

Abstract
A kinase-anchoring proteins (AKAPs) coordinate cAMP-mediated signaling by binding and localizing cAMP-dependent protein kinase (PKA), using an amphipathic helical docking motif. Peptide disruptors of PKA localization that mimic this helix have been used successfully to assess the involvement of PKA in specific signaling pathways. However, these peptides were developed as disruptors for the type II regulatory subunit (RII) even though both RI and RII isoforms can bind to AKAPs and have discrete functions. To evaluate the effects of each localized isoform, we designed peptides that specifically bind to either RI or RII. Using a peptide array, we have defined the minimal binding sequence of dual specific-AKAP 2 (d-AKAP2), which binds tightly to both RI and RII. Side-chain requirements for affinity and isoform specificity were evaluated by using a peptide substitution array where each position along the A kinase binding domain of d-AKAP2 was substituted by the other 19 l-amino acids. This array comprises 513 single-site substitution analogs of the d-AKAP2 sequence. Peptides containing single and multiple mutations were evaluated in a quantitative fluorescence binding assay and a cell-based colocalization assay. This strategy has allowed us to design peptides with high affinity (KD = 1–2 nM) and high specificity for RIα versus RIIα. These isoform-specific peptides will be invaluable tools to evaluate functional differences between localized RI and RII PKA and are RIα-specific disruptors. This array-based analysis also provides a foundation for biophysical analysis of this docking motif.
Keywords: dual-specific A kinase-anchoring protein‖Ht31‖peptide array
As we begin to unravel the complex signaling networks in the cell, some of the greatest challenges will be to determine the functional importance of isoform diversity and signaling through organized microdomains. In addition, an added layer of difficulty has become apparent in recent years. Many vital protein–protein interactions within these structured microdomains are regulated in a highly dynamic fashion, which can be, for instance, both cell type and cell cycle dependent. In the case of cAMP-dependent protein kinase (PKA), it is the regulatory subunits that control, in large part, both isoform diversity and subcellular localization. PKA is a tetramer consisting of two catalytic subunits and one regulatory subunit dimer. The regulatory subunit maintains the catalytic subunit in the inactive state and couples cAMP binding to kinase activation (1). In addition, the regulatory subunit localizes the kinase to specific subcellular locations via its interaction with A kinase-anchoring proteins (AKAPs) (2).
There are four regulatory subunit isoforms of PKA, type I (RI) (α,β) and type II (RII) (α,β). Each contains the same overall domain organization, but differs in cAMP responsiveness and particular subcellular localization. The RI isoforms are predominantly diffuse in the cytoplasm and are more sensitive to cAMP signaling (requiring lower levels of cAMP for kinase activation) whereas the RII isoforms are more localized in cells and less responsive to cAMP signaling (3–5). However, the RI isoform is also controlled via subcellular localization (6–9), suggesting that both cytosolic and localized pools of RI are important for its function. The localization of RII isoforms occurs through interactions with many different AKAPs (2), whereas RI isoforms interact with a small subset of AKAPs (9–13). In general, AKAPs bind RIα much weaker than RIIα and with a much faster off-rate in vitro (14), (L.L.B.-H, J. Cànaves, D. Blumenthal, and S.S.T., unpublished work). This finding suggests that RI targeting may be more dynamic than RII and may explain the inability to readily detect RI/AKAP interactions with traditional gel-overlay assays.
Abundant evidence demonstrates that the regulatory subunits are not functionally redundant (15). RIα, in particular, has been implicated in cancer (16) and immune cell regulation (17) and is the only isoform that when knocked out in mouse models results in embryonic lethality (18). Recent reports also suggest that RIα can function in tumor suppression (19, 20). Mutations in the RIα gene that result in haploinsufficiency of RIα are found in patients with familial cardiac myxomas and Carney complex, an autosomal dominant disorder associated with benign tumor formation throughout various tissues in the body (19, 20). Additionally, RIα compensates when other isoforms are deleted in the mouse to guarantee that there is no unregulated catalytic activity (15). Thus, of the R subunits, RIα appears to be unique and essential. To further support the functional differences between these isoforms, a recent finding revealed that a single nucleotide polymorphism (SNP) associated with morbidity and mortality was identified in the dual-specific A kinase-anchoring protein 2 (d-AKAP2), also known as AKAP10 (21). The amino acid change associated with this polymorphism is located in the kinase binding domain and effects only RIα isoform binding. These unique RI-specific functions, in addition to the SNP analysis, prompted us to design isoform-specific inhibitors of PKA localization.
The prototypic inhibitor that has been used in the past to disrupt PKA localization is the peptide known as Ht31 (22–24). The sequence for this peptide was derived from a human thyroid AKAP, and the peptide is used frequently to assess the involvement of localized PKA (2). However, this peptide has the potential to disrupt both RI- and RII- mediated localization (14, 25). In an effort to develop a RI isoform-specific localization inhibitor, a peptide derived from d-AKAP2 was used as a template to design peptides that offer both high affinity and selectivity for either RI or RII. Using an array of all single-site substitution analogs, we have evaluated a 27-aa sequence encompassing the A kinase binding (AKB) domain of d-AKAP2. The minimal sequence required for binding each isoform was evaluated by using N- and C-terminal truncations of the AKB domain and specific isoform differences were identified in the C-terminal region of the peptide. Side-chain requirements for a given affinity and selectivity were evaluated by using a peptide substitution array in which all 20 amino acids were substituted into each position along the AKB. Candidate isoform-specific peptides were synthesized and evaluated in a quantitative fluorescence binding assay and a cell-based colocalization assay. Using this approach, we successfully designed RI- and RII-specific binding peptides in addition to a null peptide; these peptides can be used to evaluate RI- and RII-localized PKA in cells.
Materials and Methods
Peptide Synthesis and Fluorescence Labeling.
The WT D-AKAP2 peptide (VQGNTDEAQEELAWKIAKMIVSDVMQQ) for the in-solution peptide binding assays was synthesized by SynPep (Dublin, CA). The following peptides were synthesized by Anaspec (San Jose, CA): VQGNTDEAQEELLWKIAKMIVSDVMQQ, VQGNTDEAQEELAWKIEKMIWSDVMQQ, VQGNTDEAQEELAWKIAKMIWSDVMQQ, and Ac-DLAWKIAKMIVSDVMQQ.
Multiple substitution peptides (PV-37, -38, -47, -48, and -49) were synthesized by Peptron (Daejeon, South Korea). All peptides contained a C-terminal Cys for conjugation of the fluorescence probe and contained an amide-protected C terminus. The peptides were HPLC-purified and the molecular mass was verified by MS. Peptide purities were >95%.
Each peptide was fluorescently labeled by using a 25-mM solution of tetramethylrhodamine-5-maleimide (catalog no. T-6027, Molecular Probes) dissolved in DMSO. The peptides were labeled by incubating with a 3-fold molar excess of the label for 16 h at 4°C in 20 mM Tris, pH 7.0, and 1 mM Tris-(2-carboxyethyl) phosphine, hydrochloride (TCEP; nonthiol reducing agent, Molecular Probes). The sample was quenched with 1 mM β-mercaptoethanol to bind to any unreacted maleimide and diluted with 0.1% trifluoroacetic acid for purification by HPLC. The labeled peptides were resolved by using a C18 column with a water/acetonitrile gradient containing 0.1% trifluoroacetic acid. The concentration of each peptide was determined by absorbance at 541 nm after diluting into 100% methanol and by using an extinction coefficient of 91,000 M−1⋅cm−1 for absorbance of the rhodamine label at 541 nm (Molecular Probes). The peptides were stored at 4°C in 50% acetonitrile.
Protein Expression and Purification.
Murine RIIα was expressed in Escherichia coli BL21 (DE3). Bovine RIα was expressed in E. coli 222. The proteins were purified as described by using a cAMP affinity resin (26). The protein concentrations were determined by using the following extinction coefficients at 280 nm, which were calculated with a standard concentration of protein calibrated by using quantitative amino acid analysis: RIα, 52,603 M−1⋅cm−1 and RIIα, 62,456 M−1⋅cm−1. The proteins were stored at 4°C in 50 mM Mes (pH 5.8), 50 mM NaCl, 2 mM EDTA, 2 mM EGTA, and 2 mM DTT.
The dimerization/docking (D/D) domain of bovine RIα (residues 1–109) and mouse RIIα (residues 1–46), fused to GFP, were subcloned into a pRSET expression vector (Invitrogen) downstream of a histidine tag. The proteins, GFP-RIα D/D and GFP-RIIα D/D, were expressed in E. coli BL21 (DE3) and purified with Talon (CLONTECH) resin. The his tag was cleaved by using thrombin, and the protein was further purified by using an S75-Sephadex (16/60) gel filtration column (Amersham Pharmacia) in 50 mM Mes (pH 5.8), 50 mM NaCl, 2 mM EDTA, 2 mM EGTA, and 2 mM DTT. The protein was stored at 4°C.
Fluorescence Anisotropy.
Binding of each fluorescently labeled peptide to the regulatory subunits was monitored by using fluorescence anisotropy. RIα and RIIα were serially diluted beginning at 1 and 0.1 μM, respectively, into 10 mM Hepes (pH 7.4), 0.15 M NaCl, 3 mM EDTA, and 0.005% Surfactant P20 (Biacore, Uppsala) containing either 10 or 1 nM of fluorescently labeled peptide for RIα and RIIα, respectively. The samples were equilibrated for at least 1 h at room temperature and fluorescence anisotropy was monitored with a Fluoromax-2 (Jobin-Yvon Horiba, Longjumeau, France) equipped with Glan-Thompson polarizers. The rhodamine-labeled peptide was excited at 541 nm (5- to 10-nm bandpass) and emission was monitored at 575 nm (5- to 10-nm bandpass). The anisotropy was calculated directly with the Fluoromax software by using the following equation:
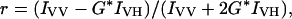 |
1 |
where r is the steady-state anisotropy, IVV is the fluorescence intensity with the excitation and emission polarizers oriented in the vertical position (0° from normal), IVH is the fluorescence intensity with the excitation polarizer in the vertical position and the emission polarizer oriented in the horizontal position (90°) relative to the excitation polarizer, and G is the monochromator grating factor that is equal to (IHV/IHH), with the first subscript indicating the position of the excitation polarizer and the second subscript indicating position of the emission polarizer. Three separate binding experiments were averaged and fit to a 1:1 binding model by using the nonlinear regression application in graphpad prism version 3.00 (GraphPad, San Diego).
Peptide Array Synthesis.
Cellulose-bound peptide libraries were automatically prepared according to standard SPOT synthesis protocols (27) by using a SPOT synthesizer (Abimed Analysentechnik, Langenfeld, Germany) as described (28, 29). In brief, the peptides were synthesized on an amino functionalized cellulose membrane as distinct spots. A β-alanine dipeptide spacer was inserted between the C terminus of the peptide and the membrane support. The peptide loading of the membranes was reduced by mixing 10% fluorenylmethoxycarbonyl-β-alanine-pentafluorophenolester (OPfp) and 90% acetylated β-alanine-OPfp active esters for the first coupling step. This peptide loading was optimized in advance by varying the fluorenylmethoxycarbonyl-β-alanine-OPfp percentage from 0.1% to 50%. The effects of peptide loading on the assay performance have been described (30). The peptide was extended stepwise by using standard fluorenylmethoxycarbonyl solid-phase peptide synthesis followed by cleavage of the side-chain protecting groups under trifluoroacetic acid conditions. Sequence files were generated with the software digen (Jerini AG). All peptides were N-terminally acetylated. For synthesis quality control, a selection of peptides that was synthesized in duplicate was cleaved from the solid support by ammonia vapor in the dry state (29). Subsequently, identity was verified by matrix-assisted laser desorption ionization–MS (Voyager-DE, Applied Biosystems).
Peptide Array Screening.
The peptide arrays (Figs. 1 and 2) were preincubated with T-TBS blocking buffer (TBS, pH 8.0/0.05% Tween 20 in the presence of blocking reagent; Roche Diagnostics chemiluminescence detection kit 1500694). Subsequently, the peptide arrays were incubated with solutions of GFP-RIα D/D domain or GFP-RIIα D/D domain (see above for expression and purification) at a final concentration of 1.0 μg/ml for 2 h in T-TBS blocking buffer. After washing three times for 10 min with T-TBS the anti-GFP antibody 3E6 (Quantum Biotechnologies, Montreal) was added to a final concentration of 1 μg/ml in T-TBS blocking buffer for 1 h followed by washing three times for 10 min with T-TBS. Finally, the arrays were incubated with a second anti-mouse IgG peroxidase-labeled antibody (catalog no. A5906, Sigma), which was applied at a concentration of 1 μg/ml in T-TBS blocking buffer for 1 h, followed by washing three times for 10 min with T-TBS. Analysis and quantification of peptide-bound GFP-RIα D/D- or GFP-RIIα D/D-antibody complexes was done by using a chemiluminescence substrate (see above) and the LumiImager (Roche Diagnostics). All steps were carried out at room temperature. Binding of the detection antibodies to the peptides was excluded by control incubations with antibody 3E6 and the anti-mouse IgG peroxidase-labeled antibody alone (data not shown). For a given membrane, binding of GFP-RIα D/D domain was performed first. Subsequently, the membrane was regenerated by using detergent (28), and complete removal of the GFP-RIα D/D domain was proven by a control incubation with antibody 3E6 and the anti-mouse IgG peroxidase-labeled antibody alone (data not shown). Afterward, binding of GFP-RII D/D domain to the peptide array was assessed.
Figure 1.

N-terminal (A) and C-terminal (B) truncations of the 27-residue AKB domain of D-AKAP2. Truncated peptides were synthesized by using SPOT synthesis on cellulose membrane as described in Materials and Methods. Binding was evaluated by incubating each membrane with GFP-RIα D/D domain and GFP-RIIα D/D domain as indicated. Bound protein was detected by using a primary antibody against GFP and enzyme-conjugated secondary antibody for amplification of signal. The membrane was then analyzed by chemiluminescence.
Figure 2.

Peptide substitution array of the 27-residue AKB domain of d-AKAP2 prepared by SPOT synthesis. All 20 amino acids (top of blot) were substituted into each position along the AKB domain (left side of blot). The sequences corresponding to the left column of each array are identical and represent the unsubstituted peptide. Other spots are single substitution analogs. Binding was evaluated by using a conjugated antibody system for both RIα (A) and RIIα (B). The residues highlighted in red show a decreased tolerance for substitutions at these positions. The red bar to the left of each blot highlights this region. Yellow boxes indicate those residues that disrupt binding to RIα while maintaining binding to RIIα. Green circles indicate those positions that enhance binding to RIα while disrupting binding to RIIα. Highlighted in blue are substituted proline residues.
Cell-Based Assay for Regulatory Subunit/AKAP Colocalization.
The targeting constructs of the AKB domain were made by fusing the C-terminal 156 residues of mouse d-AKAP2 with the N-terminal mitochondrial-targeting domain of d-AKAP1 (residues 1–30 of d-AKAP1a) to the N terminal (10, 31). After adding a Flag tag to the C terminus, the constructs were subcloned into pCI (Promega). The mutations of the AKB domain were made with the QuickChange mutagenesis method (32). Bovine RIα and mouse RIIα were fused with GFP by subcloning into pEGFPN1 (CLONTECH). Equal amounts of the targeting constructs and either GFP-RIα or GFP-RIIα constructs were cotransfected into 10T(1/2) cells with Lipofectamin (Invitrogen). The AKB domain was detected by immuno-staining with mAbs against the Flag tag (Kodak) followed by a rhodamine-conjugated secondary antibody (The Jackson Laboratory). The cells were imaged with a radiance confocal microscope (Bio-Rad).
Results
Defining the Minimal Sequence for Regulatory Subunit Binding.
The minimal sequence required for regulatory subunit binding was assessed by using N- and C-terminal truncations of the 27-residue human d-AKAP2 sequence. The truncations were synthesized by SPOT synthesis onto cellulose membrane, and binding was evaluated for both GFP-RIα and GFP-RIIα D/D domain constructs (Fig. 1). The C-terminal truncations defined clearly the C-terminal boundary for binding to the isoforms. There was an absence of binding to both regulatory subunits at a defined residue from the C terminus. For RIα, binding abruptly stopped after the C-terminal isoleucine (VQGNTDEAQEELAWKIAKMIVSDI), suggesting that the C-terminal (… MQQ) residues are not required for binding (Fig. 1B). Interestingly, this C-terminal isoleucine residue is the location of a single nucleotide polymorphism of d-AKAP2, which codes for either a valine or isoleucine at this position (21). For RIIα, binding abruptly stopped at the upstream valine position (VQGNTDEAQEELAWKIAKMIV), suggesting that more C-terminal residues (… SDIMQQ), which contain the polymorphism, are dispensable for the RIIα binding site (Fig. 1B). This finding is consistent with the polymorphic site only having an effect on binding to the RIα isoform (21). The N-terminal truncations did not result in a clear-cut boundary, but rather there was a titrateable decrease in signal for both isoforms starting at the glutamine (QEELAWKIAKMIVSDIMQQ) (Fig. 1A). This finding suggested that the N-terminal negative charges play a role in enhancing the affinity to both isoforms.
Amino Acid Residues Required for Binding.
To assess the requirement of individual side chains to bind to the regulatory subunits, peptide arrays were synthesized containing substitutions of all other 19 l-amino acids at each position in the 27-residue AKB domain of d-AKAP2, which contained valine at the allelic position. One of the key features of this array is the ability to identify which positions along the d-AKAP2 sequence are less tolerable to substitution. Distinct periodicities of the less tolerable substitutions for RIα and RIIα were immediately apparent (Fig. 2). The periodicities are indicated by the red bar for RIα binding (Fig. 2A) and RIIα binding (Fig. 2B). If projected onto a helical wheel, the substitutions that are not tolerated (Fig. 2, indicated in red) would be located on the same side of the amphipathic helix and presumably would provide direct contacts with the regulatory subunit. Despite the higher affinity of RIIα for d-AKAP2, fewer residues appear critical for binding (Fig. 2, highlighted with red bar). This finding suggests a smaller contact surface for d-AKAP2 binding to RIIα. It is consistent with recent hydrogen/deuterium exchange experiments, showing reduced protection of the AKB domain of D-AKAP2 when bound to RIIα relative to RIα (L.L.B.-H., Y. Hamuro, J. Kim, P. Sigala, S.S.T., and V. Woods, Jr., unpublished work). The periodicity of the substitution array and the reduced binding affinity for the proline substitutions (Fig. 2, blue box), which are known helix disruptors, confirmed that a peptide helical motif is essential for binding to each regulatory subunit isoform.
Selectivity Using Amino Acid Substitutions.
In addition to defining the helical motif critical for binding, the peptide array revealed several key substitutions along the helix that either enhanced binding to RIα relative to RIIα (Fig. 2, green circles) or disrupted binding to RIα while maintaining RIIα binding (Fig. 2, yellow box). For example, valine at position 21, when substituted with a tryptophan, abolished binding to RIIα but maintained affinity for RIα. In contrast, a single substitution at position 13 (A13L), abolished binding to only the RIα isoform. Several other substitutions at positions 9, 12, 21, and 25 seemed to enhance binding to RIα, while reducing binding to RIIα.
Using these isoform-selective positions as guides, several peptides were synthesized with the desired selectivity by using single or multiple substitutions and their activity was tested in a quantitative fluorescence binding assay (Fig. 3). The binding affinities of three peptides designated RIIα-specific (A13L) and RIα-specific (Q9F, V21W, and M25F) and null (A17E and V21W) were compared with the unsubstituted peptide (Fig. 3 and Table 1). The A13L substitution did not affect binding to RIIα, but dramatically affected binding to RIα, establishing this peptide as an RIIα-specific peptide, AKB(RII) (Table 1). The triple substituted RIα-specific peptide not only resulted in a considerable reduction in affinity to RIIα, but also enhanced binding to RIα by a factor of 10 (Table 1). The null peptide bound very poorly to RIα and binding to RIIα could not be detected (Fig. 3).
Figure 3.

Binding of AKB(dual) (■), AKB(RI) (Q9F, V21W, M25F) (⧫), AKB(RII) (A13L) (▴), and AKB(null) (●) peptides to full-length RIα (A) and RIIα (B). Each peptide was fluorescently labeled and incubated with the corresponding regulatory subunit for 1 h in 10 mM Hepes, 150 mM NaCl, 3 mM EDTA, pH 7.4. Fluorescence anisotropy was used to monitor bound peptide.
Table 1.
Dissociation constants (KD) with standard error (n = 3) for peptides binding to RIα and RIIα determined by using a fluorescence anisotropy binding assay
| Peptide | RIα, nM | RIIα, nM |
|---|---|---|
| AKB(dual), WT | 48 ± 4 | 2.2 ± 0.2 |
| VQGNTDEAQEELAWKIAKMIVSDVMQQ | ||
| AKB(RII), RII specific | 2,493 ± 409 | 2.7 ± 0.1 |
| VQGNTDEAQEELLWKIAKMIVSDVMQQ | ||
| AKB(RI), RI specific | 5.2 ± 0.5 | 456 ± 33 |
| FEELAWKIAKMIWSDVFQQ | ||
| AKB(null) | 998 ± 66 | >10,000 |
| VQGNTDEAQEELAWKIEKMIWSDVMQQ |
Substituted residues are underlined and in bold.
Several additional truncated and substituted peptides were synthesized and their affinity to RIα and RIIα was evaluated (Fig. 4). To determine whether the N-terminal negative charges were important for high-affinity binding as suggested from the truncation data, a truncation peptide was synthesized that did not contain the two N-terminal Glu residues, but instead contained an N-terminal α-acetylated Asp. This peptide also showed reduced binding to both regulatory subunits, confirming the requirement of at least one of the N-terminal negative charges. Also evident from these mutatations is the importance of the bulky hydrophobic tryptophan at position 21 in selectively reducing the affinity to the RII isoform. The single mutant V21W showed dramatically reduced binding to RIIα, while only showing modestly decreased affinity toward RIα (Fig. 4). This position is therefore a critical position along the helix for establishing RI/RII selectivity. In the background of V21W, further substitutions at positions 9 and 25 dramatically enhanced binding to RIα while further disrupting RIIα binding (Fig. 4, PV-37 and PV-38). RIIα also seemed to be less tolerant of the Leu-to-Phe substitution at position 12 (Fig. 4, PV-47 and PV-48). PV-49, which is identical to PV-38 except that it has an Ile instead of a Trp at position 21, bound with the greatest affinity to RIα (Fig. 4). However, this peptide also bound very tightly to RIIα. This finding again reinforced that a bulky aromatic residue at position 21 was important to select against RIIα binding. Interestingly, the Phe at positions 9 and 25 are only disruptive to RIIα binding when Trp is present at position 21 (PV-38). When Trp is replaced with Ile, the affinity for RIIα is restored and the affinity for RIα is further enhanced (PV-49). Thus Trp at position 21 (i) and the Phe at position 25 (i + 4) may interact to form additional unfavorable interactions for RIIα binding.
Figure 4.

Binding dissociation constants (KD) of selected mutant peptides of d-AKAP2. Binding was evaluated for both RIα and RIIα by using fluorescence anisotropy as described in Fig. 3. Substituted residues are bold. Tryptophan at position 21, highlighted with the arrow, is important for discriminating against binding to the RIIα subunit. PV-38 is designated the RI-specific binding peptide, AKB(RI) and is indicated by an asterisk.
Colocalization of Regulatory Subunits with AKB(Dual), AKB(RII), AKB(RI), and AKB(Null) in Cells.
To test the ability of these mutations to colocalize with selected isoforms in cells, a Flag-tagged AKB domain construct targeted to the mitochondria was prepared by using the AKB domain and a mitochondria targeting sequence from D-AKAP1 (10, 31). Mutations were then incorporated into the AKB domain of D-AKAP2 to test for selectivity in the cell. By concentrating D-AKAP2 at the mitochondria, we could easily detect colocalization of the AKB domain and the R isoforms. GFP constructs of RIα and RIIα were cotransfected into 10T (1/2) cells with Flag-tagged WT AKB(dual), AKB(RII), AKB(RI), and AKB(null). All of the AKB domains localized well to the mitochondria (Fig. 5 Bottom). The AKB(dual) was able to recruit both GFP-RIα (Fig. 5 a and i) and GFP-RIIα (Fig. 5 e and m) to the mitochondria. The RII-specific peptide, AKB(RII), recruited RII to the mitochondria but was incapable of recruiting GFP-RI (Fig. 5 b and j). In contrast, the RI-specific peptide, AKB(RI), only localized GFP-RI to the mitochondria (Fig. 5 c, g, k, and o); GFP-RII was not localized by the targeted AKB(RI) peptide. The null peptide, AKB(null), could not colocalize either GFP-RIα or RIIα (Fig. 5 d, h, l, and p).
Figure 5.

Isotype-specific mutations of AKB domain interact with RI or RII distinctively. The mitochondrial-targeting constructs containing WT AKB (q and u), RII-specific mutation (r and v), RI-specific mutation (s and w), or null mutation (t and x) were cotransfected with RIα-GFP (i–l) or RIIα-GFP (m–p), respectively. The interactions between AKB and RI or RII are reflected by the localization of the GFP tag on mitochondria (i, k, m, and n), whereas the noninteracting combinations give a diffused pattern (j, l, o, and p). (a–h) The composite images of AKB and R in the same cells.
Discussion
The dynamic signaling pathways that exist in the cell are mediated primarily by protein–protein interactions that typically involve small peptide motifs that bind to modular docking domains (33, 34). Polyproline sequences binding to Src homology 3 domains (35), C-terminal tripeptide motifs binding to PDZ domains (36), and phosphotyrosine peptides binding to Src homology 2 domains (37) are only a few examples of specific docking partners that are used to build modular networks in a highly combinatorial manner. Amphipathic helices are also extremely important for mediating protein–protein interactions in biology, not only for establishing stable dimeric and trimeric complexes through leucine zipper motifs (38), but also for dynamic interactions in signaling pathways. Some examples include nuclear export signals (39), mitochondrial targeting motifs (40), and the amhipathic helix of the PKA-specific protein kinase inhibitor (41). Although the sequences coding for the formation of antiparallel and parallel leucine zipper dimers and trimers have been elucidated (38), we do not yet understand the molecular basis for the specificity of these other signaling helices. Furthermore, the sequence requirements for the AKAP helical motifs are less rigid than the leucine zipper motifs.
The amphipathic nature of the AKAP binding motif was realized through deletion and mutagenesis studies, which suggested a structural conservation of the regulatory subunit binding site rather than a consensus sequence binding motif (22, 23, 42). The AKAP helix docks to the surface of the regulatory subunit D/D domain, which consists of an antiparallel four-helix bundle docking module (43). Subsequent structural studies showing peptides docked to the RII D/D domain confirmed the helical nature of the AKAP binding motif (44). Two different AKAP motifs, Ht31 and AKAP79, bind similarly to the surface of the RIIα D/D domain with only modest changes in the D/D domain upon binding, suggesting a preformed docking surface. There currently is no detailed structural information for the RI D/D–AKAP complex, but structural studies of the free RI D/D domain reveals differences in the surface topology of this domain relative to the RII D/D domain (45). Type I has dynamic N-terminal helical extensions, which partially occlude the AKAP binding surface and suggest that the entire process of an AKAP peptide docking to the surface is more dynamic for RIα than for RIIα (P. Banky and P. Jennings, personal communication). The AKAP binding surface of RIα is further constrained by disulfide bonds that crosslink the N-terminal extensions from each protomer with helix I and helix I′ of the docking surface. The surface charges are also quite different for RI and RII. The RII docking surface is primarily hydrophobic, whereas the RI contains several charged residues, which reduce the hydrophobic nature of this surface. The sequence and structural differences between RI and RII D/D domains suggest differences in the mechanism of AKAP binding.
As a first step to elucidate specificity of docking to RIα and RIIα, we used a peptide-based array strategy. d-AKAPs are unique in that they have the ability to bind both RI and RII subunits. Using the high-affinity binding site of d-AKAP2 as a starting point, we modulated the affinity of this d-AKAP by incorporating single or multiple mutations within the sequence to give a desired selectivity for a specific regulatory subunit isoform. Peptide arrays allowed us to rigorously define the minimal binding sequence required for regulatory subunit binding and allowed us to screen for mutations along the AKB domain that give a desired affinity and selectivity to each regulatory subunit. This strategy revealed not only differences in the size of the hydrophobic surface that is required for docking to the two isoforms, but also revealed differences in the specific amino acids that can contribute either positively or negatively to docking. Using this approach we successfully designed amphipathic helices, which are highly selective for binding RIα versus RIIα. We have designated these peptides AKB(RI) and AKB(RII), respectively, and have shown that these peptides have the desired affinity and selectivity in vitro and are highly selective in colocalizing the respective regulatory subunit in cells when targeted to the mitochondria.
The ability to modulate the binding selectivity of d-AKAP2 suggests that there are distinct modes for AKAP binding to the surface of the two isoforms. The RII/AKAP surface is dominated by hydrophobic interactions as seen from the structural data (44) and from the observed increases in binding affinity with increases in solution ionic strength (L.L.B.-H., J. Cànaves, D. Blumenthal, and S.S.T., unpublished work). This is not the case for RIα. The binding surface for RIα is different not only in surface topology, but also in its electrostatic character. It will be important to understand how the greater surface charges on the RIα docking surface and the steric constraints of the N-terminal extensions contribute to the reduced affinity for AKAPs. The peptide array data presented here demonstrate that the contact surface area between the docking surface of the R subunit and the AKAP was larger for the RIα interaction than for the RIIα interaction. However, a caveat with interpreting this data is that removing a residue completely or substituting one residue with another not only has direct effects on binding caused by side-chain changes, but also has an indirect effect on helix-forming propensity of the substituted or truncated peptide, which can also alter its binding properties. With this in mind, the N- and C-terminal truncation data suggest that at least 14 residues define the high-affinity binding sequence for RIα. Only 12 residues define the binding site for RIIα. In addition, the N-terminal region of the helix, which contains two glutamic acid residues seemed to enhance the binding affinity to both isoforms with a gradual decrease in affinity as each of these residues was separately deleted. From the peptide substitution array, the more C-terminal region of the helix seemed important for contacts with the RIIα surface, whereas for RIα the critical residues extended the length of the helix. This model is reinforced by the substitution at position 21 at the C terminus (V21W), which dramatically affected binding to RIIα, with only a modest decrease in RIα binding. This finding suggests that the binding energy for RIIα is concentrated at the C terminus. This idea is consistent with the structural data of Ht31 and AKAP79 bound to RIIα D/D, in which there is differential ordering of the ends of the helix on the surface of the D/D domain (44).
The selectivity of the triple mutant peptide (PV-38) for RIα is intriguing and suggests that interactions between Trp at position 21 (i) and Phe at position 25 (i + 4) are important for selectivity. PV-49, which is identical to PV-38 except that it contains an Ile instead of a Trp at position 21, is no longer selective for binding RIα, but rather binds tightly to both isoforms. Interactions between hydrophobic side chains are known to stabilize helices (46). Creamer and Rose (46) have calculated the free energy of interaction of hydrophobic side chains within α-helices and have found that significant interactions, both stabilizing and destabilizing, occur at (i, i + 3) and (i, i + 4) (40). According to their calculations, the Trp (i) and Phe (i + 4) pair in PV-38 would stabilize the helix, whereas the Ile (i) and Phe (i + 4) pair in PV-49 would destabilize the helix. However, because PV-38 binds much weaker to RIIα compared with PV-49, and PV-49 binds very tightly to both isoforms, enhanced helix stabilization does not directly translate into enhanced binding affinity for the R/AKAP interaction. Although further biophysical analysis is clearly required to explain this phenomenon it is nevertheless apparent that docking of RIα and RIIα is different.
Using a peptide array approach we were able to design high-affinity peptides that have absolute specificity for RIα or RIIα. These peptides will be useful probes to evaluate AKAP-mediated PKA-I and PKA-II localization and evaluate to what extent isoform diversity contributes to signaling specificity. We now have a peptide probe that can selectively disrupt targeting of RIα. This work also provides a basis for developing a theoretical understanding of the principles underlying the mechanisms for differential binding of the AKAP to each isoform. Finally, this study provides a useful framework for designing peptide mimetics that could potentially be useful for therapeutic intervention of isoform-specific mediated diseases.
Acknowledgments
We thank Patricia Jennings and Donald Blumenthal for their careful review of the manuscript and their helpful comments; Siv Garrod, Mira Sastri, and Simon Brown from the Taylor Laboratory for their expertise in purifying some of the peptides and proteins used in this study; and Marén Schlief for technical assistance in the peptide array synthesis. This work was funded by National Institutes of Health Grant DK-54441 (to S.S.T.). L.L.B.-H. was funded by National Institutes of Health Training Grant 5T32 DK-07233. Sequenom, Inc., has an ownership interest in patent applications that have been filed and are directed to the subject matter of this article. Provid Pharmaceuticals has received an equity investment from Sequenom and will receive research funding to develop novel drugs based on genetic targets discovered by Sequenom and is eligible to receive milestones and royalties on future products, the foregoing including intellectual property encompassed by the patent applications in which Sequenom has an ownership interest. Sequenom, Inc., was a financial contributor through its equity investment in and research support of Provid Pharmaceuticals in addition to the support of experiments conducted by Jerini AG.
Abbreviations
- PKA
cAMP-dependent protein kinase
- AKAP
A kinase-anchoring protein
- d-AKAP
dual-specific AKAP
- RI and RII
type I and type II regulatory subunits, respectively
- D/D
dimerization/docking
- AKB
A kinase binding
References
- 1.Taylor S S, Buechler J A, Yonemoto W. Annu Rev Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 2.Colledge M, Scott J D. Trends Cell Biol. 1999;9:216–221. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- 3.Doskeland S O, Maronde E, Gjertsen B T. Biochim Biophys Acta. 1993;1178:249–258. doi: 10.1016/0167-4889(93)90201-y. [DOI] [PubMed] [Google Scholar]
- 4.Feliciello A, Gottesman M E, Avvedimento E V. J Mol Biol. 2001;308:99–114. doi: 10.1006/jmbi.2001.4585. [DOI] [PubMed] [Google Scholar]
- 5.Skalhegg B S, Tasken K. Front Biosci. 2000;5:D678–D693. doi: 10.2741/skalhegg. [DOI] [PubMed] [Google Scholar]
- 6.Barradeau S, Imaizumi-Scherrer T, Weiss M C, Faust D M. Proc Natl Acad Sci USA. 2001;98:5037–5042. doi: 10.1073/pnas.081393598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imaizumi-Scherrer T, Faust D M, Barradeau S, Hellio R, Weiss M C. Exp Cell Res. 2001;264:250–265. doi: 10.1006/excr.2001.5164. [DOI] [PubMed] [Google Scholar]
- 8.Skalhegg B S, Tasken K, Hansson V, Huitfeldt H S, Jahnsen T, Lea T. Science. 1994;263:84–87. doi: 10.1126/science.8272870. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Degenhardt B, Tobin D, Yao Z X, Tasken K, Papadopoulos V. Mol Endocrinol. 2001;15:2211–2228. doi: 10.1210/mend.15.12.0736. [DOI] [PubMed] [Google Scholar]
- 10.Huang L J, Durick K, Weiner J A, Chun J, Taylor S S. J Biol Chem. 1997;272:8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- 11.Huang L J, Durick K, Weiner J A, Chun J, Taylor S S. Proc Natl Acad Sci USA. 1997;94:11184–11189. doi: 10.1073/pnas.94.21.11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miki K, Eddy E M. J Biol Chem. 1998;273:34384–34390. doi: 10.1074/jbc.273.51.34384. [DOI] [PubMed] [Google Scholar]
- 13.Angelo R, Rubin C S. J Biol Chem. 1998;273:14633–14643. doi: 10.1074/jbc.273.23.14633. [DOI] [PubMed] [Google Scholar]
- 14.Herberg F W, Maleszka A, Eide T, Vossebein L, Tasken K. J Mol Biol. 2000;298:329–339. doi: 10.1006/jmbi.2000.3662. [DOI] [PubMed] [Google Scholar]
- 15.Amieux P S, McKnight G S. Ann NY Acad Sci. 2002;968:75–95. doi: 10.1111/j.1749-6632.2002.tb04328.x. [DOI] [PubMed] [Google Scholar]
- 16.Cho-Chung Y S, Pepe S, Clair T, Budillon A, Nesterova M. Crit Rev Oncol Hematol. 1995;21:33–61. doi: 10.1016/1040-8428(94)00166-9. [DOI] [PubMed] [Google Scholar]
- 17.Torgersen K M, Vang T, Abrahamsen H, Yaqub S, Tasken K. Cell Signalling. 2002;14:1–9. doi: 10.1016/s0898-6568(01)00214-5. [DOI] [PubMed] [Google Scholar]
- 18.Amieux P S, Howe D G, Knickerbocker H, Lee D C, Su T, Laslo G S, Idzerda R L, McKnight G S. J Biol Chem. 2002;277:27294–27304. doi: 10.1074/jbc.M200302200. [DOI] [PubMed] [Google Scholar]
- 19.Kirschner L S, Carney J A, Pack S D, Taymans S E, Giatzakis C, Cho Y S, Cho-Chung Y S, Stratakis C A. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 20.Casey M, Vaughan C J, He J, Hatcher C J, Winter J M, Weremowicz S, Montgomery K, Kucherlapati R, Morton C C, Basson C T. J Clin Invest. 2000;106:R31–R38. doi: 10.1172/JCI10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kammerer S, Burns-Hamuro L L, Ma Y, Hamon S C, Cànaves J M, Shi M M, Nelson M R, Sing C F, Cantor C R, Taylor S S, Braun A. Proc Natl Acad Sci USA. 2003;100:4066–4071. doi: 10.1073/pnas.2628028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carr D W, Hausken Z E, Fraser I D, Stofko-Hahn R E, Scott J D. J Biol Chem. 1992;267:13376–13382. [PubMed] [Google Scholar]
- 23.Carr D W, Stofko-Hahn R E, Fraser I D, Bishop S M, Acott T S, Brennan R G, Scott J D. J Biol Chem. 1991;266:14188–14192. [PubMed] [Google Scholar]
- 24.Vijayaraghavan S, Goueli S A, Davey M P, Carr D W. J Biol Chem. 1997;272:4747–4752. doi: 10.1074/jbc.272.8.4747. [DOI] [PubMed] [Google Scholar]
- 25.Burton K A, Johnson B D, Hausken Z E, Westenbroek R E, Idzerda R L, Scheuer T, Scott J D, Catterall W A, McKnight G S. Proc Natl Acad Sci USA. 1997;94:11067–11072. doi: 10.1073/pnas.94.20.11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamuro Y, Burns L, Canaves J, Hoffman R, Taylor S, Woods V. J Mol Biol. 2002;321:703–714. doi: 10.1016/s0022-2836(02)00419-9. [DOI] [PubMed] [Google Scholar]
- 27.Frank R. Tetrahedron. 1992;48:9217–9232. [Google Scholar]
- 28.Kramer A, Schneider-Mergener J. Methods Mol Biol. 1998;87:25–39. doi: 10.1385/0-89603-392-9:25. [DOI] [PubMed] [Google Scholar]
- 29.Wenschuh H, Volkmer-Engert R, Schmidt M, Schulz M, Schneider-Mergener J, Reineke U. Biopolymers. 2000;55:188–206. doi: 10.1002/1097-0282(2000)55:3<188::AID-BIP20>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 30.Kramer A, Reineke U, Dong L, Hoffmann B, Hoffmuller U, Winkler D, Volkmer-Engert R, Schneider-Mergener J. J Pept Res. 1999;54:319–327. doi: 10.1034/j.1399-3011.1999.00108.x. [DOI] [PubMed] [Google Scholar]
- 31.Huang L J, Wang L, Ma Y, Durick K, Perkins G, Deerinck T J, Ellisman M H, Taylor S S. J Cell Biol. 1999;145:951–959. doi: 10.1083/jcb.145.5.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makarova O, Kamberov E, Margolis B. BioTechniques. 2000;29:970–972. doi: 10.2144/00295bm08. [DOI] [PubMed] [Google Scholar]
- 33.Pawson T, Scott J D. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 34.Pawson T, Raina M, Nash P. FEBS Lett. 2002;513:2–10. doi: 10.1016/s0014-5793(01)03292-6. [DOI] [PubMed] [Google Scholar]
- 35.Koch C A, Anderson D, Moran M F, Ellis C, Pawson T. Science. 1991;252:668–674. doi: 10.1126/science.1708916. [DOI] [PubMed] [Google Scholar]
- 36.Sheng M, Sala C. Annu Rev Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- 37.Pawson T, Gish G D, Nash P. Trends Cell Biol. 2001;11:504–511. doi: 10.1016/s0962-8924(01)02154-7. [DOI] [PubMed] [Google Scholar]
- 38.Alber T. Curr Opin Genet Dev. 1992;2:205–210. doi: 10.1016/s0959-437x(05)80275-8. [DOI] [PubMed] [Google Scholar]
- 39.Moroianu J. J Cell Biochem. 1999;32–33, Suppl.:76–83. doi: 10.1002/(sici)1097-4644(1999)75:32+<76::aid-jcb10>3.3.co;2-h. [DOI] [PubMed] [Google Scholar]
- 40.Polakis P G, Wilson J E. Arch Biochem Biophys. 1985;236:328–337. doi: 10.1016/0003-9861(85)90633-2. [DOI] [PubMed] [Google Scholar]
- 41.Hauer J A, Barthe P, Taylor S S, Parello J, Padilla A. Protein Sci. 1999;8:545–553. doi: 10.1110/ps.8.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glantz S B, Li Y, Rubin C S. J Biol Chem. 1993;268:12796–12804. [PubMed] [Google Scholar]
- 43.Newlon M G, Roy M, Morikis D, Hausken Z E, Coghlan V, Scott J D, Jennings P A. Nat Struct Biol. 1999;6:222–227. doi: 10.1038/6663. [DOI] [PubMed] [Google Scholar]
- 44.Newlon M G, Roy M, Morikis D, Carr D W, Westphal R, Scott J D, Jennings P A. EMBO J. 2001;20:1651–1662. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banky P, Newlon M G, Roy M, Garrod S, Taylor S S, Jennings P A. J Biol Chem. 2000;275:35146–35152. doi: 10.1074/jbc.M003961200. [DOI] [PubMed] [Google Scholar]
- 46.Creamer T P, Rose G D. Protein Sci. 1995;4:1305–1314. doi: 10.1002/pro.5560040706. [DOI] [PMC free article] [PubMed] [Google Scholar]