

Abstract
Myosin heavy chain (MHC) is a large, multidomain protein important for both cellular structure and contraction. To examine the functional role of two C-terminal domains, the end of the coiled-coil rod and the nonhelical tailpiece, we have generated constructs in which residues within these domains are removed or mutated, and examined their behavior in Caenorhabditis elegans striated muscle. Genetic tests demonstrate that MHC lacking only tailpiece residues is competent to support the timely onset of embryonic contractions, and therefore viability, in animals lacking full-length MHC. Antibody staining experiments show that this truncated molecule localizes as wild type in early stages of development, but may be defective in processes important for thick filament organization later in embryogenesis. Ultrastructural analysis reveals thick filaments of normal morphology in disorganized arrangement, as well as occasional abnormal assemblages. In contrast, molecules in which the four terminal residues of the coiled coil are absent or mutated fail to rescue animals lacking endogenous MHC. Loss of these four residues is associated with delayed protein localization and delayed contractile function during early embryogenesis. Our results suggest that these two MHC domains, the rod and the tailpiece, are required for distinct steps during muscle development.
INTRODUCTION
The mechanism by which the proteins of the striated muscle thick filament are organized into the highly ordered contractile apparatus is an important unresolved question in the biology of the muscle cell. A key aspect of the process, how a cell specifies where and when to build a thick filament, is poorly understood. Control of filament placement presumably involves regulation of the distribution and activity of at least some of the individual protein components found within the thick filament. In invertebrates, the major structural proteins of the thick filament are myosin heavy chain (MHC) and paramyosin, which is homologous to the C-terminal two-thirds of the MHC coiled-coil domain or “rod” (Kagawa et al., 1989). In this article, we investigate the role of two MHC domains, the C-terminal rod and the tailpiece, in MHC localization and function in Caenorhabditis elegans striated muscle.
The rod has long been recognized as the filament-forming domain of the MHC molecule (for review, see Squire, 1981). Interaction of rod residues is thought to play a large part in driving filament assembly and specifying filament structure in striated muscle. A potential mechanism for temporal and spatial control of filament assembly is regulation of the accessibility or activity of the rod residues required for filament initiation. In smooth muscle, such regulation has been proposed to occur through the action of a C-terminal tailpiece domain: a small, nonhelical region that contains phosphorylation sites. Phosphorylation of the tailpiece promotes the formation of a folded, assembly-incompetent confomer (Castellani and Cohen, 1987). In addition to regulation of assembly competence, the smooth muscle MHC tailpiece may play a structural role, influencing molecular packing within the filament (Rovner et al., 2002).
Tailpiece sequences are often associated with nonmuscle myosins that form dynamic structures. Studies using primarily biochemical techniques suggest that the tailpiece may have different functions in nonmuscle myosins and may even serve more than one role in a given molecule. Like smooth muscle myosin, the tailpiece domain of nonmuscle myosins has been implicated in filament assembly and structure. For example, in human nonmuscle myosin II, tailpiece phosphorylation acts to decrease assembly of rod fragments in vitro (Murakami et al., 1998). In Acanthamoeba, the tailpiece has been proposed to mediate the initial association of assembling myosin II molecules (Sinard et al., 1989), thereby driving assembly and specifying minifilament structure. There are contradictory data as to whether phosphorylation regulates assembly of Acanthamoeba myosin into filaments (Collins et al., 1982; Sinard and Pollard, 1989). Phosphorylation of the tailpiece has also been proposed as a mechanism for regulating activity of the MHC motor domain once filaments are formed (Sathyamoorthy et al., 1990).
A tailpiece domain is present in the MHC and paramyosin proteins of C. elegans body-wall muscle. Interestingly, these homologous sequences are at opposite ends of the two molecules: the MHC tailpiece is at the extreme C terminus, whereas the paramyosin “tailpiece” is at the extreme N terminus. The paramyosin tailpiece is phosphorylated at multiple sites by an endogenous kinase, and a phosphorylation motif (S_S_A) was identified (Schriefer and Waterston, 1989). Several copies of this motif are found in the tailpiece domain of the body-wall MHC isoforms. MHC is also phosphorylated (Dey et al., 1992), but the location of the phosphorylated residues is unknown.
The C. elegans body-wall muscle cells provide a system with well-defined genetics and morphology in which to test the function of MHC domains (for reviews, see Waterston, 1988; Moerman and Fire, 1997). Two isoforms of MHC (MHC A and MHC B) assemble upon a core composed of paramyosin and associated proteins (Deitiker and Epstein, 1993). MHC A, encoded by the myo-3 gene, is present in the central 1.8-μm region of the 10-μm-long thick filament. This region includes the part of the bipolar filament in which myosin molecules associate in an antiparallel (tail-to-tail) manner (Miller et al., 1983). Mutations in the myo-3 locus that eliminate MHC A cause the complete lack of normal thick filaments, resulting in paralysis and death (Waterston, 1989). Thus, MHC A is required for an essential aspect of filament formation. The major isoform, MHC B, is encoded by unc-54 (Epstein et al., 1974) and is present in the filament arms, where parallel addition occurs. Mutations that eliminate MHC B reduce the number of thick filaments present. The resulting filaments can be of normal length, and they contain MHC A along their entire length (Epstein et al., 1986). Increasing the expression of the minor isoform, MHC A, can restore an unc-54 (MHC B-deficient) animal to near wild type (Fire and Waterston, 1989). Thus, in addition to being required to initiate the filament center, MHC A can add in parallel to form the filament arms.
In this article, we use the advanced molecular genetics of C. elegans to examine the function of the nonhelical tailpiece and the C-terminal rod. Performing these experiments in vivo allows the assessment of MHC function in the context of the muscle cell, with its full complement of potential interacting proteins, including endogenous kinases. Our results demonstrate that very small perturbations of the coiled coil have dramatic effects on early myosin localization and the onset of contractile function. In contrast, removal of tailpiece residues has no discernible effect on these early events of protein localization and filament formation. Instead, defects observed during later stages of embryogenesis and in adults suggest that the tailpiece domain may be required to establish or maintain proper filament position within the sarcomere. These observations suggest that the functions of the rod and the tailpiece are distinct, and are required at different times during development in C. elegans striated muscle.
MATERIALS AND METHODS
Transgenic Nematode Strains
MHC A construct, pUCAA plasmid: RW3680 unc-54(e190); stEx79. RW3825 unc-54(e190); myo-3(st386); stEx79. RW3838 myo-3(st386); stEx79. RW3681 unc-54(e190); stEx80. RW3685 myo-3(st386); stEx80. RW3826 unc-54(e190); myo-3(st386); stEx80.
Δ30, pRA7: RW3896 unc-54(e190); myo-3(st386); stEx148. RW3897 unc-54(e190); myo-3(st386); stEx149. RW3873 unc-54(e190); stEx148. RW3874 unc-54(e190); stEx149. RW3883 myo-3(st386); stEx149. RW3884 myo-3(st386); stEx148. RW3875 unc-54(e190); stEx150. RW3889 myo-3(st386); stEx150.
BHtagΔ30, pPP3: RW3885 unc-54(e190); stEx152. RW3905 myo-3(st386); stEx152. RW3906 unc-54(e190); myo-3(st386); stEx152. RW3907 myo-3(st386); stEx153. RW3908 unc-54(e190); myo-3(st386); stEx153. RW3886 unc-54(e190); stEx153. RW3909 myo-3(st386); stEx154. RW3887 unc-54(e190); stEx154.
Δ34, pRA4: RW3829 unc-54(e190); stEx129. RW3867 myo-3(st386); stEx129. RW3868 unc-54(e190); myo-3(st386)/eDf1; stEx129.
BHΔ34 previously called chimera 6 (Hoppe and Waterston, 1996): New transgenic lines carrying the rol-6::GFP coinjection marker were generated for these experiments. RW3824 unc-54(e190); stEx111. RW3781 myo-3(st386); stEx111. RW3820 unc-54(e190); myo-3(st386)/eDf1; stEx111. RW3823 unc-54(e190); stEx112. RW3817 unc-54(e190); myo-3(st386)/eDf1; stEx112. RW3782 myo-3(st386); stEx112. RW3783 myo-3(st386); stEx113. RW3818 unc-54(e190); stEx113. RW3819 unc-54(e190); myo-3(st386)/eDf1; stEx113. RW3816 unc-54(e190); stEx114. RW3784 myo-3(st386); stEx114.
BHtagΔ34, pRA2: RW3828 myo-3(st386); stEx120. RW3830 unc-54(e190); myo-3(st386)/eDf1; stEx120. RW3833 unc-54(e190); stEx120.
Chimera 3: RW3768 unc-54(e190); stEx55. RW3827 unc-54(e190); myo-3(st386); stEx55.
Chimera 4.tag: RW3811 unc-54(e190); stEx118. RW3860 unc-54(e190); myo-3(st386); stEx118
BHtagΔKIRA: RW3942 unc-54(e190); stEx200; RW3949 myo-3(st386); stEx200; RW3954 unc-54(e190); myo-3(st386)/eDf1; stEx200
BHtagPIRA: RW3946 unc-54(e190); stEx203. RW3957 myo-3(st386); stEx203. RW3956 unc-54(e190); myo-3(st386)/eDf1; stEx203. RW3947 unc-54(e190); stEx204. RW3955 myo-3(st386); stEx204. RW3958 unc-54(e190); myo-3(st386)/eDf1; stEx204.
BHtagPIAA: RW3944 unc-54(e190); stEx201. RW3951 myo-3(st386); stEx201. RW3952 unc-54(e190); myo-3(st386)/eDf1; stEx201. RW3945 unc-54(e190); stEx202. RW3950 myo-3(st386); stEx202. RW3953 unc-54(e190); myo-3(st386)/eDf1; stEx202.
DNA Construct
The previously published chimera 6 construct (Hoppe and Waterston, 1996) has been renamed BHΔ34. The premature stop codon resulted from an error introduced during a polymerase chain reaction step used to generate chimera 4, which contains MHC B sequences in the tailpiece domain. Therefore, the last amino acid residue before the stop codon is from MHC B, but the remainder of the rod residues are from MHC A. The other Δ34 constructs were made by moving the sequences encoding the premature stop codon from BHΔ34 to the myo-3 gene or a hemagglutinin (HA)-tagged chimeric construct, by using standard cloning techniques and previously described genomic cassettes (Hoppe and Waterston, 1996). The addition of the HA epitope tag to the MHC B head has been described previously (Hoppe and Waterston, 2000). To make the Δ30 constructs, oligo ACAAGATTCGTGCATaAtaaTCCATGGaTCCACCAGATGGTTT and its complement were used with the Stratagene QuikChange kit to replace S1940 and A1941 codons with consecutive ochre codons in the 1.4-kb SphI-KpnI subclone encoding the myo-3 C terminus. The same protocol was used with the following oligonucleotides to generate the constructs containing mutations within the C-terminal rod: ΔKIRA, CTGTCAAAGATGCGTAACTCAGCTTCgATGGCTCCACCA; PIRA, TCAAAGATGCGTAACccaATTCGTGCcTCAGCTTCCATGGC; and PIAA, TCAAAGATGCGTAACccaATTgccGCcTCAGCTTCCATGGC. Oligonucleotide bases in lowercase do not match wild-type sequence and include silent mutations to eliminate hairpins. Fragments generated by polymerase chain reaction were verified by DNA sequencing. Complete constructs encoding truncated MHC A or chimeric proteins were made by moving the mutant subclone as described for Δ34.
Injections
Transformed lines were generated as described previously (Mello et al., 1991). A 30:10:1 ratio of Bluescript/pPHgfp-1/myosin (at 200 ng/μl in 10 mM Tris, 1 mM EDTA, pH 8) was used to produce extrachromosomal arrays with low myosin copy number. The coinjection marker expresses green fluorescent protein (GFP) in the hypodermis under the control of the rol-6 promoter (Hoppe and Waterston, 2000). Arrays were selected for robust expression by injection into unc-54(e190) animals and screening for restoration of motility. Arrays with lower expression levels were identified by poor rescue of unc-54(0), or by injection into myo-3(st386)/eDf1 and screening rescued myo-3 homozygotes for wild-type muscle structure by using polarized light microscopy. To test whether a construct could support viability when it is the only myosin heavy chain expressed, arrays with robust expression were first crossed into an unc-54(e190); myo-3(st386)/eDf1; stExN background. Animals were then picked singly to identify any viable double mutant animals, which do not segregate the recessive larval lethal phenotype associated with the eDf1 balancer chromosome.
Brood Analysis
Transgenic unc-54(e190); myo-3(st386)/eDf1; stExN hermaphrodites were picked singly to freshly seeded plates and allowed to lay for 1 d. After an additional day, the transgenic progeny (identified using a fluorescent dissecting microscope) were sorted and counted. Arrested animals that showed no GFP signal were picked onto microscope slides and scored again using a compound fluorescent microscope. Viable transgenic animals were picked singly to plates, and their progeny examined to determine parental genotype.
Antibody Staining
Embryos were fixed with paraformaldehyde and methanol and stained using the methods of Hresko et al. (1994). Adults were fragmented, fixed with methanol, and stained as described previously (Francis and Waterston, 1985). Monoclonal antibodies 5–6, 5–8, and 5–14 were gifts of Henry Epstein and Irving Ortiz (Baylor College of Medicine, Houston, TX). The HA epitope was detected using the rat monoclonal anti-HA high affinity (Roche Diagnostics, Indianapolis, IN).
Time-Lapse Recording
Gravid adult hermaphrodites were cut in half with a razor blade in M9 buffer. The embryos in a small amount of buffer were transferred to a 2% agarose pad on a microscope slide and covered and gently flattened with a Vaseline-lined coverslip. The embryos were videotaped overnight (∼16 h) using Nomarski optics and then photographed on a fluorescence microscope to record the presence of GFP expression in any arrested individuals. The progression of development up until the twofold stage (designated as 450 min in Figure 3) was scored by monitoring morphology rather than actual time elapsed. Subsequent to the twofold stage, elapsed time was used to determine the point at which movement began. Because temperature was not strictly controlled, all times are approximate. The strains carrying genomic mutations were RW3667 myo-3(st386)/eDf1, CB190 unc-54(e190), and RW3858 unc-54(e190); myo-3(st386)/eDf1.
Figure 3.

Animals expressing only MHC with alterations in the C-terminal rod show delayed onset of movement during embryogenesis. The black bars indicate the window of time in which videotaped embryos of each genotype first twitched. The time line, measured in minutes from first cleavage, represents data from Sulston et al. (1983). Our observations of each embryo began at the 1.5-fold stage, which was designated as 420 min (see MATERIALS AND METHODS). For the top four strains, which express MHC from genomic loci, N represents the number of individuals scored. The bottom nine strains express MHC only from extrachromosomal arrays. For rescuing arrays, N represents only animals that elongated successfully, thus excluding presumptive mosaic animals that had intermediate phenotypes. In cases where inviable transgenic double mutants segregate from a heterozygous strain, N represents the number of arrested individuals that expressed the coinjection marker and/or showed delayed movement throughout the length of the body, confirming the expression of the transgene in the arrested animal.
Electron Microscopy
Samples were prepared as in Waterston et al. (1977): adult worms were fixed with 3% glutaraldehyde, 0.1 M sodium phosphate (pH 7.4) at 0°C for 4 h., cut in half, and fixed overnight in the same solution. After postfixation in 1% osmium tetroxide in 0.1 M sodium phosphate (pH 7.4) for 1 h at 4°C, the samples were embedded in agarose, dehydrated, and embedded as described previously (Hall, 1995) using a Pelco eponate 12 kit.
RESULTS
To investigate the possible structural or regulatory roles of the C-terminal MHC residues in thick filament formation and function, we generated various mutant constructs containing point mutations or deletions within the tailpiece and/or C-terminal rod residues. Extrachromosomal arrays expressing mutant or chimeric protein were isolated (see MATERIALS AND METHODS) and crossed into various genetic backgrounds to test their ability to function in different aspects of thick filament formation. Filament initiation function was assessed by testing for rescue of the lethal phenotype of myo-3(0) mutations, which eliminate MHC A. Competence in thick filament elongation and contractile function was assessed by the ability to restore motility in unc-54(0) animals, which lack MHC B. Last, constructs were tested in the double mutant background to determine the requirement for C-terminal residues in animals that express no other MHC in their body-wall muscles.
Constructs were made primarily using the myo-3 gene, which encodes MHC A, the minor isoform that is essential for thick filament initiation, and therefore for viability (see INTRODUCTION). All chimeric constructs, made by combining sequences from MHC A and MHC B, contain all or most of the MHC A rod and are therefore able to supply MHC A-specific initiation function in a myo-3(0) mutant (Hoppe and Waterston, 1996). Chimeric and HA-tagged constructs were included to test the function of different MHC domains and to allow construct-specific antibody staining experiments (see below).
Truncated Constructs Define a Four-Amino-Acid Region of Coiled Coil Required for Viability
The C-termini of the constructs are shown in Figure 1A, and diagrams of the sequence content of the complete constructs are shown in Figure 1B. The Δ30 and BHtagΔ30 (chimeric) constructs lack all potential phosphorylation motifs, but based upon paircoil scores (Berger et al., 1995) are likely to have an intact rod. These constructs were used to examine the behavior of MHC lacking only the tailpiece domain. The use of truncations, rather than point mutations, is likely to produce a loss-of-function phenotype and requires no assumptions as to the precise site or timing of potential phosphorylation events. The more severe truncation, present in all Δ34 constructs, removes the tailpiece and four residues within presumptive coiled coil. The Δ34 constructs were used to determine the effects of disrupting the extreme C terminus of the coiled-coil domain. The results of the genetic tests are shown in Figure 1B. All truncated constructs were able to produce lines of rescued myo-3(0) homozygous animals. Similarly, all constructs rescued the paralysis of unc-54(0) mutants. These genetic tests demonstrate that truncated proteins lacking up to 34 C-terminal residues have sufficient function to restore viability and motility in single-mutant animals lacking either MHC A or MHC B.
Figure 1.

Truncated constructs define a small region of predicted coiled coil that is essential for viability. (A) Top line shows the N-terminal nonhelical peptide in paramyosin that is phosphorylated by an endogenous kinase (see INTRODUCTION). The proposed phosphorylation motif S_S_A is underlined. The C termini of full-length MHC A (black) and MHC B (gray) are shown as aligned by the program Megalign, with the C termini of mutant and chimeric constructs shown below. The number of C-terminal residues removed in each construct follows the delta (Δ) symbol. Those residues designated as nonhelical score p = 0 in paircoil (Berger et al., 1995), and those designated as rod score p = 1.0. The Δ34 constructs contain unc-54 (MHC B) sequences that result in the presence of a single MHC B amino acid residue at the C terminus (see MATERIALS AND METHODS). Amino acid residues changed by missense mutation are marked by a shaded rectangle. (B) Constructs were tested for the ability to rescue single mutant animals lacking one MHC isoform, and for the ability to rescue double mutant animals lacking both isoforms. Schematic drawings of the myosin constructs (left) show MHC A residues in black and MHC B residues in gray. The oval represents the head domain, the thick line represents the coiled-coil rod, and the small angled C-terminal box represents the tailpiece. BH denotes the presence of the MHC B head, and the boxed HA label or “tag” indicates the addition of the hemagglutinin epitope. Some of the single mutant data have been published (Hoppe and Waterston, 1996). ND, not done; P, partial rescue.
To determine whether functional thick filaments could be formed containing only truncated myosin, we tested the transgenes for rescue in the double-mutant background unc-54(0); myo-3(0). MHC A and chimeric constructs that lack all of the potential phosphorylation motifs (Δ30) rescue the double mutant animals to viability and are thus competent to function as the sole myosin heavy chain. Some of the rescued lines have good motility and egg-laying function, but do not seem completely wild type in movement. The observed slowness may be due to a number of factors, including inappropriate levels of myosin accumulation or a small impairment in MHC function. However, these results demonstrate that the nonhelical tailpiece and its candidate phosphorylation motifs do not play an essential role in thick filament formation or function.
In contrast, the Δ34 constructs could not be isolated in the double-mutant background (Figure 1B). Therefore, the four residues of predicted coiled coil that are present in Δ30 but absent in Δ34 are essential for some aspect of myosin function. The two full-length chimeric constructs, chimera 3 and chimera4.tag, which contain the MHC A rod but have some MHC B sequences, both rescue the double mutant. Thus, the MHC A head and tailpiece residues do not have isoform-specific function that is required for viability. Furthermore, the presence of the HA tag does not affect rescuing activity.
The Four-Amino-Acid Region KIRA Contains Highly Conserved Charged Residues
Genetic tests of the truncated constructs Δ30 and Δ34 define an essential region of the MHC A C-terminal coiled coil containing the residues KIRA. To examine the importance of these residues in striated muscle of other species, we compared the C-termini of striated muscle myosins from a variety of organisms (Figure 2). The two charged residues within the four-residue region (lysine and arginine) are highly conserved, suggesting an important role for this part of the coiled coil in all striated muscle. In contrast, the length of the potential tailpiece domain, as well as the placement of possible phosphorylatable residues (serines and threonines) within this domain, varies among species. Therefore, the role of the tailpiece, or at least the mechanism of domain action, does not seem to be broadly conserved. Invertebrate myosins tend to have a longer tailpiece that contains a number of phosphorylatable residues. Interestingly, all vertebrate striated-muscle myosins examined contain a phosphorylatable residue within the four-amino-acid region of coiled coil defined by our truncated constructs.
Figure 2.

C-terminal rod residues, but not the tailpiece, are conserved in striated muscle myosins. The C-terminal sequences from a diverse array of striated muscle myosins selected from the public database (invertebrate striated, fish and rat skeletal, human and chicken cardiac) are aligned at the beginning of rod zone 40 (McLachlan and Karn, 1982). The essential MHC A region defined by constructs Δ30 and Δ34 (horizontal bar) contains conserved charged residues (arrows) that are likely to be coiled coil (paircoil score p = 1.0; Berger et al., 1995). Boxes indicate serine and threonine residues that might be potential phosphorylation sites. The accession numbers are P12844, M61229, P24733, AAK73348, A24922, A46762, and P29616.
The Four-Amino-Acid Region Is Essential for Function of Full-Length MHC
Our experiments with truncated constructs indicate that removal of the tailpiece and four residues of coiled coil disrupts an essential myosin function, whereas removal of the tailpiece alone does not. To determine whether the four coiled-coil residues, KIRA, are essential for MHC function in molecules that contain an intact tailpiece domain, we generated three constructs in which either one or both conserved basic residues (K, R) were altered or deleted (Figure 1A). The BHtagΔKIRA construct contains an in-frame deletion that removes the four-residue region. The resulting protein is thus slightly shorter and has a small shift in the position of the tailpiece relative to the remainder of the molecule. The other two constructs contain point mutations that alter the properties of the KIRA region but leave the overall length of the molecule intact. The BHtagPIRA construct changes K1936 to P, eliminating a positive charge and prematurely disrupting the coiled coil. The BHtagPIAA construct, containing mutations K1936P and R1938A, eliminates two positive charges and prematurely disrupts coiled coil.
Genetic tests revealed that the two constructs in which both basic residues were altered or deleted failed to rescue double mutant animals (Figure 1B). Thus, mutations within the four residues of C-terminal coiled coil result in the disruption of an essential myosin function despite the presence of an intact tailpiece. The inability of the tailpiece domain to compensate for disruption of the four-residue region of coiled coil suggests that the two domains play distinct roles in thick filament formation or function.
The BHtagPIRA construct, in which the K1936P change prematurely disrupts coiled coil and removes one of the conserved basic residues, exhibits partial function in vivo. Most double mutant animals expressing only BHtagPIRA die (Table 1). However, some animals of this genotype survive and reproduce, although most of their transgenic progeny die. These data suggest that the mutational changes in BHtagPIRA reduce protein function to a level near the threshold of activity required for survival. Again, the tailpiece domain in this full-length construct cannot compensate for a point mutation within the C-terminal rod.
Table 1.
Animals expressing only MHC in which the essential KIRA residues are removed or mutated have the Pat phenotype
| Genotypes segregating in progeny | Expected % phenotype in progeny if no rescue | BHtagΔ34 stEx120 | BHtagΔKIRA stEx200 | BHtagPIRA stEx204 | BHtagPIAA stEx201 | |
|---|---|---|---|---|---|---|
| unc-54; myo-3/eDf1; stExN | Viable, motile | 50 | 44% (25) | 47% (38) | 45% (30) | 49% (23) |
| unc-54; myo-3; stExN | Viable, motile | 0 | 0 | 0 | 7% (5) | 0 |
| Dead, arrested | 25 | 33% (19) | 29% (23) | 21% (14) | 23% (11) | |
| unc-54; eDf1; stExN | Larval lethal | 25 | 23% (13) | 24% (19) | 27% (18) | 28% (14) |
The phenotypes of transgenic progeny from heterozygous hermaphrodites [unc-54(0); myo-3(0)/eDf1; stExN] carrying arrays with robust expression of a mutant construct (designated stExN) were scored and counted (see MATERIALS AND METHODS). The transgenic progeny were identified by expression of the GFP cotransformation marker. The first column lists the three genotypes that should segregate in a 2:1:1 ratio in the progeny of the heterozygous parent. The second column describes the phenotypes associated with each genotype, and the expected frequency of that phenotype in the brood if the myosin construct present in the parent cannot rescue an animal lacking endogenous body-wall MHC. The three constructs that remove or replace both of the conserved basic residues within the KIRA sequence fail to rescue double mutant animals. The PIRA construct, which contains a single amino acid change within the KIRA sequence, can rescue double mutant animals, but fewer than half survive. Animals homozygous for the eDf1 balancer chromosome elongate normally but die as larvae. Similar data were collected from: Δ34 array stEx129; BHΔ34 arrays stEx111, stEx112, stEx114; BHtagPIRA array stEx203; and BHtagPIAA array stEx202 (our unpublished data).
The Four-Amino-Acid Region Is Required for MHC Function in Early Muscle Development
To define the functional deficit of the constructs that do not support viability, we analyzed the broods from heterozygous animals to determine the terminal phenotype of the transgenic double mutant animals (see MATERIALS AND METHODS). Like the myo-3 mutants that lack MHC A, animals expressing only Δ34 constructs arrest elongation at the twofold stage of embryogenesis and die as misshapen L1 larvae (Table 1). These results suggest that small deletions in the rod cause a severe deficit in early myosin function. Double mutant animals expressing constructs with mutations within the KIRA sequence that disrupt both conserved basic residues show a similar terminal phenotype (Table 1). However, some of the double mutant animals expressing these constructs arrest between the two- and threefold stages. The ability of these constructs to support elongation beyond the twofold stage suggests that the presence of the tailpiece residues can to a small degree ameliorate the elongation defect associated with the Δ34 construct.
Disruption of the Four-Residue Region Delays the Onset of Movement in Embryos
The Pat phenotype (paralyzed, arrested elongation at the twofold stage) observed in animals expressing only Δ34 has been associated with mutations that have a range of defects in early movement (Williams and Waterston, 1994). To examine the movement of animals expressing only Δ34 myosin, we used time-lapse videomicroscopy to view developing embryos from heterozygous hermaphrodites carrying arrays selected for robust expression (see MATERIALS AND METHODS). Embryos expressing any of the Δ34 constructs alone showed delayed initiation of movement (Figure 3). Whereas wild-type animals twitch at the 1.5-fold stage and begin coordinated rolling at the twofold stage (Williams and Waterston, 1994), all transgenic double mutant embryos exhibited no movement during these early stages and arrested elongation at the twofold stage.
Double mutant embryos expressing Δ34 constructs did eventually move. In most Δ34 transgenic lines, the late movements began as weak, isolated twitches that slowly increased in frequency and strength, such that most animals showed late (660–900 min), weak movement of the head and tail, but did not roll. The BHΔ34 array stEx120 produced the strongest movement in double mutant animals. In this line, some individuals eventually twitched vigorously and showed clear attempts to roll. In all cases, however, the late movement did not lead to further elongation, and therefore did not change the terminal arrest phenotype (Figure 4). Similar analysis of myo-3(0) mutants revealed that the movement phenotype of embryos expressing only MHC B is similar to that of embryos expressing Δ34 (Figure 3). Double mutant animals that express no body-wall myosin show no movement at any time, and have the Pat phenotype.
Figure 4.
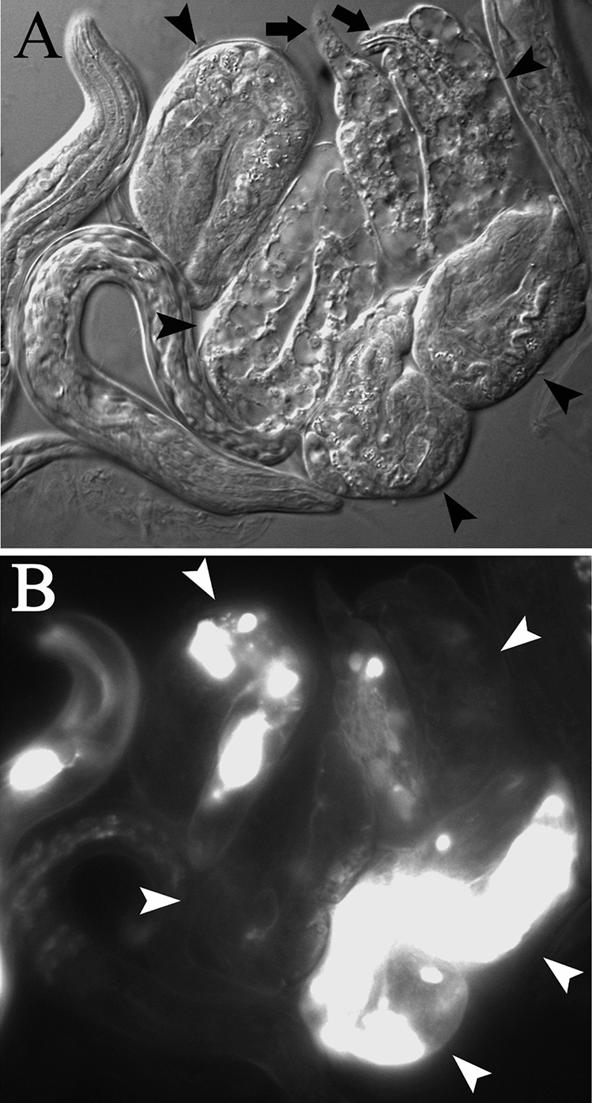
Late movement in animals expressing only Δ34 does not produce further elongation. (A) Nomarski micrograph of larvae from the BHtagΔ34-expressing strain unc-54(e190); myo-3(st386)/eDf1; stEx120 after time-lapse video recording (see MATERIALS AND METHODS). Five larvae that arrested elongation at the twofold stage (arrowheads) are flanked by their siblings that elongated normally. Arrows point to the tips of the head and tail in one Pat larva. (B) GFP coinjection marker indicates the presence of the transgene in some of the Pat larvae.
Animals expressing only MHC with an intact tailpiece and mutations within the four-residue region (ΔKIRA, PIRA, PIAA) exhibited similar defects in motility. The observed delay in the onset of twitching was not as great in animals expressing these constructs compared with those expressing only Δ34. However, the observed movement was qualitatively similar: motility slowly progressed from twitches to movements of the head and tail, but no rolling occurred.
In contrast, animals expressing only Δ30, which lacks the tailpiece, have a movement phenotype similar to control lines expressing full-length MHC. Some variability in the onset of twitching is apparent in any line expressing MHC from extrachromosomal arrays compared with the lines expressing MHC from genomic loci (Figure 3). The second Δ30 transgene (stEx149), as well as the chimera4.tag array, showed a slightly wider range in the time at which movement was first detected. In both these lines, a few animals that did not twitch until reaching the twofold stage went on to move well and elongate properly. Therefore, these viable animals initiated movement later than some nonviable individuals that expressed only MHC with mutations within the KIRA residues. Thus, movement before the twofold stage is apparently not essential for proper morphogenesis or viability. In addition, these data indicate that lethality is not caused by a simple delay in the onset of contractions. Instead, developmental arrest is associated with defects in both the onset of twitching and the subsequent progression to coordinated movement.
To assess the possible effects of gene dosage on early movement, we videotaped embryos where myosin levels were altered by genomic mutation or by transgene expression (Figure 3). Animals heterozygous for myo-3(0), which have reduced MHC A, as well as unc-54(0) homozygotes, which express no MHC B, showed little or no delay in the onset of movement compared with wild type. Double mutant animals that express full-length MHC A from transgenic arrays showed a small variation in onset of movement. Because the onset of twitching and progression to coordinated movement are not dramatically changed by variations in the level of full-length myosin, the delayed movement and subsequent lethality associated with constructs that have mutations in the C-terminal rod cannot be explained by a simple deficit in myosin expression.
Defects in Early Localization of Δ34 Protein
To elucidate the defect in Δ34 that leads to poor movement and lethality, we used immunocytochemistry to examine the localization of truncated proteins in developing embryos. Antibody staining of homozygous myo-3 lines rescued by Δ34 revealed delayed localization of the truncated protein into discrete bands. At the 1.5-fold stage, when movement normally begins, the Δ34 protein did not exhibit the pattern of strong bands evident in wild type and in controls (Figure 5). Instead, the protein localized weakly to bands, with much stain remaining diffuse in the cytoplasm. Animals expressing reduced or increased levels of full-length myosin do not show this phenotype (Figure 5). Thus, in the absence of full-length MHC A, the truncated myosin appears to assemble more slowly. However, these rescued animals, which contain endogenous MHC B, elongate normally and survive.
Figure 5.

Δ34 protein shows delayed localization into discrete bands during early stages of muscle cell organization. Each column shows embryos from a given strain. Each row depicts a different developmental stage: 1.5-fold (A–D), twofold (E–H), or threefold (I–L). The staining pattern in unc-54(e190) is similar to that in wild-type, despite the lack of MHC B and the resultant reduction in total myosin. The third column shows full-length MHC A that is overexpressed from a transgene in myo-3; stEx80 animals (array chosen for robust rescue; see MATERIALS AND METHODS). Increased MHC A results in stray filamentous staining (arrowhead in G) and occasional aberrant expansion of stain within or near the contractile apparatus (K). However, strong bands of stain are evident at the 1.5-fold stage (C). Distribution of Δ34 protein in myo-3; stEx129 animals is distinct, with diffuse stain outlining the nuclei (D and H). Occasional abnormal round accumulations that are found within the muscle cell body (L) may be due to inappropriate levels of MHC accumulation. Animals are stained with MHC A-specific antibody (5–6), which recognizes endogenous protein in wild-type and unc-54 mutants, and transgenic protein in the remaining genotypes. In wild type, four muscle quadrants run longitudinally along the length of the animal. In most areas, four longitudinal bands of stain are apparent across the width of a single muscle quadrant (between the arrowheads in I). The stages refer to the length of the elongating animal compared with the surrounding eggshell: at 1.5-fold, the posterior tip (arrowhead in A) has reached half the length of the egg as it elongates and moves toward the anterior end (arrow), positioned at the opposite pole of the egg. Bar, 5 μm. Myosin localization during embryogenesis has been described previously (Epstein et al., 1993; Hresko et al., 1994; Williams and Waterston, 1994).
To control for the possibility that the apparent delay in localization of the truncated protein was a result of errors in staging or an imbalance in myosin expression, we examined the localization of the truncated protein in animals expressing endogenous MHC A but no MHC B. In this background, we could directly compare the localization of full-length MHC A and truncated MHC in the same muscle cells (Figure 6). BHtagΔ30 protein colocalizes with endogenous MHC A at all stages, and its early localization appears wild type. In contrast, BHtagΔ34 myosin shows a delay in localization compared with MHC A during early stages of embryogenesis, similar to the delay seen with the Δ34 construct in the myo-3 mutant (Figure 5). Thus, the presence of full-length MHC A protein does not discernibly improve the movement of Δ34 protein into A-bands. Conversely, we did not detect any dominant-negative effect of the truncated protein disrupting the assembly of wild-type MHC A. These data argue that an intact rod is required at the level of the individual molecule for proper protein localization in early stages of muscle assembly. However, the tailpiece does not play a detectable role in early protein localization.
Figure 6.

Comparison of the distribution of full-length and truncated proteins within the same muscle cells at three developmental stages (A–C, 1.5-fold; D–F, twofold; and G–I, threefold) shows delayed localization of Δ34, but not Δ30 constructs. Strains in which a truncated or chimeric construct rescues the unc-54(0) mutation were stained with antibody 5–14 (which recognizes an epitope in the MHC A head) to visualize endogenous MHC A. All transgenic proteins, which contain the tagged MHC B head, were detected using anti-HA antibody. In early stages, BHtagΔ34 staining is strong outside the contractile apparatus (arrowheads A′ and D′) compared with MHC A (A and D). Insets show staining in the perinuclear region. The chimera4.tag staining colocalizes with that of MHC A at all stages (B, E, and H), demonstrating that the HA tag, and the MHC B head and tailpiece sequences do not discernibly alter localization. The BHtagΔ30 protein localizes normally at 1.5-fold (C′). Later, embryos expressing Δ30 constructs often show areas in which staining of Δ30 and MHC A are abnormally expanded within or near the plane of the contractile apparatus (arrows in F, F′, I, I′). Transgenic arrays shown are stEx120, stEx118, and stEx154. Bar, 5 μm.
To determine the behavior of Δ34 protein in the double mutant background, where absence of the tailpiece and four rod residues results in lethality and a long delay in the onset of contractile function, we examined the localization of BHtagΔ34 protein in embryos by using isoform-specific antibodies (Figure 7). The staining pattern observed is very similar to that seen in the single mutants. Again, truncated protein showed a delay in localization into discrete bands compared with Δ30 constructs and a full-length control. The localization of BHtagΔ34 protein in the double mutants improves over time, even following the failure in elongation, such that the normal number of stained bands across the quadrant is apparent in some regions of older arrested animals. These results suggest that the C-terminal rod residues absent in Δ34 are required for timely localization but that sequences within the remainder of the myosin molecule also contribute to protein localization, although their action is not sufficient to support viability.
Figure 7.

Localization of Δ30 and Δ34 proteins is distinct at early developmental stages in animals expressing only truncated MHC. Three stages of embryogenesis are represented: A–D, 1.5-fold; E–H, twofold; and I–L, threefold. Like wild-type, embryos expressing only a Δ30 construct have well-defined stripes of myosin staining at the 1.5-fold stage (arrowheads in A and B), the time at which movement commences in wild type. A full-length chimeric protein expressed in the double mutant shows the same pattern (D). Later in development, animals expressing only Δ30 often show regions of expanded stain, indicating aberrant accumulations of Δ30 MHC within or near the contractile apparatus (arrows in F, I, and J). Expression of a single full-length construct can also result in abnormal, irregularly shaped accumulations (L, arrow) or stray filamentous staining (L, arrowhead) in later developmental stages. In contrast, a Δ34 construct in double mutant animals shows delayed localization of the protein in early stages of muscle cell organization. At the 1.5-fold stage, embryos expressing only BHtagΔ34 have no prominent bands (C). Instead, staining appears dotted or broken (arrowhead), and diffuse staining outside the contractile apparatus is strong. These animals arrest elongation at the twofold stage, but the localization of truncated myosin changes as development continues. In younger arrested animals (G), the bands of staining grow stronger compared with diffuse staining in the cell body. Older arrested animals (K) show strong localization of the protein to bands, in some places showing the four bands per quadrant characteristic of wild type (arrowheads), despite the aberrant shape of the animals. Δ30 expressed from transgene stEx148 was stained with antibody 5.6. The three tagged constructs, BHtagΔ30 stEx153, BHtagΔ34 stEx120, and chimera4.tag stEx118, were stained with anti-HA. The transgenic double mutant embryos from the strain unc-54(e190); myo-3(st386)/eDf1; stEx120 were recognized by their failure to stain with the MHC A head-specific antibody 5–14. Bar, 5 μm.
Animals expressing only MHC that contains mutations in the four-residue region of the rod but has an intact tailpiece domain have a terminal phenotype similar to those expressing only Δ34 (Table 1), but have less severe defects in early movement (Figure 3). In antibody-staining experiments we did not find a convincing delay in the localization of these proteins into bands compared with endogenous MHC A within the same cell (our unpublished data). Therefore, unlike the Δ34 proteins, the functional deficit associated with mutations that alter only residues within the four-residue-region of the C-terminal rod cannot be consistently detected at the level of the light microscope. Nonetheless, these proteins are unable to support the timely initiation and progression of motility required for proper development.
Defective Localization of Δ30 Protein in Later Stages of Embryogenesis
Although Δ30 protein shows no apparent delay in localization during early stages of embryogenesis (Figures 5 and 6), all lines expressing Δ30 showed a variably penetrant defect in localization in later stages. In these lines, some embryos at the twofold stage or later contained regions where myosin staining had broadened such that the banded pattern that was apparent at earlier stages was no longer visible (Figures 6, F and I, and 7, I and J). The abnormally localized myosin appeared either as an inward extension of the membrane-associated contractile apparatus, or as a relatively discrete round accumulation. In lines that express both Δ30 and full-length MHC A, the endogenous protein also appeared in regions of expanded stain (Figure 6). Thus, molecules lacking only the tailpiece seem to influence the localization of full-length molecules. These observations suggest that Δ30 protein may be defective in a function required at the level of the filament during later events in sarcomere formation or maintenance.
Tailpiece Residues Are Not Required for Proper Filament Morphology
Animals expressing only Δ30, which lacks the tailpiece, show no delay in early myosin localization or the onset of muscle contraction. These observations suggest that the tailpiece does not play an important role in the assembly of myosin into filaments. To test this hypothesis further, we examined the ability of Δ30 protein to show the MHC A-specific localization to the central portion of the myosin-containing A-bands of the contractile apparatus. To obtain a level of total myosin expression close to that of wild type, a Δ30 array selected for poor rescue of unc-54 (which lacks the major myosin, MHC B) was crossed into the myo-3 background, which lacks MHC A but expresses wild-type levels of endogenous MHC B. In these animals the isoform-specific staining pattern, as well as overall organization of the contractile apparatus, appeared wild-type (Figure 8). These data argue that the MHC A tailpiece sequences are not required for proper localization of the MHC A isoform within the filament, or for proper placement of filaments within the sarcomere.
Figure 8.

Isoform-specific localization does not require the tailpiece. Adults in which relatively low levels of Δ30 expression replace endogenous MHC A [myo-3(st386); stEx150] were stained with directly labeled isoform-specific antibodies. Isoform-specific localization and overall cell structure are normal, as viewed in this portion of a muscle quadrant in which parts of three cells are visible. The left side of the micrograph shows MHC B staining along the outside edges of each A-band, and the central stripe of MHC A is shown on the right (compare with Miller et al., 1983). Bar, 5 μm.
To determine whether loss of tailpiece residues affects filament morphology, we examined cross sections of adult animals by transmission electron microscopy (TEM). Most filaments in animals expressing only Δ30 seemed normal in diameter and orientation, but were poorly organized (Figure 9, C and D). Defects in organization varied, even between muscle cells within the same animal. Common abnormalities included large clusters of filaments that had no apparent division into individual sarcomeres, and groups of filaments displaced interiorly from the membrane. Occasionally abnormal assemblages of filaments were seen, but the majority of filaments appeared normal in cross section. These observations suggest that the tailpiece residues are not required for proper filament structure but may be required at the level of the filament for establishment or maintenance of sarcomere structure.
Figure 9.

Thick filaments containing truncated protein have normal morphology by TEM analysis. (A) In wild-type adults, thick filaments (viewed in cross section, arrowhead) are organized around the M-line structure (m) within the contractile apparatus. An area of thin filaments devoid of thick filaments surrounds the dense body (db), which anchors thin filaments. The contractile apparatus is attached to the muscle cell membrane that is adjacent to the hypodermis, which secretes the outer cuticle. (B) Animals [myo-3(st386); stEx114] in which an array with robust expression of BHΔ34 replaces MHC A (the essential minor isoform) contain well-organized, normal thick filaments despite the increased level of total myosin, and the presence of the truncated myosin that exhibits delayed localization. Some ectopic filaments are found above the dense bodies (arrowheads). Given the structure of obliquely striated muscle (Waterston, 1988), these are likely to be filaments from an adjacent sarcomere that are abnormally long. (C and D) In animals expressing only Δ30 [unc-54(e190); myo-3(st386); stEx148], most thick filaments are uniform and normal in appearance, but are poorly organized. (C) Some thick filaments are found associated with M-line structures, but are unevenly and loosely packed with respect to each other. Some filaments are located at a more interior position, apparently not attached to the membrane (arrowheads). These stray filaments overlie two dense bodies that have no A-band between them. (D) In some areas there appear to be tangles of filaments that are not oriented on the long axis of the cell. Bar, 200 nm.
We were unable to do a similar TEM analysis of the morphology of filaments containing only Δ34 because these animals arrest during embryogenesis. However, we did examine muscle in which BHΔ34 replaced the essential isoform, MHC A. In these animals, thick filament morphology appeared wild type, and only relatively minor defects in filament organization were apparent (Figure 9B). Therefore, the delayed localization of Δ34 constructs does not have dramatic effects on final filament or sarcomere structure in animals that also express full-length MHC B.
DISCUSSION
An Intact Rod Is Required for Proper Assembly
Our experiments using C-terminally truncated myosin constructs have defined a four amino-acid region at the C terminus of the MHC A rod that is essential for viability (Figure 1). The observation that a small deletion within the coiled coil causes a marked disruption of early myosin function suggests that these residues are critical for early events in filament formation (Figure 3 and Table 1). The abnormal persistence of diffuse Δ34 protein (which lacks the tailpiece and four residues of coiled coil) during early muscle development suggests that loss of these rod residues delays proper localization and assembly into filamentous structures (Figures 5–7). The presence of Δ34 MHC did not disrupt the localization pattern of endogenous MHC within the same muscle cell. Therefore, loss of these rod residues seems to delay the organization of individual molecules but does not lead to dominant-negative effects, such as the initiation of ectopic structures, that incorporate full-length MHC.
Although the four terminal residues of the coiled coil are essential for viability, removal of these residues clearly results in a molecule that has only partial loss of function. The Δ34 constructs, which lack these residues, are able to rescue mutants that lack either single MHC isoform. Animals in which Δ34 replaces MHC A, the essential isoform, can have near-normal muscle cell organization and contain thick filaments of normal morphology (Figure 9). Therefore, in the presence of full-length MHC B, the four residues of MHC A coiled coil are not required for proper filament initiation or filament structure. Although animals expressing only Δ34 did not commence embryonic movement at the correct time, movement did eventually occur, as did the late localization of the truncated protein to filamentous structures. The motility phenotype and antibody staining results are consistent with the hypothesis that the Δ34 protein eventually forms functional filaments. Although we cannot rule out the possibility that filaments containing only Δ34 are abnormal, the tools required to examine the appropriate developing embryos at the ultrastructural level do not currently exist.
Comparison of the C. elegans MHC A sequence to other striated muscle myosins in the public databases revealed that two basic amino acids within the four-residue region are conserved in both vertebrate and invertebrate MHCs (Figure 2). Mutant constructs in which these basic residues are altered or deleted behave genetically like Δ34 (Figure 1 and Table 1). That is, these proteins have sufficient activity to rescue single mutant animals but do not support viability of double mutant animals that lack endogenous MHC. Although the precise manner in which loss of these charged residues within the rod affects myosin function is unknown, previous studies have shown that small perturbations in charge can have profound effects on assembly in vivo (Gengyo-Ando and Kagawa, 1991; Nock et al., 2000). Therefore, the functional defect may result directly from loss of two highly conserved charged residues that play a key role in the intermolecular interactions involved in driving filament assembly or specifying filament structure.
A number of studies in various systems point to the importance of the C-terminal rod in myosin assembly. In vertebrate striated muscle myosin, a 29-residue region of the coiled coil near the C terminus of the rod, the assembly-competent domain (ACD), has been proposed to be essential for ordered assembly of rod fragments in vitro and of truncated myosin expressed in COS cells (Sohn et al., 1997). Our studies did not alter or remove any residues within the ACD, so the role of the ACD in C. elegans has not yet been tested. Sohn et al. (1997) tested C-terminally truncated constructs in which the four-residue region defined in our study was removed and found no effect on assembly competence in vitro or in cell lines. Their observations are consistent with our data, which indicate that loss of the four terminal residues of coiled coil causes a partial loss of assembly function. Our genetic and antibody staining experiments were able to detect a delay in the assembly of the Δ34 protein because of the lethal consequences of delayed assembly in vivo and our ability to compare the localization of truncated protein to that of endogenous MHC A within the same muscle cell at different developmental stages.
MHC A-specific Function
Our current experiments confirm that the essential MHC A-specific activity is limited to the rod sequences. A chimeric molecule that contains only the MHC A rod has sufficient activity to rescue double mutant animals expressing no endogenous MHC (Figure 1). However, the observation that the Δ34 construct is able to provide MHC A-specific function in the single myo-3(0) mutant demonstrates that the presence of full-length MHC B allows viability when the only MHC A present has partial function. In a previous study, two regions of the MHC A rod sufficient for the essential MHC A-specific function were mapped by testing chimeric myosins in the myo-3(0) single-mutant background (Hoppe and Waterston, 1996). Our current results raise the possibility that the regions defined previously are important for MHC A isoform-specific function but may not have sufficient activity to support viability in the double-mutant background. Consistent with this possibility, studies of the MHC A-specific interaction with paramyosin led to the proposal that sequences along the length of the rod contribute to isoform-specific activity (Hoppe and Waterston, 2000).
The Tailpiece Domain Is Not Required for Filament Formation
In contrast to the effects of small deletions in the coiled coil, removal of the tailpiece alone does not discernibly alter early myosin function in animals that do not express full-length MHC in the body-wall muscle. Early myosin localization (Figures 5–7) and the onset of contraction (Figure 3) seem normal in animals expressing only Δ30 myosin, which lacks the tailpiece domain. In addition, tailpiece sequences are not required for isoform-specific localization within the thick filament (Figure 8) or for normal filament morphology (Figure 9). Because alterations within the adjacent rod region produce a dramatic defect in early myosin function in vivo, we believe it is unlikely that our failure to detect similar defects in the Δ30 protein is due to a lack of sensitivity in our assays. Instead, our data suggest that the two adjacent domains, the rod and the tailpiece, are required for distinct steps during muscle development in vivo. A very similar conclusion was reached in the in vitro analysis of proteolytically cleaved Acanthamoeba myosin II (Sathyamoorthy et al., 1990).
The ability of MHC A lacking the tailpiece (Δ30) to localize correctly and support the timely onset of contractile function suggests that the tailpiece residues do not play an important role in promoting assembly of the filament per se. Therefore, our data do not support a model in which tailpiece sequences in C. elegans striated muscle act to drive filament assembly or specify filament structure, as has been proposed for the tailpiece in Acanthamoeba myosin II (Sinard et al., 1989) and for smooth muscle MHC (Rovner et al., 2002). Our results are consistent with the observation that the assembly of Drosophila MHC into thick filaments of the distinct morphologies found in different skeletal muscle cell types is not dependent upon the identity of the tailpiece residues (Wells et al., 1996).
Our failure to detect defects in Δ30 localization also suggests that the tailpiece is unlikely to play an important role in regulating assembly of MHC into filaments. In smooth muscle, phosphorylation of tailpiece residues has been associated with the formation of a folded, assembly-incompetent form of the MHC molecule. Similarly, phosphorylation within the tailpiece domain can reduce assembly of nonmuscle MHCs in vitro (Murakami et al., 1998). If the tailpiece played a similar inhibitory role in C. elegans, we might expect loss of the tailpiece domain to lead to premature assembly of MHC into ectopic or otherwise aberrant structures. Instead, myosin stain in embryos expressing only Δ30 appears normal during early stages of muscle development (Figure 7). Furthermore, the Δ30 MHC A protein shows the appropriate isoform-specific staining pattern within the A-band in adult muscle cells (Figure 8). Therefore, our data do not support a model in which the C. elegans body-wall MHC tailpiece regulates the assembly competence of the individual molecule.
Possible Roles for the Tailpiece Residues
Our experiments with truncated MHC do not support a requirement for the tailpiece in early events of thick filament formation. Furthermore, because animals in which truncated MHC A (Δ30) replaces endogenous MHC A have normal isoform-specific localization and wild-type cellular organization (Figure 8), we can conclude that the tailpiece need not be present throughout the filament, nor in the filament center where initiation is thought to occur, for normal sarcomere patterning and maintenance. Therefore, our data suggest that any requirement for the tailpiece may be at the level of the individual filament rather than at the level of the single molecule.
Antibody staining experiments reveal that animals expressing truncated myosin lacking tailpiece residues (Δ30) exhibit defects in myosin localization in later embryogenesis (Figures 6 and 7). Therefore, it is possible that the tailpiece is required for the establishment or maintenance of filament position within the contractile apparatus. Two observations suggest that loss of the tailpiece may affect maintenance of sarcomere structure, rather than specification of its pattern. First, initial localization of Δ30 during embryogenesis appears normal, and the appearance of disordered myosin stain occurs when the embryo is actively moving. Second, electron microscopy (Figure 9) reveals that animals expressing only Δ30 contain filaments that are located internally in the cell, rather than being attached to the membrane adjacent to the hypodermis. Because the patterning of the contractile apparatus in C. elegans is thought to occur at this membrane (Hresko et al., 1994; Williams and Waterston, 1994), the ectopic filaments may have been displaced from their initial location. In Drosophila, loss of sarcomere structure upon contraction has been demonstrated in animals expressing an embryonic MHC isoform in adult flight muscle (Wells et al., 1996).
Two possible models for tailpiece function are suggested by the apparent loss of sarcomere structure after the onset of contractile activity. First, the tailpiece may mediate proper attachment to, or stabilization of, the M-line, which anchors thick filaments. Second, the tailpiece domain may be required for appropriate regulation of the contractile activity of the filament. A similar role has been proposed for the tailpiece of Acanthamoeba myosin II (Ganguly et al., 1990). Consistent with this possibility, mutations in C. elegans twitchin (encoded by unc-22) that disrupt contractile regulation lead to defects in sarcomere structure (Moerman et al., 1988; Benian et al., 1989).
Although the tailpiece does not play an essential role in early muscle development, our data suggest that the domain does affect the activity of myosin molecules during early stages. The early movement and terminal arrest phenotypes are more severe in animals expressing only Δ34 (which lacks the tailpiece and four rod residues) than in animals expressing only constructs that have mutations within the C-terminal rod and an intact tailpiece domain. The ability of the tailpiece to improve motility and embryonic elongation suggests that the presence of the tailpiece domain contributes to early myosin function. There are three plausible mechanisms by which tailpiece sequences may be acting during these stages: 1) the domain may directly contribute to early localization of the molecule, although this role is not essential or detectable in our studies of Δ30 protein; 2) the presence of the tailpiece may stabilize the coiled coil at the C terminus of the rod, which plays a key role in localization and assembly; and 3) the tailpiece contributes to some other early function, such as filament placement or contractile activity. Because our genetic and antibody staining experiments have proven to be sensitive (discussed above), our data favor the latter two models. However, further studies are required to elucidate tailpiece function in C. elegans. Because the organization of the contractile apparatus is not completely wild type in any strain expressing a single full-length MHC construct (our unpublished observations), these experiments must involve different approaches to test the possible role of the tailpiece in filament organization.
ACKNOWLEDGMENTS
We thank Henry Epstein and Irving Ortiz for providing antibodies. We thank Bob Waterston, Chelly Hresko, and Larry Schriefer for scientific discussions, and Doug Coulter and Daniela Gerhard for critical reading of the manuscript. We thank Tim Schedl and laboratory members for generosity with equipment, time, and ideas. We thank Sandhya Koushika and Mike Nonet for materials and instruction in preparation of worms for electron microscopy, and Marilyn Levy for sectioning, materials, and expertise. We are grateful for the helpful suggestions provided by anonymous reviewers. This work was funded by National Science Foundation grant 9905687. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E02–11–0728. Article and publication date are at www.molbiolcell.org/cgi/doi/10.1091/mbc.E02–11–0728.
REFERENCES
- Benian GM, Kiff JE, Neckelmann N, Moerman DG, Waterston RH. Sequence of an unusually large protein implicated in regulation of myosin activity in C. elegans. Nature. 1989;342:45–50. doi: 10.1038/342045a0. [DOI] [PubMed] [Google Scholar]
- Berger B, Wilson DB, Wolf E, Tonchev T, Milla M, Kim PS. Predicting coiled coils by use of pairwise residue correlations. Proc Natl Acad Sci USA. 1995;92:8259–8263. doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani L, Cohen CR. Phosphorylation favors folding in a catch muscle myosin. Proc Natl Acad Sci USA. 1987;84:4058–4062. doi: 10.1073/pnas.84.12.4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JH, Kuznicki J, Bowers B, Korn ED. Comparison of the actin binding and filament formation properties of phosphorylated and dephosphorylated Acanthamoebamyosin II. Biochemistry. 1982;21:6910–6915. doi: 10.1021/bi00269a045. [DOI] [PubMed] [Google Scholar]
- Deitiker PR, Epstein HF. Thick filament substructures in Caenorhabditis elegans: evidence for two populations of paramyosin. J Cell Biol. 1993;123:303–311. doi: 10.1083/jcb.123.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey CS, Deitiker PR, Epstein HF. Assembly-dependent phosphorylation of myosin and paramyosin of native thick filaments in Caenorhabditis elegans. Biochem Biophys Res Commun. 1992;186:1528–1532. doi: 10.1016/s0006-291x(05)81580-3. [DOI] [PubMed] [Google Scholar]
- Epstein HF, Waterston RH, Brenner S. A mutant affecting the heavy chain of myosin in Caenorhabditis elegans. J Mol Biol. 1974;90:291–300. doi: 10.1016/0022-2836(74)90374-x. [DOI] [PubMed] [Google Scholar]
- Epstein HF, Casey DL, Ortiz I. Myosin and paramyosin of Caenorhabditis elegansembryos assemble into nascent structures distinct from thick filaments and multi-filament assemblages. J Cell Biol. 1993;122:845–858. doi: 10.1083/jcb.122.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein HF, Ortiz I, Mackinnon LA. The alteration of myosin isoform compartmentation in specific mutants of Caenorhabditis elegans. J Cell Biol. 1986;103:985–993. doi: 10.1083/jcb.103.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Waterston RH. Proper expression of myosin genes in transgenic nematodes. EMBO J. 1989;8:3419–3428. doi: 10.1002/j.1460-2075.1989.tb08506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis GR, Waterston RH. Muscle organization in Caenorhabditis elegans: localization of proteins implicated in thin filament attachment and I-band organization. J Cell Biol. 1985;101:1532–1549. doi: 10.1083/jcb.101.4.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly CM, Atkinson AL, Attri AK, Sathyamoorthy V, Bowers B, Korn ED. Regulation of the actin-activated ATPase activity of Ancanthamoebamyosin II by copolymerization with phosphorylated peptides derived from the carboxyl-terminal end of the heavy chain. J Biol Chem. 1990;265:9993–9998. [PubMed] [Google Scholar]
- Gengyo-Ando K, Kagawa H. Single charge change on the helical surface of the paramyosin rod dramatically disrupts thick filament assembly in Caenorhabditis elegans. J Mol Biol. 1991;219:429–441. doi: 10.1016/0022-2836(91)90184-8. [DOI] [PubMed] [Google Scholar]
- Hall DH. Caenorhabditis elegans: Modern Biological Analysis of an Organism. San Diego: Academic Press; 1995. [Google Scholar]
- Hoppe PE, Waterston RH. A region of the myosin rod important for interaction with paramyosin in Caenorhabditis elegansstriated muscle. Genetics. 2000;156:631–643. doi: 10.1093/genetics/156.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe PE, Waterston RH. Hydrophobicity variations along the surface of the coiled-coil rod may mediate striated muscle myosin assembly in Caenorhabditis elegans. J Cell Biol. 1996;135:371–382. doi: 10.1083/jcb.135.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hresko MC, Williams BD, Waterston RH. Assembly of body wall muscle and muscle cell attachment structures in Caenorhabditis elegans. J Cell Biol. 1994;124:491–506. doi: 10.1083/jcb.124.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa H, Gengyo K, McLachlan AD, Brenner S, Karn J. Paramyosin gene (unc-15) of Caenorhabditis elegans: molecular cloning, nucleotide sequence and models for thick filament structure. J Mol Biol. 1989;207:311–333. doi: 10.1016/0022-2836(89)90257-x. [DOI] [PubMed] [Google Scholar]
- McLachlan AD, Karn J. Periodic charge distributions in the myosin rod amino acid sequence match cross-bridge spacings in muscle. Nature. 1982;299:226–231. doi: 10.1038/299226a0. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DM 3d, Ortiz I, Berliner G C, Epstein H F. Differential localization of two myosins within nematode thick filaments. Cell. 1983;34:477–490. doi: 10.1016/0092-8674(83)90381-1. [DOI] [PubMed] [Google Scholar]
- Moerman DG, Benian GM, Barstead RJ, Schriefer LA, Waterston RH. Identification and intracellular localization of the unc-22 gene product of Caenorhabditis elegans. Genes Dev. 1988;2:93–105. doi: 10.1101/gad.2.1.93. [DOI] [PubMed] [Google Scholar]
- Moerman DG, Fire A. Muscle. Structure, function, and development. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 417–470. [PubMed] [Google Scholar]
- Murakami N, Chauhan VP, Elzinga M. Two nonmuscle myosin II heavy chain isoforms expressed in rabbit brains: filament forming properties, the effects of phosphorylation by protein kinase C and casein kinase II, and location of the phosphorylation sites. Biochemistry. 1998;37:1989–2003. doi: 10.1021/bi971959a. [DOI] [PubMed] [Google Scholar]
- Nock S, Liang W, Warrick HM, Spudich JA. Mutational analysis of phosphorylation sites in the Dictyosteliummyosin II tail: disruption of myosin function by a single charge change. FEBS Lett. 2000;466:267–272. doi: 10.1016/s0014-5793(99)01796-2. [DOI] [PubMed] [Google Scholar]
- Rovner AS, Fagnant PM, Lowey S, Trybus KM. The carboxyl-terminal isoforms of smooth muscle myosin heavy chain determine thick filament assembly properties. J Cell Biol. 2002;156:113–123. doi: 10.1083/jcb.200107131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyamoorthy V, Atkinson MA, Bowers B, Korn ED. Functional consequences of the proteolytic removal of regulatory serines from the nonhelical tailpiece of Acanthamoebamyosin II. Biochemistry. 1990;29:3793–3797. doi: 10.1021/bi00467a028. [DOI] [PubMed] [Google Scholar]
- Schriefer L, Waterston RH. Phosphorylation of the N-terminal region of Caenorhaabditis elegans. J Mol Biol. 1989;207:451–454. doi: 10.1016/0022-2836(89)90267-2. [DOI] [PubMed] [Google Scholar]
- Sinard JH, Pollard TD. The effect of heavy chain phosphorylation and solution conditions on the assembly of Acanthamoebamyosin-II. J Cell Biol. 1989;109:1529–1535. doi: 10.1083/jcb.109.4.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinard JH, Stafford WF, Pollard TD. The mechanism of assembly of Acanthamoebamyosin-II minifilaments: minifilaments assemble by three successive dimerization steps. J Cell Biol. 1989;109:1537–1547. doi: 10.1083/jcb.109.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn RL, Vikstrom KL, Strauss M, Cohen C, Szent-Gyorgyi AG, Leinwand LA. A 29 residue region of the sarcomeric myosin rod is necessary for filament formation. J Mol Biol. 1997;266:317–330. doi: 10.1006/jmbi.1996.0790. [DOI] [PubMed] [Google Scholar]
- Squire JM. The Structural Basis of Muscle Contraction. New York: Plenum Press; 1981. [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Fishpool RM, Brenner S. Mutants affecting paramyosin in Caenorhabditis elegans. J Mol Biol. 1977;117:679–697. doi: 10.1016/0022-2836(77)90064-x. [DOI] [PubMed] [Google Scholar]
- Waterston RH. Muscle. In: Wood WB, editor. The Nematode Caenorhabditis elegans. Cold Spring Harbor, NY: Cold, Spring Harbor Laboratory Press; 1988. pp. 281–335. [Google Scholar]
- Waterston RH. The minor myosin heavy chain, mhcA, of Caenorhabditis elegansis necessary for the initiation of thick filament assembly. EMBO J. 1989;8:3429–3436. doi: 10.1002/j.1460-2075.1989.tb08507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells L, Edwards KA, Bernstein SI. Myosin heavy chain isoforms regulate muscle function but not myofibril assembly. EMBO J. 1996;15:4454–4459. [PMC free article] [PubMed] [Google Scholar]
- Williams BD, Waterston RH. Genes critical for muscle development and function in Caenorhabditis elegansidentified through lethal mutation. J Cell Biol. 1994;124:475–490. doi: 10.1083/jcb.124.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]