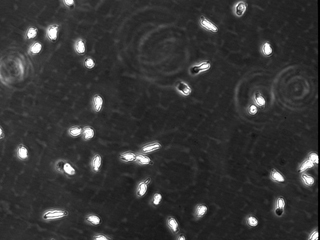
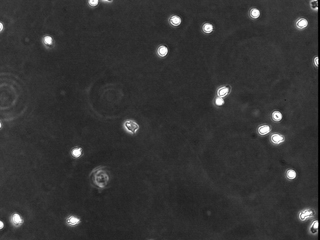
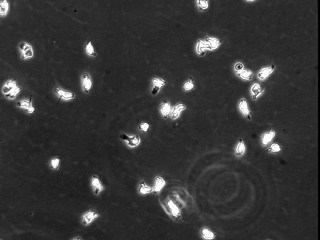
To determine whether calpain modulates neutrophil adhesion and polarization, primary human neutrophils from healthy donors were pretreated with different cell-permeant calpain inhibitors in the absence of exogenous activators. Unstimulated neutrophils typically do not spread on Fbg and do not form a polarized morphology. Calpain inhibition, in the absence of exogenous stimulation, induced neutrophil spreading and polarization (Fig. 1). Similar effects on morphology were obtained with three different compounds (ALLN, ALLM, and PD150606), as well as with calpastatin peptide, a biologically active peptide derived from the endogenous inhibitor of calpain activity, calpastatin. The changes in morphology did not occur in the presence of other protease inhibitors, including cathepsin inhibitor I or the proteosome inhibitor clasto-lactacystin β-lactone. Strikingly, using isoform-specific calpain inhibitors, this morphologic change was seen only after inhibition of μ-calpain but not m-calpain. Calpain inhibition also caused increased adhesion on 2.5 μg/ml Fbg (data not shown). The polar morphology and increased adhesion induced by calpain inhibitors was comparable to the effects observed in neutrophils treated with IL-8 and fMLP. Taken together, these data suggest that the calpain activity in resting neutrophils may function as a negative regulator of cell spreading and polarization.
Figure 1.

Calpain inhibition affects neutrophil morphology and adhesion. Neutrophils were incubated for 20 min in CCM-1 containing vehicle (a), 50 μg/ml (130 μM) ALLN (b), 50 μg/ml (125 μM) ALLM (c), 75 μM compound 73 (d), 1 μM PD150606 (e), 79 μM calpastatin peptide (f), 75 μM compound 42 (g), 200 μM cathepsin inhibitor I (h), or 50 μM clasto-lactacystin β-lactone (i). Cells were then plated onto 2.5 μg/ml Fbg for 30 min at 37°C. Morphology was compared with cells treated with 1.25 nM IL-8 (j) or 100 nM fMLP (k) as described in Materials and Methods. All samples were subsequently imaged by time-lapse videomicroscopy. A representative frame is shown. Bar = 50 μm.
Neutrophils Exhibit Constitutive Calpain Activity.
We analyzed the relative activities of the calpain isoforms in neutrophils by casein zymography. As shown in Fig. 2A, the predominant active isoform in neutrophils is μ-calpain in contrast to mouse embryonic fibroblasts, which display primarily m-calpain activity (7). Further, to determine calpain activity levels in resting neutrophils, we performed flow cytometry of live cells by using CMAC,t-BOC-LM, a fluorescent indicator that on cleavage by calpain causes increased fluorescence (8, 22, 23). In accordance with a previously published report (24), we found that calpain is constitutively active in resting neutrophils (Fig. 2B). Calpain activity was down-regulated in the presence of the calpain inhibitors ALLN, ALLM, compound 73, and compound 42 in a dose-dependent manner (Fig. 2B and data not shown). Further, by Western blot analysis, we found that resting neutrophils exhibit significant talin cleavage (data not shown). Together, the findings indicate that resting neutrophils exhibit basal levels of calpain activity that can be inhibited.
Figure 2.

Neutrophils exhibit constitutive calpain activity and predominantly express μ-calpain activity. (A) m- and μ-calpain activities were detected by casein zymography (7, 18). Samples included 2 μg (1.02 units) of purified porcine μ-calpain, neutrophil lysates, mouse embryonic fibroblast lysates, and 2.2 μg (0.675 units) of purified porcine m-calpain, as described in Materials and Methods. (B) Neutrophils were incubated for 30 min in CCM-1 containing 10 μM CMAC,t-BOC-LM and vehicle, 50 μg/ml ALLN, 50 μg/ml ALLM, 75 μM compound 73, or 75 μM compound 42. Fluorescence was quantified by using a FACSVantage cell sorter. Shown is the relative fluorescence intensity ± SD (n = 3).
Calpain Inhibition Increased Neutrophil Migration Speeds.
Neutrophil migration rates in the presence and absence of calpain inhibitors were examined to determine the effect of calpain inhibition on neutrophil motility (Fig. 3). Unstimulated neutrophils pretreated with vehicle control remained poorly adherent with minimal migration (45.0 ± 47 μm/hr). Pretreatment with calpain inhibitors increased cell migration rates dramatically (ALLN 515.3 ± 62.6, ALLM 516.1 ± 70.3, and calpastatin peptide 277.3 ± 40.7 μm/hr). These speeds were comparable with chemokinetic responses of neutrophils to IL-8 (428.9 ± 94.9 μm/hr) and fMLP (390.0 ± 35.2 μm/hr). Further, this enhanced chemokinesis upon calpain inhibition was shown to be induced in a dose-dependent manner (Fig. 3C). Treatment with cathepsin or proteosome inhibitors had no effect on neutrophil chemokinesis. In addition, transduction of a CID:HIV-TAT (CID-TAT) fusion protein also significantly enhanced neutrophil polarization and migration relative to cells transduced with a GFP-TAT control protein (362.2 ± 113 and 95.9 ± 39.1, respectively; Fig. 4). Interestingly, the μ-calpain-specific inhibitor compound 73 (576.7 ± 70.9 μm/hr), but not the m-calpain specific inhibitor compound 42 (228.3 ± 86.9 μm/hr) induced neutrophil polarization and chemokinesis. This finding suggests that μ-calpain is the isoform responsible for the chemokinetic effects seen in neutrophils. These data suggest that the high calpain activity constitutively present in resting neutrophils may suppress neutrophil adhesion and migration and act to maintain their unstimulated spherical phenotype.
Figure 3.

Effects of calpain inhibition on neutrophil migration. (A) Neutrophils were pretreated with EtOH, DMSO, 50 μg/ml ALLN, 50 μg/ml ALLM, 200 μM cathepsin inhibitor I, 79 μM calpastatin peptide, 75 μM cmpd73, 75 μM cmpd42, or 50 μM clasto-lactacystin β-lactone (lacta) and plated onto 2.5 μg/ml Fbg as above. Cells were imaged by time-lapse videomicroscopy, and the path of each cell was analyzed to determine cell speed as described. Data shown are the mean of three to four experiments ± SD. *, Significant differences from appropriate control (P < 0.05 by a paired, two-tailed Student's t test). See Movies 1–6 (for vehicle, IL-8, compound 73, compound 42, calpastatin peptide, and ALLN treatments), which are published as supporting information on the PNAS web site, www.pnas.org. (B) Cell paths as a function of time with each cell starting at (0,0) are shown for EtOH (Ba), ALLN (Bb), and IL-8 (Bc). (C) Neutrophils were treated with various concentrations of ALLN as above. Cell paths and resultant speeds were determined and plotted as a function of increasing ALLN concentration ± SD (n = 3).
Figure 4.

CID-TAT enhances neutrophil polarization and migration. Neutrophils were incubated in CCM-1 containing 3 μM CID-TAT or 3 μM GFP-TAT for 5 min. Cells were then plated on 2.5 μg/ml Fbg for 10 min, and cells were imaged by time-lapse videomicroscopy as in Fig. 3. Representative frames are shown for GFP-TAT (a) and CID-TAT (b). (c) Cell speeds were determined, and the means of three experiments are shown ± SD.
Calpain Modulates Neutrophil Chemotaxis to IL-8.
To further characterize the role of calpain in the migration of neutrophils, we examined the effects of calpain inhibition on the random and directional migration of neutrophils by using a transwell assay. In agreement with the observed increase in cell speeds after calpain inhibition, pretreatment of neutrophils with ALLN or the μ-calpain-specific inhibitor, but not the m-calpain-specific inhibitor, increased the random migration of neutrophils across a 3-μm pore filter coated with Fbg (Fig. 5). The effect may be isoform-specific because no effect was seen with the m-calpain inhibitor. However, chemotactic migration to IL-8 was not enhanced in the presence of calpain inhibitors relative to control migration. These findings suggest that directional migration to a chemoattractant may be disrupted after calpain inhibition.
Figure 5.

Calpain inhibition increases neutrophil migration across a membrane. Neutrophils were pretreated with vehicle (EtOH or DMSO), 50 μg/ml ALLN, 75 μM compound 73, or 75 μM compound 42 as in Fig. 1 and incubated in the absence (medium) or presence (IL-8) of a chemoattractant gradient across a 3-μm pore membrane. Migration is expressed relative to vehicle control. Shown is the mean of four experiments ± SEM. Statistical significance was determined by using a paired, two-tailed Student's t test. An “a” indicates significant difference as compared with control in the absence of chemoattractant (P < 0.05). A “b” indicates that no statistical differences were seen as compared with vehicle in the presence of a gradient of IL-8 (P > 0.05).
To determine whether calpain modulates chemotactic migration, live imaging studies were performed to observe neutrophil chemotaxis in a gradient of IL-8 or fMLP. A gradient of chemoattractant was established by using a micropipette tip filled with IL-8 or fMLP as described (3). Control neutrophils demonstrated a rapid chemotactic migration to IL-8 (Fig. 6 Aa–Ad) or fMLP (data not shown). Chemotactic migration to the pipette tip was reduced after calpain inhibition with 50–150 μg/ml ALLN (Fig. 6 Be–Bh), 100–150 μg/ml ALLM, and 80 μM PD150606 (data not shown). The effects on chemotaxis were reversible and dose dependent, requiring higher concentrations of inhibitor to disrupt chemotaxis than those required to induce chemokinesis. A minimum of 50 μg/ml ALLN was required to affect chemotaxis, with maximal reduction occurring at 150 μg/ml (data not shown). In contrast, a concentration of 50 ng/ml ALLN was sufficient to enhance chemokinesis (Fig. 3C). The average migration speed in the presence of calpain inhibitors was comparable to that of the control cells that migrated directionally to IL-8 (data not shown), but the cells failed to respond to the gradient of IL-8 and instead demonstrated a random chemokinetic migration pattern. Similar results were observed with fMLP (data not shown). Directional persistence, a measure of the time a cell migrates in one direction using a random walk model (20), is decreased when calpain is inhibited in a chemotactic gradient (Fig. 6C). Together, the findings suggest that optimal chemotactic migration toward IL-8 and fMLP requires calpain activity.
Figure 6.

Effect of calpain inhibition on neutrophil chemotaxis to IL-8. Neutrophils were pretreated with EtOH (Aa–Ad) or 150 μg/ml ALLN (Ae–Ah) as above. IL-8 was loaded into a FemptoTip, and a chemotactic gradient was generated. Neutrophils were then imaged at ×200 by time-lapse videomicroscopy. (A) Images were captured at 15-s intervals for 20 min. Bar = 50 μm. See Movies 7 and 8, which are published as supporting information on the PNAS web site, for vehicle and ALLN treatments. (B) As in Fig. 4, cell centroid positions were determined, and the cell paths are indicated relative to the tip of the micropipet (0,0). Filled squares indicate the starting points of the cells. (C) Directional persistence was determined by tracking at least 90 cells for each condition. The mean directional persistence in seconds ± SEM is shown. *, P < 0.05 by the paired, two-tailed Student's t test.
GPCR Signaling Is Not Required for the Effect of Calpain Inhibition on Chemokinesis.
To investigate the possibility that IL-8 receptor signaling modulates the activity of calpain, we examined the effects of PTx, the G-protein inhibitor, on neutrophil chemokinesis induced by calpain inhibition. Although IL-8-induced neutrophil migration was blocked by PTx, migration induced by calpain inhibition was not diminished by PTx (Fig. 7). These findings suggest that GPCR are not required for the effects of calpain on chemokinesis and that calpain functions downstream of GPCR during chemokinetic migration.
Figure 7.

Calpain may act downstream of GPCRs. Neutrophils were pretreated for 1 h in CCM-1 containing 0.5 mg/ml PTx or vehicle at 37°C. Subsequently, 50 μg/ml ALLN or vehicle was added, and cells were plated on 2.5 μg/ml Fbg for 25 min. IL-8 was added during the last 5 min to appropriate dishes, and cells were imaged as in Fig. 4. Neutrophils were tracked, and cell speeds were determined. The means of three experiments ± SD are shown. *, Significant differences by two-tailed Student's t test (P < 0.05).
Calpain Inhibition Increases Cdc42 and Rac Activities.
To determine how calpain modulates neutrophil migration, we examined the effects of calpain inhibition on the activity of the Rho GTPases Cdc42 and Rac. Using a pull down assay, we found that both Cdc42 and Rac activities were low in resting neutrophils (Fig. 8). On stimulation with IL-8 or fMLP, the activities of both GTPases were increased. Additionally, on inhibition of calpain activity with ALLN, the activities of both Cdc42 and Rac were also increased. These data suggest that the constitutive activity of calpain found in resting neutrophils may inhibit the activation of Cdc42 and Rac, thereby providing a global inhibitory signal that inhibits neutrophil protrusion and migration.
Figure 8.

Calpain inhibition enhances Cdc42 and Rac activity. Neutrophils were incubated in CCM-1 containing vehicle or 50 μg/ml ALLN for 20 min, and 1.25 nM IL-8 or 100 nM fMLP was added during the last 5 min as indicated. The cells were then spun and lysed, and 300 μg of lysate were incubated with GST beads containing the PAK binding domain. The samples were the analyzed by SDS/PAGE, transferred to nitrocellulose, and probed for Rac or Cdc42. Shown is a representative blot from three experiments.
Discussion
Chemotactic migration involves both the sensing of chemoattractant gradients and the subsequent activation of signaling cascades that allow for directed migration (25–27). Neutrophil chemotaxis and extravasation are important for effective inflammatory responses. The essential role of chemoattractants and the activation of seven-transmembrane GPCRs during the chemotactic migration of leukocytes have been clearly documented (28, 29). However, the signaling mechanisms that regulate neutrophil chemotaxis remain elusive. Our data suggest that calpain regulates the directed migration of neutrophils along gradients of chemoattractant by regulating the activities of Cdc42 and Rac.
To our knowledge, there have been no previous demonstrations that calpain inhibition can enhance cell speed. In contrast to all other cell types examined to date, calpain inhibition promotes neutrophil polarization and migration. In fibroblasts, calpain inhibition limits migration rates by impairing cell detachment. The differential regulation of integrin at the rear of migrating fibroblasts and neutrophils may account for this discrepancy (30). The primary fate of integrin at the rear of migrating fibroblasts is a severing of the link between integrin and the cytoskeleton, with integrin footprints remaining behind on the substratum as a cell migrates (31, 32). Calpain inhibition in fibroblasts reduces the amount of integrin left behind on the substratum after detachment (5, 33). In contrast, the majority of integrin is endocytosed and recycled at the rear of neutrophils (34, 35). Accordingly, inhibition of calpain does not tether neutrophils or limit their migration speed. Further, because fibroblasts display higher levels of m-calpain and neutrophils have more μ-calpain, the possibility exists that these isoforms play distinct roles during cell migration.
Impaired cell spreading has been observed with many cell types in the presence of calpain inhibitors (6, 9, 10). This result is also true for Capn4−/− embryonic fibroblasts, which have reduced spreading on a fibronectin substratum (7). However, neutrophils display enhanced spreading after calpain inhibition. The differential effects on cell spreading and migration may reflect the context in which calpain is inhibited. Unlike fibroblasts, which express low baseline calpain activity, calpain is active in resting neutrophils. An optimum level of calpain activity may be required for cell spreading and migration, with decreased cell spreading observed at both high and low calpain activities, suggesting a biphasic relationship between calpain activity and maximum cell spreading and migration.
Our data suggest that calpain functions as a negative regulator of neutrophil polarization and migration in resting neutrophils. Treatment with calpain inhibitors or CID-TAT, but not inhibitors of other proteases, promotes pseudopod formation and rapid neutrophil chemokinesis in the absence of exogenous activators. These findings suggest that constitutive calpain activity in resting neutrophils may function to suppress neutrophil protrusion and spreading (Fig. 9). Evidence supporting this finding is provided by our observation that resting neutrophils exhibit constitutive levels of μ-calpain activity. Previous work in fibroblasts shows that the chemokine interferon-inducible protein 10 down-regulates calpain activity (22). It is possible that IL-8 also down-regulates calpain activity. Further evidence to support this possibility is the requirement of GPCR for IL-8-mediated chemokinesis but not for chemokinesis induced by calpain inhibition. Together, these findings suggest that IL-8 may affect neutrophil spreading and polarization by modulating the activity of calpain and that this modulation is likely to take place downstream of GPCRs.
Figure 9.

Schematic of calpain regulation during neutrophil migration.
A role for calpain in protrusion and leading edge formation has been suggested by recent investigations (7, 9). In Capn4−/− embryonic fibroblasts, the cells form prominent filopodia. However, a clear role for calpain in pseudopod formation and directed cell migration has not previously been reported. Our results clearly demonstrate that inhibition of calpain promotes pseudopod formation and cell polarization. Further, inhibition of calpain up-regulates the activities of the Rho GTPases Cdc42 and Rac. These findings suggest that calpain acts upstream of Rac and Cdc42 to inhibit their activity, thereby acting as a negative regulator of cell protrusion and migration by modulating the activities of Cdc42 and Rac (Fig. 9).
Establishment of cell asymmetry is essential for polarized cell migration. In contrast to the effects observed with chemokinesis, calpain inhibition reduced chemotaxis and directional persistence to IL-8. The apparent discrepant effects of calpain inhibition on neutrophil chemokinesis and chemotaxis can be explained by postulating a role for calpain in mediating a negative regulatory pathway critical for optimum chemotaxis along a gradient of chemoattractant. Recent studies support a central role for the temporal and spatial regulation of positive and negative regulators of chemotaxis, including regulators of 3-phosphoinositides, phosphatidylinositol 3-kinase, and PTEN (36–39). It is intriguing to speculate that calpain activity is asymmetrically distributed in chemotaxing cells, with localized changes in activity required for efficient chemotaxis. Previous studies have demonstrated that calpain activity is enhanced by PIP2 (4), suggesting that calpain activity may be regulated by localized changes in lipid composition downstream of GPCRs. We hypothesize that calpain activity in neutrophils is regulated asymmetrically during chemotaxis as a result of GPCR signaling via polyphosphoinositides. Loss of PIP2 at the leading edge in response to a gradient of chemoattractant may decrease calpain activity, whereas calpain activity at the cell's rear remains high. This asymmetric calpain activity may serve to direct chemotaxis by regulating protrusion events, allowing Cdc42 and Rac activation at the leading edge while preventing their activation at the rear.
Supplementary Material
Acknowledgments
We thank Julie Sedgwick and Kristyn Jansen for phlebotomy services and Nic Lehmann-Ziebarth for his invaluable assistance in cell tracking. We also thank Drs. N. Forsberg and S. Dowdy for constructs. This work was supported by National Institutes of Health Grants RO1CA85862-01 and 1PO1AI50500-01 and American Heart Association Grant O255769N.
Abbreviations
- Fbg
fibrinogen
- fMLP
formyl-Met-Leu-Phe
- GPCR
G protein-coupled chemoattractant receptor
- ALLN
N-acetyl-Leu-Leu-Nle-CHO
- ALLM
N-acetyl-Leu-Leu-methioninal
- PTx
pertussis toxin
- CID
calpastatin inhibitory domain
- PIP
phosphatidylinositol phosphate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Servant G, Weiner O D, Neptune E R, Sedat J W, Bourne H R. Mol Biol Cell. 1999;10:1163–1178. doi: 10.1091/mbc.10.4.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ueda M, Sako Y, Tanaka T, Devreotes P, Yanagida T. Science. 2001;294:864–867. doi: 10.1126/science.1063951. [DOI] [PubMed] [Google Scholar]
- 3.Servant G, Weiner O D, Herzmark P, Balla T, Sedat J W, Bourne H R. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saido T C, Shibata M, Takenawa T, Murofushi H, Suzuki K. J Biol Chem. 1992;267:24585–24590. [PubMed] [Google Scholar]
- 5.Huttenlocher A, Palecek S P, Lu Q, Zhang W, Mellgren R L, Lauffenburger D A, Ginsberg M H, Horwitz A F. J Biol Chem. 1997;272:32719–32722. doi: 10.1074/jbc.272.52.32719. [DOI] [PubMed] [Google Scholar]
- 6.Kulkarni S, Saido T C, Suzuki K, Fox J E. J Biol Chem. 1999;274:21265–21275. doi: 10.1074/jbc.274.30.21265. [DOI] [PubMed] [Google Scholar]
- 7.Dourdin N, Bhatt A K, Dutt P, Greer P A, Arthur J S, Elce J S, Huttenlocher A. J Biol Chem. 2001;276:48382–48388. doi: 10.1074/jbc.M108893200. [DOI] [PubMed] [Google Scholar]
- 8.Glading A, Uberall F, Keyse S M, Lauffenburger D A, Wells A. J Biol Chem. 2001;276:23341–23348. doi: 10.1074/jbc.M008847200. [DOI] [PubMed] [Google Scholar]
- 9.Potter D A, Tirnauer J S, Janssen R, Croall D E, Hughes C N, Fiacco K A, Mier J W, Maki M, Herman I M. J Cell Biol. 1998;141:647–662. doi: 10.1083/jcb.141.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stewart M P, McDowall A, Hogg N. J Cell Biol. 1998;140:699–707. doi: 10.1083/jcb.140.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu D M, Uckun F M. Clin Cancer Res. 2000;6:2064–2069. [PubMed] [Google Scholar]
- 12.Dick L R, Cruikshank A A, Destree A T, Grenier L, McCormack T A, Melandri F D, Nunes S L, Palombella V J, Parent L A, Plamondon L, et al. J Biol Chem. 1997;272:182–188. doi: 10.1074/jbc.272.1.182. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Ortega-Vilain A C, Patil G S, Chu D L, Foreman J E, Eveleth D D, Powers J C. J Med Chem. 1996;39:4089–4098. doi: 10.1021/jm950541c. [DOI] [PubMed] [Google Scholar]
- 14.Cox E A, Sastry S K, Huttenlocher A. Mol Biol Cell. 2001;12:265–277. doi: 10.1091/mbc.12.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang J, Forsberg N E. Proc Natl Acad Sci USA. 1998;95:12100–12105. doi: 10.1073/pnas.95.21.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ezhevsky S A, Ho A, Becker-Hapak M, Davis P K, Dowdy S F. Mol Cell Biol. 2001;21:4773–4784. doi: 10.1128/MCB.21.14.4773-4784.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becker-Hapak M, McAllister S S, Dowdy S F. Methods. 2001;24:247–256. doi: 10.1006/meth.2001.1186. [DOI] [PubMed] [Google Scholar]
- 18.Arthur J S, Mykles D L. Methods Mol Biol. 2000;144:109–116. doi: 10.1385/1-59259-050-0:109. [DOI] [PubMed] [Google Scholar]
- 19.Palecek S P, Loftus J C, Ginsberg M H, Lauffenburger D A, Horwitz A F. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- 20.Maheshwari G, Lauffenburger D A. Microsc Res Tech. 1998;43:358–368. doi: 10.1002/(SICI)1097-0029(19981201)43:5<358::AID-JEMT2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto H, Sedgwick J B, Vrtis R F, Busse W W. Am J Respir Cell Mol Biol. 2000;23:379–388. doi: 10.1165/ajrcmb.23.3.3707. [DOI] [PubMed] [Google Scholar]
- 22.Shiraha H, Glading A, Gupta K, Wells A. J Cell Biol. 1999;146:243–254. doi: 10.1083/jcb.146.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosser B G, Powers S P, Gores G J. J Biol Chem. 1993;268:23593–23600. [PubMed] [Google Scholar]
- 24.Knepper-Nicolai B, Savill J, Brown S B. J Biol Chem. 1998;273:30530–30536. doi: 10.1074/jbc.273.46.30530. [DOI] [PubMed] [Google Scholar]
- 25.Devreotes P N, Zigmond S H. Annu Rev Cell Biol. 1988;4:649–686. doi: 10.1146/annurev.cb.04.110188.003245. [DOI] [PubMed] [Google Scholar]
- 26.Stossel T P. Science. 1993;260:1086–1094. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]
- 27.Downey G P. Curr Opin Immunol. 1994;6:113–124. doi: 10.1016/0952-7915(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 28.Murphy P M. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 29.Premack B A, Schall T J. Nat Med. 1996;2:1174–1178. doi: 10.1038/nm1196-1174. [DOI] [PubMed] [Google Scholar]
- 30.Cox E A, Huttenlocher A. Microsc Res Tech. 1998;43:412–419. doi: 10.1002/(SICI)1097-0029(19981201)43:5<412::AID-JEMT7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 31.Palecek S P, Schmidt C E, Lauffenburger D A, Horwitz A F. J Cell Sci. 1996;109:941–952. doi: 10.1242/jcs.109.5.941. [DOI] [PubMed] [Google Scholar]
- 32.Regen C M, Horwitz A F. J Cell Biol. 1992;119:1347–1359. doi: 10.1083/jcb.119.5.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palecek S P, Huttenlocher A, Horwitz A F, Lauffenburger D A. J Cell Sci. 1998;111:929–940. doi: 10.1242/jcs.111.7.929. [DOI] [PubMed] [Google Scholar]
- 34.Lawson M A, Maxfield F R. Nature. 1995;377:75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- 35.Pierini L M, Lawson M A, Eddy R J, Hendey B, Maxfield F R. Blood. 2000;95:2471–2480. [PubMed] [Google Scholar]
- 36.Hannigan M, Zhan L, Li Z, Ai Y, Wu D, Huang C K. Proc Natl Acad Sci USA. 2002;99:3603–3608. doi: 10.1073/pnas.052010699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Funamoto S, Meili R, Lee S, Parry L, Firtel R A. Cell. 2002;109:611–623. doi: 10.1016/s0092-8674(02)00755-9. [DOI] [PubMed] [Google Scholar]
- 38.Weiner O D, Neilsen P O, Prestwich G D, Kirschner M W, Cantley L C, Bourne H R. Nat Cell Biol. 2002;4:509–513. doi: 10.1038/ncb811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iijima M, Devreotes P. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}