

Abstract
The focus of human genetics in recent years has shifted toward identifying genes that are involved in the development of common diseases such as cancer, diabetes, cardiovascular diseases, and Alzheimer's disease. Because many complex diseases are late-onset, the frequencies of disease susceptibility alleles are expected to decrease in the healthy elderly individuals of the population at large because of their contribution to disease morbidity and/or mortality. To test this assumption, we compared allele frequencies of 6,500 single-nucleotide polymorphisms (SNPs) located in ≈5,000 genes between DNA pools of age-stratified healthy, European-American individuals. A SNP that results in an amino acid change from Ile to Val in the dual-specific A kinase-anchoring protein 2 (d-AKAP2) gene, showed the strongest correlation with age. Subsequent analysis of an independent sample indicated that the Val variant was associated with a statistically significant decrease in the length of the electrocardiogram PR interval. The Ile/Val SNP is located in the A-kinase-binding domain. An in vitro binding assay revealed that the Ile variant bound ≈3-fold weaker to the protein kinase A (PKA)-RIα isoform than the Val variant. This decreased affinity resulted in alterations in the subcellular distribution of the recombinantly expressed PKA-RIα isoform. Our study suggests that alterations in PKA-RIα subcellular localization caused by variation in d-AKAP2 may have a negative health prognosis in the aging population, which may be related to cardiac dysfunction. Age-stratified samples appear to be useful for screening SNPs to identify functional gene variants that have an impact on health.
Keywords: single-nucleotide polymorphism‖protein kinase A‖health risk factor‖ cardiac dysfunction‖morbidity
Past efforts to identify susceptibility genes for common diseases have considered association studies using markers of candidate genes in samples of cases and controls or linkage analyses using markers of genome variation in samples of pedigrees. Both of these approaches have critical limitations. Candidate gene studies are generally limited to a small number of genes already suggested to be involved in the disease pathway. They provide little opportunity for novel gene discovery in a disease field. Linkage analyses to study common diseases have proven to be very expensive, and in very few cases have they resulted in the identification of a disease susceptibility gene. The shared public and private efforts to identify and validate single-nucleotide polymorphisms (SNPs) genome-wide (1) and improvements in SNP measurement technologies have enhanced the feasibility of genome-wide association studies. Such studies are expected to be much more powerful than traditional linkage analysis methods for identifying genes involved in common diseases (2).
Rather than searching for genes based on their influence in a specific disease, our aim is to screen for susceptibility genes that have a significant influence on late-onset diseases in the general population. Instead of considering clinical samples for a conventional case-control study, we chose to sample healthy individuals ascertained through blood donation centers. Our hypothesis is that older individuals carrying disease susceptibility alleles are more likely to be absent from such a sample because they are less likely to be eligible for donating blood. Hence, the frequency of a disease susceptibility allele is expected to be lower in samples of older blood donors. Similarly, a protective allele would be seen at a higher frequency in the older sample. We define variants with a decreased frequency due to morbidity and, to a small extent, mortality as disease susceptibility alleles. The identification of longevity genes by comparing genetic markers between young and elderly individuals, usually octogenarians or centenarians, has become more popular in recent years (3). The well known age-related associations of Alzheimer's disease and risk of cardiovascular disease with the ɛ4 allele of the apolipoprotein E (ApoE) gene provides the initial proof of concept (4, 5). However, our approach is clearly distinct from the previous approaches because our samples are primarily selected as being healthy and not by age.
In this paper, we report the results of a screen comparing allele frequencies of 6,500 SNPs located in ≈5,000 genes between samples of young and elderly healthy individuals. This resulted in the identification of a gene encoding a functional variant with an impact on morbidity that may be involved in the etiology of cardiac dysfunction.
Materials and Methods
Study Sample.
All subjects involved in our studies signed a written informed consent and the institutional ethics committees of participating institutions approved the experimental protocols. Subjects for the disease susceptibility genome screen were part of a sample that was recruited during a routine blood donation from private blood collection centers in San Bernardino and Rancho Mirage, California. The staff of the blood collection agencies invited all healthy blood donors to participate, and helped the subjects fill out a consent form and a simple personal data collection form before sample collection. The data collection form included information about age, sex, body size, personal and family disease history, and ethnic background of both parents. Information about the identity of the study participants was not recorded. Subjects would be excluded if they failed to meet the blood donation eligibility guidelines established by the American Red Cross. A summary of the samples collected is presented in Table 1. Ethnicity was defined for each subject if they identified both parents as having the same ethnic/geographic background, otherwise they are indicated as “Other.” For the purpose of identifying disease susceptibility associated SNPs, we derived from this collection a discovery cohort consisting of male and female European Americans divided into young (18–39 years) and old (>60 years) groups. These groups and others used in this study are shown in Table 2.
Table 1.
Summary of the blood donor collection stratified by gender
| Female (n = 6,202) | Male (n = 4,766) | |
|---|---|---|
| Age, years | 39.8 (13.7)* | 43.2 (14.5)* |
| 18–39 | 0.53 (3,256)† | 0.44 (2115) |
| 40–59 | 0.39 (2,394) | 0.41 (1,953) |
| >60 | 0.09 (550) | 0.15 (698) |
| Ethnicity | ||
| African | 0.03 (167) | 0.02 (108) |
| Asian | 0.01 (92) | 0.02 (94) |
| European | 0.74 (4,582) | 0.77 (3,687) |
| Hispanic | 0.12 (770) | 0.10 (470) |
| Other | 0.10 (591) | 0.09 (407) |
| Donor disease | 0.06 (364) | 0.06 (292) |
Age is summarized by the mean and standard deviation (in parentheses).
All categorical variables are summarized as the column proportion and count (in parentheses).
Table 2.
Composition of age-, gender-, and ethnicity-stratified groups
| Group | Gender | n | Age range, years | Mean age (SD) |
|---|---|---|---|---|
| EA-YF | Female | 276 | 18–39 | 27.0 (6.68) |
| EA-YM | Male | 276 | 18–39 | 27.1 (6.65) |
| EA-OF | Female | 184 | 60–69 | 64.1 (2.84) |
| EA-OM | Male | 367 | 60–79 | 66.7 (4.78) |
| HI-YF | Female | 359 | 18–39 | 29.0 (6.42) |
| HI-YM | Male | 173 | 18–39 | 28.9 (6.67) |
| HI-OF | Female | 61 | 50–78 | 55.6 (5.66) |
| HI-OM | Male | 64 | 50–89 | 57.9 (7.49) |
| AF | Female/male | 97/97 | 18–76 | 37.4 (11.49) |
| AS | Female/male | 62/64 | 18–65 | 34.4 (12.18) |
EA, European American; HI, Hispanic American; AF, African American; AS, Asian American; YF, young female; YM, young male; OF, old female; OM, old male.
A follow-up study of the SNPs significantly associated with age in the genome scan was carried out in a sample of 417 white twin pairs from the adult twin registry at St. Thomas Hospital (London). Participants in this collection were enrolled without regard to health status as described (6). For this study, 97 traits were selected to explore possible disease associations. The selected traits have connections to many disease areas, including cardiovascular diseases, diabetes, hypertension, obesity, and osteoporosis.
DNA Extraction and Genotyping.
DNA from 10 ml of blood from each blood donor was extracted by using a desalting method (Gentra Systems), and quantitated fluorimetrically (Fluoroskan Ascent CF, Labsystems, Chicago) by using Pico green. Age-, gender-, and ethnicity-specific pools were generated by pooling equimolar amounts of each DNA sample to a final concentration of 5 ng/μl. All PCR and MassEXTEND reactions were conducted by using standard conditions, and both alleles were analyzed to calculate allele frequencies as described elsewhere (7–9).
Statistical Analysis.
Estimates of allele frequencies derived from pooled DNA were based on independent mass spectrometry measurements of four analyte aliquots derived from a single PCR reaction (8, 9). The median standard deviation for these values was ≈0.01. For comparing allele frequencies between the young and old pools, females and males were analyzed separately. The statistic used to test the difference in allele frequencies between pools was of the form
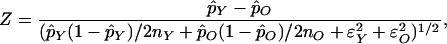 |
which follows a standard normal distribution. Here p̂Y and p̂O are the allele frequency estimates and ɛ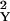 and ɛ
and ɛ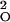 are estimates of measurement variability calculated from measurement replicates in the young and old pools, respectively. In this study, we made no attempt to correct for additional sources of variation or for multiple testing. Rather, SNPs were identified that had P values < 0.05 among all measured SNPs, followed by a second, independent measurement of all significant SNPs based on three separate PCRs of each DNA pool. The results of the second round of measurement were analyzed in a manner similar to the first round, and were compared for consistency. SNPs that showed statistically significant differences between young and old groups from pooled DNA analyses were individually genotyped for final validation.
are estimates of measurement variability calculated from measurement replicates in the young and old pools, respectively. In this study, we made no attempt to correct for additional sources of variation or for multiple testing. Rather, SNPs were identified that had P values < 0.05 among all measured SNPs, followed by a second, independent measurement of all significant SNPs based on three separate PCRs of each DNA pool. The results of the second round of measurement were analyzed in a manner similar to the first round, and were compared for consistency. SNPs that showed statistically significant differences between young and old groups from pooled DNA analyses were individually genotyped for final validation.
Estimates of allele frequencies using individual genotype data were found by using the gene counting method. Comparisons of allele frequencies as well as genotype frequencies between groups were carried out by using a χ2 test of independence.
The SNPs found to be associated with age were further analyzed for association with disease-related quantitative traits in the twin collection. The analysis was conducted by using a quantitative transmission-disequilibrium test as described by Abecasis and colleagues (10) to take advantage of the twin-based sample and to control for admixture and other nongenetic sources of variation. We implemented the form of the test that does not require the estimation of variance components. In our experience with this method, the inclusion of variance components usually has little effect on the resulting inferences. We did not use formal statistical procedures to account for multiple testing, but report the distribution of the resulting P values.
Sequence Alignment of the A-Kinase Binding (AKB) Domain of Duel-Specific A-Kinase-Anchoring Protein 2 (d-AKAP2).
Orthologous sequences were identified with PSI-blast (11) against the NCBI nonredundant database by using mouse d-AKAP2 (SwissProt O88845) as query sequence and a BLOSUM62 weight matrix (12). Sequences unequivocally identified as orthologues from Homo sapiens (SwissProt O43572), Sus scrofa (SwissProt P57770), Drosophila melanogaster (SW:Q9V3I3), and Caenorhabditis elegans (SwissProt Q10955) were obtained from SwissProt. Partial sequences from Anopheles gambiae (GenBank accession no. AJ281189) and Gallus gallus (GenBank accession no. AJ398916) were derived from GenBank EST sequences.
Recombinant d-AKAP2 Variant Construction and Functional Interactions with PKA Subunits.
GST fusion constructs were made by fusing the 40 C-terminal amino acids of d-AKAP2 to the C terminus of GST as described by Huang et al. (13) and subcloned between the NdeI and BamHI sites of pRSET (Invitrogen). The constructs were transfected into BL21 cells and expressed for 6 h at 20°C. The cells were lysed in PBS with 5 mM BME and 0.1% Triton X-100. Three microliters of supernatant were added to 200 μl of this buffer and 10 μl of glutathione beads. After three washes, bovine RIα (53 μg, 20 μM) and murine RIIα (2.4 μg, 2 μM) were added to the beads, respectively, and the total volume was adjusted to 40 μl. After incubating for 30 min at 4°C, the beads were washed three times, and separated in a 10% acrylamide gel.
Twenty-seven-residue peptides containing the two AKB domain variants of d-AKAP2 with a C-terminal cysteine were synthesized by SynPep (Dublin, CA). The peptides were HPLC purified and the molecular mass verified by mass spectrometry. Both peptides were labeled with tetramethylrhodamine-5-maleimide (Molecular Probes) at the cysteine residue and HPLC purified. RIα and RIIβ were serially diluted into 10 mM Hepes/150 mM NaCl/3 mM EDTA/0.005% polysorbate 20 containing 10 nM fluorescently labeled peptide. For RIIα tests, 1 nM labeled peptide was used because the binding affinity to RIIα was greater than that to RIα and RIIβ. The regulatory subunit concentrations were 1,000 − 0.5 nM for RIα, 500 − 0.1 nM for RIIβ, and 100 − 0.02 nM for RIIα. Fluorescence anisotropy was monitored by using a Fluoromax-2 (Spex, Jobin Yvon Horiba) equipped with polarizers. The fluorophore was excited at 541 nm (5- to 10-nm bandpass) and emission was monitored at 575 nm (5- to 10-nm bandpass). For each protein, fluorescence anisotropy was monitored for three separate binding experiments. The data were averaged and fit to a 1:1 binding model by using the nonlinear regression application in graphpad prism version 3.00 (GraphPad Software).
Subcellular Localization of d-AKAP2 Variants.
The 30-aa mitochondrial-anchoring domain of d-AKAP1 (14) was fused to the C-terminal 156 residues of mouse and human d-AKAP2, respectively, followed by a Flag-tag and subcloned into pcDNA4 (Invitrogen). The N terminus, including the dimerization/docking (D/D) domain of RIα (residues 1–109) and full-length RIIα were each fused with GFP and cloned into pEGFPN1 (BD CLONTECH). Equal molar ratios of the Flag-tagged d-AKAP2 constructs and either RIIα-GFP or RIα-D/D-GFP constructs were mixed and transfected into 10T(1/2) cells by using cytofectene (Bio-Rad). The AKB domain was detected by immuno-staining with monoclonal antibodies against the Flag-tag (Kodak) followed by rhodamine-conjugated secondary antibody (The Jackson Laboratory). The cells were imaged by using a Zeiss microscope equipped with a digital camera. Each channel was exposed for the same amount of time.
Results
Genome-Wide SNP Analysis for Morbidity Gene Discovery.
The limitations for conducting genome-wide association studies by genotyping each subject with a dense panel of genetic markers include high costs, use of relatively large amounts of DNA and the extensive time requirements for such an endeavor. We have pursued a strategy that utilizes estimates of allele frequencies in DNA pools to be able to screen large numbers of SNPs in a quick and affordable manner (8, 9, 15). To apply this to disease susceptibility gene discovery, we divided the European-American individuals by age (under 40 and over 60 years) and by sex (Table 2). The fraction of included subjects reporting any health problem was only 1.8% and 2.9% in young females and males, and 3.8% and 3.5% in old females and males, respectively. In this study, we used a collection of 6,500 exonic SNPs located in ≈5,000 genes or EST clusters. The majority of assays for these SNPs, originally identified in an in silico discovery project, were developed in collaboration with the National Cancer Institute (7).
There is some concern with the strategy of using samples collected through blood donation centers in a rapidly growing, highly mobile and ethnically diverse population such as Southern California to compare allele frequencies between young and old groups. One concern is potential inaccuracies in the self-reporting of ethnic or geographic background, particularly age-dependent biases. Another concern is that the older and younger generations may represent different compositions of geographic heritage. Such confounders could lead to differences in allele frequencies for reasons unrelated to health and lead to false inferences. To address these concerns, we obtained estimates of allele frequencies from a subset of 271 markers randomly selected from the larger SNP panel in the four European-American pools, as well as pools of subjects of reported African-American, Asian-American, and Hispanic-American ethnicity (described in Table 2). We used these estimates to calculate Nei's genetic distance between pools (16), which is illustrated in the neighbor-joining tree (17) in Fig. 1. The lengths of the branches are proportional to the pairwise genetic distances between pools. The distances between the old and young groups of European Americans are roughly equal and relatively small compared with the distances between the three other ethnic pools. This argues against the role of age-dependent population admixture as the explanation for the differences in allele frequencies observed between young and old groups.
Figure 1.

Neighbor-joining tree of genetic distances among the four European-American discovery groups (YF, YM, OF, and OM) and DNA pools from three additional ethnic groups. HI, Hispanic American; AF, African American; AS, Asian American; YF, young female; YM, young male; OF, old female; OM, old male. The lengths of branches are proportional to the pairwise genetic distances between pools.
Identification of d-AKAP2 as a Candidate Gene.
We identified >50 markers of the 6,500 tested markers that show a reproducibly significant allele frequency change between the two age groups in at least one gender (P < 0.05). The SNP that demonstrated the strongest association with age in both genders is located within the d-AKAP2 gene.
d-AKAP2 codes for dual-specific A-kinase anchor protein 2, which is part of a family of scaffold proteins known as A-kinase anchoring proteins (AKAPs) (13, 18). AKAPs bind the regulatory subunit of cAMP-dependent PKA, and target the kinase to various intracellular locations, localizing cAMP-mediated activation of the kinase (19). PKA is a broad specificity kinase and phosphorylates numerous proteins that function in many essential cellular processes such as metabolism, gene transcription, cell division, and neuronal transmission. In the inactive state, PKA is a tetramer consisting of two catalytic (C) and two regulatory (R) subunits. The dual specificity of d-AKAP2 is defined by its ability to bind both the RI and RII isoforms of PKA (13).
An A → G polymorphism in the 3′ UTR of d-AKAP2 showed a significant decrease of ≈8% (P < 0.01) of the G allele in the older sample of both genders (Table 3). The marker was individually genotyped and the frequency differences between young and old individuals calculated from the genotypes were very similar to the pool results. There was a slight skewing of frequencies in the pools likely resulting from uneven PCR amplification of the two alleles. This led to an underestimation of the G allele frequency in all pools but did not impact the significance of the differences between young and old (Table 3).
Table 3.
Comparison of allele frequencies between young and old groups
| Allele frequency
|
Difference | P value | ||
|---|---|---|---|---|
| Young | Old | |||
| Pool data | ||||
| 3′ UTR | ||||
| European female | 0.322 | 0.240 | 0.082 | 0.007 |
| European male | 0.362 | 0.286 | 0.076 | 0.002 |
| Genotype data | ||||
| 3′ UTR | ||||
| European female | 0.274 | 0.212 | 0.062 | 0.034 |
| European male | 0.304 | 0.232 | 0.072 | 0.004 |
| I646V | ||||
| European female | 0.402 | 0.318 | 0.084 | 0.009 |
| European male | 0.429 | 0.369 | 0.060 | 0.030 |
| Hispanic female | 0.445 | 0.316 | 0.129 | 0.008 |
| Hispanic male | 0.436 | 0.375 | 0.061 | 0.229 |
| In10 | ||||
| European female | 0.877 | 0.871 | 0.006 | 0.147 |
| European male | 0.880 | 0.865 | 0.015 | 0.360 |
Sample sizes are the same as those of the corresponding groups in Table 2. Allele frequencies for each SNP are given for the G allele.
To identify common polymorphisms in the d-AKAP2 gene, we sequenced the 15 exons and 100–200 bp of the flanking sequence (Fig. 4, which is published as supporting information on the PNAS website, www.pnas.org). The analysis of 36 chromosomes revealed only two additional polymorphisms: An A → G SNP in intron 10, six nucleotides downstream from exon 10 (In10), and an A → G SNP in exon 14, leading to an amino acid substitution Ile to Val at position 646 (I646V). Individual genotyping of the European-American samples showed that the intron 10 SNP exhibits no morbidity-association. The I646V polymorphism, however, was found to be significantly different between young and old in both males (P = 0.03) and females (P = 0.009) (Table 3). There was no significant difference between young males and females and between old males and females. We applied the Bayesian, coalescent theory-based method (20) to construct haplotypes at these three tightly linked sites for each subject. The estimates of the disequilibrium (D′) between the 3′ UTR and the I646V and In10 SNPs were 0.991 and 0.255 (r2 = 0.55 and 0.03), respectively. The distance between the markers in strong disequilibrium, 3′ UTR and I646V which both showed association, is ≈4 kb, whereas the intron 10 SNP is located ≈23 kb upstream of I646V (Fig. 4).
As expected, the changes in genotype frequencies between age groups for the three sites showed a similar level of statistical significance as the changes in allele frequencies (data not shown). At the 3′ UTR and the I646V variable sites, GG homozygotes were reduced and AA homozygotes increased in the older sample of both genders. This further supports the hypothesis that the G allele, which determines the Val allele at I646V, is associated with a negative health impact.
We also genotyped Hispanic-American samples for the I646V variation. Because we had only a small number of Hispanic-American individuals over 60 years of age, we extended the older sample to all individuals older than 50 years of age (Table 2). Although females showed a statistically significant allele frequency difference between old and young (P = 0.008), the males did not, which may be due to the relatively small number of male individuals (Table 3). However, the allele frequency differences in both genders were comparable to those observed in European Americans. The frequency of the G allele decreased by 0.129 in females and 0.061 in males, the GG homozygote by 0.087 in females and 0.072 in males (data not shown). These results further support the association of the Val allele with morbidity and/or mortality, and therefore the involvement of this gene in one or several disease processes. We subsequently verified another nonsynonymous d-AKAP2 variation, retrieved from dbSNP. The G–A transversion in exon 4 results in an Arg to His substitution at position 249 (R249H). The Arg was found to be in complete linkage disequilibrium with the Ile at position 646, occurring together in every case, and therefore shows the same age effect.
Association of d-AKAP2 Genotypes with a Cardiac Trait.
In an effort to identify traits correlated with the observed age association of the I646V SNP, we used a cohort of 417 fasting white twin pairs with extensive coverage for a variety of disease-related traits. Of the 97 traits analyzed, only the PR interval was statistically significant at a nominal level of 0.05. The estimate from the quantitative transmission-disequilibrium test model of the average effect of the G allele (Val) was to decrease the PR interval 6.3 units (P = 0.007). The genotype means in the subset of 207 informative twin pairs were 157 ± 23.4, 152 ± 26.9, and 146 ± 25.4 (mean ± SD) for genotypes AA, GA, and GG, respectively.
Functional Analysis of the d-AKAP2 Amino Acid Variant.
The variable position resulting from the SNP (I64V) is located in the AKB domain of d-AKAP2. This domain is the docking site for PKA (13) and is highly conserved among species (Fig. 2a). Similar to other AKAPs the AKB domain is predicted to form an amphipathic helix with hydrophobic amino acids on one face of the helix (21, 22). The length of the hydrophobic side chains on the hydrophobic face of the helix can alter PKA binding specificity (23, 24). The AKB domain of d-AKAP2 interacts with RI and RII isoforms of PKA and determines the extent of subcellular localization of PKA. The presence of two potential RGS (regulator of G protein signaling) motifs suggests the involvement of d-AKAP2 in G protein-mediated signal transduction (13, 18, 25). A putative PDZ-binding motif (TKL) at the C terminus is a protein-protein recognition module that plays a central role in organizing diverse cell signaling assemblies (26). The R249H variant is located in a spacer separating two halves of a potential RGS domain. The conservation of this position and surrounding sequence is very low compared with the AKB domain. Because there is no interaction partner identified for this spacer, the potential functional effect of R249H was not analyzed further.
Figure 2.

Characterization of the Ile/Val variant in the AKB domain of d-AKAP2. (a) Sequence alignment of the highly conserved AKB domain from several species. The residues comprising the hydrophobic face of the helix are highlighted as black boxes. The variable position I646V is part of the hydrophobic face (arrow). (b) Pull-down assays using the C-terminal, AKB-containing portion of d-AKAP2 fused to GST of mouse (Mo) and human (Hu). The Val variants at position 24 (V24) showed higher affinity to RIα as compared with the Ile forms (I24). Equal levels of GST-AKB proteins were estimated by SDS gel electrophoresis. An excess of GST-AKB was used so that the amount of R subunit bound to the glutathione beads reflects the binding affinities. Two separate experiments were performed to verify that GST-AKB proteins are in excess: when half the amount of GST-AKB was used, a similar amount of the R subunit was pulled down; when half of the amount of R protein was used, approximately half as much was bound. RIα and RIIα were used at concentrations of 2 and 20 μM, respectively. (c) Quantitative binding assay using a fluorescently labeled peptide of both the AKB variants. Binding of RIα to both the Ile (■) and Val (▴) variants is illustrated. The Val allele results in an increase in binding to the RIα isoform. The curves through the data are a fit to a 1:1 binding model. A deletion mutant of RIα (▵ 1–91) (♦), which lacks the AKAP binding domain, does not bind to either peptide. (Inset) The dissociation constants for the regulatory subunit isoforms.
To determine whether the d-AKAP2 Ile/Val variation resulted in an alteration in its binding properties to PKA, the binding of each variant to the regulatory subunits of PKA was investigated. Binding of each AKB domain variant to PKA was first examined by using an in vitro pull-down assay, in which the 40 C-terminal residues of d-AKAP2 containing the AKB were fused to glutatione S-transferase (GST) (Fig. 2b). The Ile/Val substitution resulted in an isoform-specific difference in PKA binding. The RIα isoform of PKA bound with a significantly higher affinity to the Val variant (Fig. 2b). The higher affinity was seen for both the mouse and the human AKB domains. The residue differences between mouse and human downstream of the Ile/Val position had no effect on the binding properties. Moreover, there was no difference in binding of the variants to the RIIα isoform (Fig. 2b).
To assess the magnitude of the affinity difference, binding of both variants to the R isoforms was analyzed in a quantitative assay. Peptides of each AKB variant were synthesized and fluorescently labeled. Binding of these peptides to the regulatory subunit isoforms was monitored by an increase in steady state fluorescence anisotropy (Fig. 2c). There was no difference in binding of the AKB variants to either the RIIα or RIIβ isoforms. In addition, the RII isoforms bound tighter to the variants than the RIα isoform (Fig. 2c Inset). However, as indicated by the pull-down experiments, RIα displayed differential binding to the AKB variants. The Val variant had a nearly three-fold increase in binding affinity when compared with the Ile variant. The interaction was specific because no binding was observed to a deletion construct of RIα that lacked the AKAP binding domain (delta1–91RIα).
To determine whether the observed in vitro affinity differences resulted in a difference in cellular compartmentalization, we examined the association of the AKB variants and the PKA regulatory subunits in cells. In this assay, the AKB domains of d-AKAP2 from mouse (Val) and human (Ile) were fused to the mitochondrial anchoring domain of d-AKAP1, respectively, and tethered to the outer mitochondrial membrane (27, 28). The binding of the AKB domain to the PKA regulatory subunits was detected as the cotransfected PKA regulatory domains colocalized to the mitochondria. As expected, both the Val and Ile AKB variants can target RIIα to the mitochondria effectively, in accordance with their similar affinity in vitro (Fig. 3 h and k). However, for RIα there was a difference in colocalization between the variants. The Val variant effectively targeted RIα to the mitochondria (Fig. 3b). The Ile variant, however, was unable to target RIα, which was evenly diffused in the cytosol (Fig. 3e) demonstrating that the Ile variant of d-AKAP2 has impaired ability to sequester RIα.
Figure 3.

Association of the AKB domain of d-AKAP2 with RIα and RIIα in 10T(1/2) cells. Through fusion with a mitochondrial targeting domain, the AKB domain directs different amounts of RI and RII to the mitochondria. The Val variant (V24) effectively binds RIα (a–c) and targets RIα-D/D-GFP to mitochondria (b). The Ile variant (I24) binds to RIα less effectively (d–f) and appears to be distributed evenly in the cells (e). In contrast, both variants show no apparent differences for RIIα (g–l) binding; they all colocalize at mitochondria (h and k).
Discussion
We have identified SNPs in the d-AKAP2 gene that are associated with morbidity by using a genome-wide association study from an age-stratified healthy population and 6,500 gene-based SNPs. The combined genetic and biochemical evidence points to the Ile/Val variant as the functional polymorphism. The Val variant is the apparent deleterious allele in a European-American cohort, and this finding is replicated in Hispanic Americans. The variant maps to the conserved AKB domain of the d-AKAP2 gene. The I646V variation impacts the binding to PKA in an isoform-specific manner both in vitro and in cells. The Ile variant binds three-fold weaker to the RIα isoform than to the Val variant. At the cellular level, this affinity difference results in a dramatic decrease in compartmentalization of RIα for the Ile variant. In the accompanying manuscript by Burns-Hamuro et al. (29), we provide further evidence that the Ile/Val variant is critical for binding to only the RIα isoform of PKA.
How the RIα specific binding differences of the d-AKAP2 variants translate into a health risk is likely to be complex. Compartmentalization by AKAPs is known to play an important role in the specificity and regulation of cAMP-mediated signaling of PKA (19). The cellular evidence presented here by using the mitochondrial-targeted form of d-AKAP2 suggests that the Ile variant has reduced ability to compartmentalize RIα PKA. Therefore, decreased compartmentalization of type I PKA may have an advantageous health effect. This effect could be direct by decreasing PKA signaling through d-AKAP2-Ile anchored PKA or indirect by redistributing the local pools of RIα in cells that have an important impact on health.
Increasing evidence suggests that the RI and RII isoforms of PKA have distinct functions (30). The RI isoform has been implicated in a variety of biological functions such as cell proliferation (31), tumor suppression (32, 33), immune regulation (34), and embryonic development (35, 36). In addition, the RIα isoform plays a significant role in maintaining cAMP regulation of PKA as evidenced by the embryonic lethality of mice deficient in the gene (35). Interestingly, these mice have defects in cardiac morphogenesis.
The correlation of the I646V variant with the EKG PR interval indicates that this polymorphism might be a predisposing factor for a cardiac phenotype. Individuals homozygous for the Val variant exhibit shorter atrium depolarization intervals (PR) than individuals homozygous for Ile. This phenotypic correlation combined with literature reports supporting a role for AKAP-mediated PKA signaling in normal cardiac function suggest plausible hints of how the Val variant may lead to pathogenesis.
PKA phosphorylates numerous substrates in response to β-adrenergic stimulation that influence the contractility of cardiac myocytes (37). Some of these substrates include the l-type Ca2+ channel, the ryanodine receptor, and phospholamban of the sarcoplasmic reticulum. AKAP-mediated targeting of PKA in cardiac myocytes has also been directly implicated in regulating cell contractility (38). Three AKAPs have been shown to interact with PKA in cardiac myocytes; muscle-selective AKAP (mAKAP), AKAP18 and Yotiao. mAKAP targets PKA to the perinuclear region of differentiated myocytes, coordinating both PKA and phosphodiesterase activity in a single complex (39). AKAP18 couples PKA to l-type Ca2+ channels, which enhances Ca2+ influx through the channel after β-adrenergic stimulation (40). Yotiao, an AKAP associated with N-methyl-d-aspartate (NMDA) receptors (41), has been shown to interact with the KCNQ1-KCNE1 K+ channel subunits in human hearts (42). Mutations in this channel associated with hereditary long QT syndrome abolish Yotiao interactions with the channel, and attenuate the response mediated by cAMP (42).
Direct involvement of PKA in developing heart disease comes from a recent study in which transgenic mice overexpressing the catalytic subunit of PKA developed dilated cardiomyopathy with reduced cardiac contractility and increased risk of arrhythmias (43). These cardiac abnormalities correlated with PKA-mediated hyperphosphorylation of the ryanodine receptor and Ca2+ release from the sarcoplasmic reticulum (43). RIα is likely to play a role in cardiac physiology because it is the predominant isoform associated with the sarcolemma in cardiac myocytes (44, 45). In fact, mutations in the RIα gene are associated with both familial cardiac myxomas and Carney complex, implicating this isoform in cardiac growth and differentiation (32, 33).
The PKA signaling pathways associated with d-AKAP2 are not known. However, d-AKAP2 contains a PDZ binding motif (TKL) at the C terminus (Fig. 2a), which could serve as a targeting domain to membrane-bound receptors, ion channels or ion exchangers (26), and two RGS domains, which could coordinate upstream G α signaling with downstream PKA signaling. d-AKAP2 might therefore be part of a signaling complex associated with a cardiac ion channel or ion exchanger. The d-AKAP2 variants could impact the phosphorylation state of the ion channel or ion exchanger by recruiting different amounts of PKA-RIα and might thereby modulate heart contraction. This hypothesis fits the observed association with an EKG phenotype. The shorter depolarization intervals for Val/Val individuals might be due to increased activation of ion channels in cardiac myocytes. However, the identification of this phenotypic association does not exclude the possibility that other phenotypic traits might be associated with this variant. Because it was identified by a general morbidity approach, it is possible that this functional variant has an impact on several biological pathways and can potentially lead to multiple clinical phenotypes.
In summary, we have demonstrated that screening for SNPs by using age-stratified healthy individuals is a useful and efficient way to identify functional variants in genes associated with morbidity. Because it is based on samples from the general population, this strategy may be able to identify major genetic components with significant health impacts and high penetrance. Through subsequently analyzing significant morbidity markers in another well-phenotyped healthy twin population, we were able to associate a cardiac phenotype with the d-AKAP2 genotypes. This genetics approach may be able to establish early entry points into understanding critical pathways that affect human health.
Supplementary Material
Acknowledgments
We thank T. D. Spector from St. Thomas Hospital, J. L. Fridey and D. B. Perez from the Blood Bank of San Bernardino, CA, and D. Witthaus and N. Brown from the Blood Bank Center in Rancho Mirage, CA, for their kind collaboration.The genetic analysis was partially supported by National Institutes of Health Grant CA83429 (to A.B.). The functional work was supported by National Institutes of Health Grant DK-54441 (to S.S.T.). L.L.B.-H. was supported by National Institutes of Health Training Grant 5T32 DK-07233. Sequenom, Inc., has an ownership interest in patent applications that have been filed and are directed to the subject matter of this paper.
Abbreviations
- d-AKAP2
dual-specific A kinase-anchoring protein 2
- SNP
single-nucleotide polymorphism
- AKB
A kinase binding
References
- 1.Holden A L. BioTechniques (Suppl.) 2002;22–24:26. [PubMed] [Google Scholar]
- 2.Risch N, Merikangas K. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 3.Puca A A, Daly M J, Brewster S J, Matise T C, Barrett J, Shea-Drinkwater M, Kang S, Joyce E, Nicoli J, Benson E, Kunkel L M, Perls T. Proc Natl Acad Sci USA. 2001;98:10505–10508. doi: 10.1073/pnas.181337598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haviland M B, Lussier-Cacan S, Davignon J, Sing C F. Am J Med Genet. 1995;58:315–331. doi: 10.1002/ajmg.1320580405. [DOI] [PubMed] [Google Scholar]
- 5.Schachter F, Faure-Delanef L, Guenot F, Rouger H, Froguel P, Lesueur-Ginot L, Cohen D. Nat Genet. 1994;6:29–32. doi: 10.1038/ng0194-29. [DOI] [PubMed] [Google Scholar]
- 6.Andrew T, Hart D J, Snieder H, de Lange M, Spector T D, MacGregor A J. Twin Res. 2001;4:464–477. doi: 10.1375/1369052012803. [DOI] [PubMed] [Google Scholar]
- 7.Buetow K H, Edmonson M, MacDonald R, Clifford R, Yip P, Kelley J, Little D P, Strausberg R, Koester H, Cantor C R, Braun A. Proc Natl Acad Sci USA. 2001;98:581–584. doi: 10.1073/pnas.021506298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bansal A, van den Boom D, Kammerer S, Honisch C, Adam G, Cantor C R, Kleyn P, Braun A. Proc Natl Acad Sci USA. 2002;99:16871–16874. doi: 10.1073/pnas.262671399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohlke K L, Erdos M R, Scott L J, Fingerlin T E, Jackson A U, Silander K, Hollstein P, Boehnke M, Collins F S. Proc Natl Acad Sci USA. 2002;99:16928–16933. doi: 10.1073/pnas.262661399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abecasis G R, Cardon L R, Cookson W O. Am J Hum Genet. 2000;66:279–292. doi: 10.1086/302698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaffer A A, Aravind L, Madden T L, Shavirin S, Spouge J L, Wolf Y I, Koonin E V, Altschul S F. Nucleic Acids Res. 2001;29:2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henikoff S, Henikoff J G. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang L J, Durick K, Weiner J A, Chun J, Taylor S S. Proc Natl Acad Sci USA. 1997;94:11184–11189. doi: 10.1073/pnas.94.21.11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang L J, Wang L, Ma Y, Durick K, Perkins G, Deerinck T J, Ellisman M H, Taylor S S. J Cell Biol. 1999;145:951–959. doi: 10.1083/jcb.145.5.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sham P, Bader J S, Craig I, O'Donovan M, Owen M. Nat Rev Genet. 2002;3:862–871. doi: 10.1038/nrg930. [DOI] [PubMed] [Google Scholar]
- 16.Nei M. Am Nat. 1972;106:283–292. [Google Scholar]
- 17.Saitou N, Nei M. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Sunahara R K, Krumins A, Perkins G, Crochiere M L, Mackey M, Bell S, Ellisman M H, Taylor S S. Proc Natl Acad Sci USA. 2001;98:3220–3225. doi: 10.1073/pnas.051633398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards A S, Scott J D. Curr Opin Cell Biol. 2000;12:217–221. doi: 10.1016/s0955-0674(99)00085-x. [DOI] [PubMed] [Google Scholar]
- 20.Stephens M, Smith N J, Donnelly P. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carr D W, Hausken Z E, Fraser I D, Stofko-Hahn R E, Scott J D. J Biol Chem. 1992;267:13376–13382. [PubMed] [Google Scholar]
- 22.Glantz S B, Li Y, Rubin C S. J Biol Chem. 1993;268:12796–12804. [PubMed] [Google Scholar]
- 23.Miki K, Eddy E M. J Biol Chem. 1999;274:29057–29062. doi: 10.1074/jbc.274.41.29057. [DOI] [PubMed] [Google Scholar]
- 24.Angelo R G, Rubin C S. J Biol Chem. 2000;275:4351–4362. doi: 10.1074/jbc.275.6.4351. [DOI] [PubMed] [Google Scholar]
- 25.De Vries L, Zheng B, Fischer T, Elenko E, Farquhar M G. Annu Rev Pharmacol Toxicol. 2000;40:235–271. doi: 10.1146/annurev.pharmtox.40.1.235. [DOI] [PubMed] [Google Scholar]
- 26.Harris B Z, Lim W A. J Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- 27.Huang L J, Durick K, Weiner J A, Chun J, Taylor S S. J Biol Chem. 1997;272:8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- 28.Chen Q, Lin R Y, Rubin C S. J Biol Chem. 1997;272:15247–15257. doi: 10.1074/jbc.272.24.15247. [DOI] [PubMed] [Google Scholar]
- 29.Burns-Hamuro L L, Ma Y, Kammerer S, Reineke U, Self C, Cook C, Olson G L, Cantor C R, Braun A, Taylor S S. Proc Natl Acad Sci USA. 2003;100:4072–4077. doi: 10.1073/pnas.2628038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skalhegg B S, Tasken K. Front Biosci. 2000;5:D678–D693. doi: 10.2741/skalhegg. [DOI] [PubMed] [Google Scholar]
- 31.Cho Chung Y S, Pepe S, Clair T, Budillon A, Nesterova M. Crit Rev Oncol Hematol. 1995;21:33–61. doi: 10.1016/1040-8428(94)00166-9. [DOI] [PubMed] [Google Scholar]
- 32.Casey M, Vaughan C J, He J, Hatcher C J, Winter J M, Weremowicz S, Montgomery K, Kucherlapati R, Morton C C, Basson C T. J Clin Invest. 2000;106:R31–R38. doi: 10.1172/JCI10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirschner L S, Carney J A, Pack S D, Taymans S E, Giatzakis C, Cho Y S, Cho-Chung Y S, Stratakis C A. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 34.Torgersen K M, Vang T, Abrahamsen H, Yaqub S, Tasken K. Cell Signaling. 2002;14:1–9. doi: 10.1016/s0898-6568(01)00214-5. [DOI] [PubMed] [Google Scholar]
- 35.Amieux P S, Howe D G, Knickerbocker H, Lee D C, Su T, Laslo G S, Idzerda R L, McKnight G S. J Biol Chem. 2002;277:27294–27304. doi: 10.1074/jbc.M200302200. [DOI] [PubMed] [Google Scholar]
- 36.Amieux P S, McKnight G S. Ann NY Acad Sci. 2002;968:75–95. doi: 10.1111/j.1749-6632.2002.tb04328.x. [DOI] [PubMed] [Google Scholar]
- 37.Xiao R P, Cheng H, Zhou Y Y, Kuschel M, Lakatta E G. Circ Res. 1999;85:1092–1100. doi: 10.1161/01.res.85.11.1092. [DOI] [PubMed] [Google Scholar]
- 38.Fink M A, Zakhary D R, Mackey J A, Desnoyer R W, Apperson-Hansen C, Damron D S, Bond M. Circ Res. 2001;88:291–297. doi: 10.1161/01.res.88.3.291. [DOI] [PubMed] [Google Scholar]
- 39.Dodge K L, Khouangsathiene S, Kapiloff M S, Mouton R, Hill E V, Houslay M D, Langeberg L K, Scott J D. EMBO J. 2001;20:1921–1930. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraser I D, Tavalin S J, Lester L B, Langeberg L K, Westphal A M, Dean R A, Marrion N V, Scott J D. EMBO J. 1998;17:2261–2272. doi: 10.1093/emboj/17.8.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westphal R S, Tavalin S J, Lin J W, Alto N M, Fraser I D, Langeberg L K, Sheng M, Scott J D. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 42.Marx S O, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks A R, Kass R S. Sceince. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 43.Antos C L, Frey N, Marx S O, Reiken S, Gaburjakova M, Richardson J A, Marks A R, Olsen E N. Circ Res. 2001;89:997–1004. doi: 10.1161/hh2301.100003. [DOI] [PubMed] [Google Scholar]
- 44.Robinson M L, Wallert M A, Reinitz C A, Shabb J B. Arch Biochem Biophys. 1996;330:181–187. doi: 10.1006/abbi.1996.0240. [DOI] [PubMed] [Google Scholar]
- 45.Reinitz C A, Bianco R A, Shabb J B. Arch Biochem Biophys. 1997;348:391–402. doi: 10.1006/abbi.1997.0401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.