

Abstract
Fluorescence resonance energy transfer and fluorescence polarization anisotropy are used to investigate single molecules of the enzyme staphylococcal nuclease. Intramolecular fluorescence resonance energy transfer and fluorescence polarization anisotropy measurements of fluorescently labeled staphylococcal nuclease molecules reveal distinct patterns of fluctuations that may be attributed to protein conformational dynamics on the millisecond time scale. Intermolecular fluorescence resonance energy transfer measurements provide information about the dynamic interactions of staphylococcal nuclease with single substrate molecules. The experimental methods demonstrated here should prove generally useful in studies of protein folding and enzyme catalysis at single-molecule resolution.
Single-molecule spectroscopy can provide information about complex biological molecules and systems that is difficult to obtain from ensemble measurements (1–6). For example, one can observe the time trajectories of single molecules in biochemical reactions that cannot be synchronized in ensemble experiments. In a population of molecules heterogeneous in a particular property, single-molecule spectroscopy can also resolve and quantitatively compare distinct subpopulations that would be indistinguishable at the ensemble level.
Fluorescence resonance energy transfer (FRET) measurements between single pairs of acceptor and donor fluorophores can yield information about structural relationships and distance fluctuations between regions of a single biomolecule or between components of an interacting system of biomolecules (7–10). In addition, the rotational dynamics of a single fluorophore can be probed by monitoring fluctuations in fluorescence polarization (11–14). Here we develop the techniques of single-pair FRET (spFRET) and single-molecule fluorescence polarization anisotropy (smFPA) and show how they can be used to observe the conformational fluctuations and catalytic reactions of enzymes at single-molecule resolution.
Staphylococcal nuclease (SNase) is a 19 kDa Ca2+-dependent enzyme that catalyzes the hydrolysis of DNA and RNA into mono- and dinucleotides (15). Its catalytic mechanism, thermodynamic stability, and folding pathway have been studied extensively at the ensemble level (16–21). To probe the conformational dynamics of SNase and its interactions with substrate at single-molecule resolution, three experimental methods were used. First, intramolecular spFRET was measured between donor and acceptor fluorophores attached to single SNase proteins. Second, single-molecule fluorescence polarization anisotropy measurements were performed by using SNase labeled singly with one type of fluorophore. Third, intermolecular spFRET was measured between donor-labeled SNase and acceptor-labeled DNA substrate.
Using intramolecular spFRET measurements on single SNase protein molecules, we observe interesting dynamics including gradual fluctuations in the FRET efficiencies. A combination of smFPA measurements, simulations, and spectral fluctuation measurements shows that distance fluctuations must contribute to the observed FRET efficiency fluctuations. We also use smFPA and spFRET to study protein–inhibitor binding and show that these methodologies are sensitive enough to distinguish between the free and inhibitor-bound states of the enzyme. In the intermolecular spFRET part of this work, we study the interactions between single SNase proteins and single-stranded DNA (ssDNA) substrates and develop methods that are useful for studying complex aspects of enzyme catalysis such as processivity and the statistics of enzymatic events. In the course of this work, we have also developed data analysis methods such as autocorrelation analysis of spFRET and smFPA time-trace data that will be generally useful in single-molecule experiments.
MATERIALS AND METHODS
Protein Conjugation.
Tetramethylrhodamine (TMR, donor) and Cy5 (acceptor) were selected as the FRET pair for their well-separated emission wavelengths (>100 nm) and large Förster radius [assuming isotropic and rapid rotation for both TMR and Cy5 (κ2 = 2/3) and applying the available spectral data, the calculated 50% energy transfer distance, or R0, is 53 Å, according to the equation
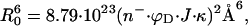 |
where n is the index of refraction of the medium, ϕD is the donor quantum yield, κ2 is the orientational factor, and J is the spectral overlap of donor emission and acceptor absorption in M−1⋅cm3]. TMR maleimide (Molecular Probes) was conjugated to the Cys28 of the K28C mutant of SNase in 50 mM glycine, pH 7.0/2% dimethyl sulfoxide over 2 h at 25°C. The conjugate was purified by size-exclusion chromatography with Sephadex G-25 resin. Dye attachment was verified by denaturing polyacrylamide gel electrophoresis. Cy5 succinimidyl ester (Amersham Pharmacia) was conjugated to TMR–SNase under the same labeling conditions and purified and characterized as described above. High-resolution mass spectrometry showed that about 15–20% of protein molecules were conjugated to only one Cy5 fluorophore with a smaller percentage of double labeling. The Km and Vmax values of TMR- and TMR/Cy5-dual labeled SNase were indistinguishable from those of the unlabeled protein.
DNA Conjugation.
The sequence of 5′-end-Cy5-labeled oligonucleotide was 5′XACGTCACGCTAGTCAGTCATCTTGCACATGTCCTTGAGA, where X represents Amino Modifier C6 dT (Glen Research, Sterling, VA). The thymidine analog was labeled with Cy5 succinimidyl ester in 0.1 M sodium carbonate buffer, pH 9.0, over 2–12 h at 25°C. The conjugate was first passed through a NAP5 column (Pharmacia) with water, then purified by RP-HPLC using a gradient of 5 → 30% acetonitrile in 50 mM triethylammonium acetate. A FPLC Superose 12 column (eluent of 20 mM NaH2PO4/150 mM NaCl, pH 6.3) was used to remove residual free dye. 3′-end-labeled oligonucleotide was prepared in an identical manner using 3′ Amino Modifier C7 CPG (Glen Research).
Protein Immobilization.
Glass coverslips were derivatized with N-[(3-trimethoxysilyl)propyl] ethylenediamine triacetic acid (United Chemical Technologies, Bristol, PA). Specific immobilization of proteins by means of hexahistidine tag was established as follows: (i) more than 90% of immobilized proteins could be removed by flowing over the coverslip 100 mM EDTA, pH 7.9 buffer, and (ii) without the prior application of Ni2+ to the derivatized glass, a 10-fold reduction in SNase binding was observed.
Experimental Setup.
A laser scanning confocal microscope with a focused laser spot (0.4 μm) was used to image surface-immobilized protein molecules and measure their individual emission time trajectories (9, 12, 13). For spFRET measurements, donor and acceptor emissions were separated by a 630-nm dichroic mirror and collected through a 585-nm band pass filter and 650-nm long pass filter, respectively. smFPA measurements of fluorophore rotational dynamics were performed by splitting the emission from a single fluorophore with a polarizing beam-splitting cube into two orthogonally polarized components, Is and Ip, and simultaneously recording them on separate detectors (12, 13). To measure the dipole effects in absorption, the excitation beam was alternated between two orthogonal polarizations by using an electro-optic modulator. Argon ion laser light (15 μW) at 514 nm was used for studying the energy transfer, as well as the rotational dynamics and spectral fluctuations of the donor, whereas 15 μW of HeNe laser light at 632 nm was used to study the rotational dynamics and spectral fluctuations of the acceptor. The spectral fluctuations of donor and acceptor fluorophores conjugated to SNase were measured by splitting the fluorophore emission spectrum with a dichroic mirror with a cutoff at the peak emission and simultaneously recording the two emission halves on separate detectors.
spFRET Analysis.
The instantaneous FRET efficiency E(t) was calculated from the donor and acceptor emission intensities, Id and Ia, according to the equation E = [1 + γId/Ia]−1, where γ is a correction factor determined to be 0.8 in the following manner. Another expression for E is [1 + Id0φa/Ia0φd]−1 where Id0 and Ia0 are the true donor intensity and the sensitized emission intensity of the acceptor in the presence of energy transfer. The measured intensities are reduced by factors of ηa and ηd because of overall instrument detection efficiencies. It can be shown that the correction factor γ (=ηaφa/ηdφd) for the expression E = [1 + γId/Ia]−1 is equivalent to ΔIa/ΔId , where ΔIa and ΔId are the acceptor and donor intensity changes, respectively, on acceptor photobleaching. ΔIa/ΔId was determined from 45 acceptor photobleaching events, and its distribution was centered at 0.8. The E(t) fluctuation time traces were processed as follows. A median filter (five points) was used to remove spikes in the data caused by triplet state-induced blinking. Dark states were discounted by skipping points with Id + Ia < 10 counts or E < 0.3 (gradual fluctuations that lead to E < 0.3 were not observed, so this screening step does not remove them). The time average of E(t), Ē, and its autocorrelation function were calculated from the remaining data points. The autocorrelation function was fit with aE2exp(-t/τE), where aE and τE are the E(t) fluctuation amplitude and time scale.
smFPA Analysis.
Rotational fluctuations were quantified by calculating the effective emission dipole angle θem(t). That is, although smFPA measurements do not directly report the in-plane dipole orientation when the fluorophore is rotating, it is possible to define an effective average dipole angle parameter θem(t) in the following way:
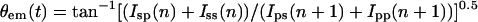 |
1 |
where Iij are a set of four consecutive data points taken at times n and n + 1. The first index denotes the excitation polarization and the second denotes the emission polarization. The angle parameter θem(t) can be thought of as the average in-plane cone angle center of the rotating emission dipole measured with circularly polarized light at the mid-time point between n and n + 1. The time average of θem(t) and its autocorrelation function were calculated. The autocorrelation function was fit with aR2exp(-t/τR), where aR and τR are the θem(t) fluctuation amplitude and time scale.
RESULTS AND DISCUSSION
A laser scanning confocal microscope with a focused laser spot (0.4 μm) was used to image surface-immobilized protein molecules and measure their individual emission time trajectories (9, 12, 13). SNase proteins were synthesized for imaging with C-terminal hexahistidine tags and a Lys28 to Cys mutation (22, 23), labeled at Cys28 with the energy donor TMR and nonspecifically labeled with the energy acceptor Cy5. The labeled proteins were immobilized on Ni2+-diaminetriacetic acid-derivatized glass plates at a density of less than 1 protein per 2 μm2 area in 40 mM glycine, 10 mM CaCl2, pH 9.5 (buffer A). In some cases, an oxygen-scavenging system [50 μg/ml glucose oxidase, 10 μg/ml catalase, 18% (wt/wt) glucose, 1% (wt/vol)] β-mercaptoethanol) was added to prolong fluorophore lifetimes (1).
Protein-conjugated donor fluorophores were selectively excited with circularly polarized 514 nm Ar+ light. The sample stage was raster-scanned as the donor and acceptor emissions were simultaneously recorded on separate detectors. The two resulting images were combined to form a composite image with donor and acceptor signals false colored in green and red, respectively. Each spot in the composite of Fig. 1a represents a single protein. During the course of the scan, most molecules initially show large acceptor emission with no or very small donor emission. Then abrupt photobleaching of the acceptor occurs, concurrent with an increase in donor emission. This anticorrelated switch in emission intensities is evidence that the single donor fluorophore was transferring energy to the acceptor fluorophore(s) before photobleaching of the acceptor (9).
Figure 1.

(a) Dual-color composite image of doubly labeled SNase molecules immobilized by means of histidine tag in buffer A at 23°C. The 514-nm excitation laser spot (Ar+ light, 15 μW) was scanned from top to bottom and left to right. Each spot represents a single protein. Donor emission is colored green and acceptor emission is colored red. (b) Emission time trace of a single double-labeled SNase molecule. The resolution of the measurement is 5 msec. The instantaneous donor and acceptor emission intensities, Id and Ia (squares and circles, respectively), are related to the energy transfer efficiency by the expression E(t) = [1 + γId/Ia]−1, where γ is a correction factor determined to be 0.8. The SNase molecule shown here displays a very high degree of energy transfer (high acceptor intensity, low donor intensity) from 0 to 80 msec. At 80 msec, photodestruction of the acceptor occurs and donor emission simultaneously increases. The inverse correlation is direct evidence for spFRET.
When an individual SNase molecule is positioned under the laser excitation spot, it is possible to record the donor and acceptor emissions as a function of time with 5-msec resolution; a representative time trace is shown in Fig. 1b. The inverse relationship between donor and acceptor emission intensities at 80 msec is again indicative of spFRET. In the subsequent analysis of spFRET, we restrict our attention to those that show the simultaneous acceptor photobleaching and donor signal recovery event to screen out SNase molecules with no acceptor or multiple acceptors. In principle, this does not completely exclude the possibility of multiple acceptor labeling; the photobleaching of one of multiple acceptors that are in close proximity to the donor will not show up as a detectable event since the energy pumped through the donor will be redistributed among the remaining acceptors.
In addition to abrupt photophysical events such as photobleaching and blinking, the emission time traces of many doubly labeled SNase molecules were found to display gradual fluctuations in the donor and acceptor emissions, ranging in time scale from 10 msec to 1 sec (Figs. 2 a–c). These temporal fluctuations may reflect conformational changes in the protein side chains or backbone that give rise to changes in energy transfer efficiency, E(t), according to the relationship E(t) = [1 + (R(t)/R0)6]−1, where R(t) is the time-dependent distance between donor and acceptor fluorophores and Ro is the Förster radius. However, fluctuations in E(t) can also arise from other factors, namely fluorophore reorientation and spectral shift. To quantify the contribution of these latter factors to E(t) fluctuations, the emission characteristics of SNase molecules labeled singly with TMR or Cy5 were measured.
Figure 2.

(a) Emission time trace of a doubly labeled TMR/Cy5 SNase molecule immobilized on glass in buffer A with an oxygen scavenging system (donor emission is dotted, acceptor emission is solid). A median filter was applied to reduce the noise caused by triplet state-induced fluorophore blinking. There are large and gradual fluctuations in Id and Ia that occur over tens of milliseconds. (b) FRET efficiency time trace calculated from a according to E(t) = [1 + γId/Ia]−1. The inset shows the autocorrelation of E(t) together with an exponential fit. τE is the characteristic time scale of the E(t) fluctuations displayed in this trace. (c) Histogram of E(t) fluctuation time constants τE for 100 doubly labeled SNase molecules. The values range from 10 msec to 1 sec, with the average being 41 msec. (d) Histogram of E(t) fluctuation time constants τE for doubly labeled SNase in the presence of active-site inhibitor pTp (50 mM pTp, Kd = 100 nM). These time constants are considerably larger (average = 133 msec) than those measured for free SNase. (e) Scatter plot of E(t) fluctuation amplitudes aE vs. mean energy transfer efficiencies Ē for free SNase (squares) and pTp-bound SNase (circles). The scatter in Ē may reflect the nonspecific nature of Cy5 labeling. The solid line represents the maximum possible contribution of dipole fluctuations of Cy5 to aE for free SNase. Only the molecules that display large E(t) fluctuations (>70% of the total) are shown. The others are concentrated around the bottom right corner (not shown) and are likely caused by Cy5 labeling at a site close to Cys28.
smFPA measurements of fluorophore rotational dynamics were performed by splitting the emission from a single fluorophore with a polarizing beam-splitting cube into two orthogonally polarized components, Is and Ip, and simultaneously recording them on separate detectors (13). To measure the dipole effects in absorption, the excitation beam was alternated between two orthogonal polarizations. A typical smFPA time trace for a single TMR fluorophore attached to Cys28 of SNase is shown in Fig. 3a. As previously demonstrated for fluorophores tethered to DNA, immobile fluorophores generate correlated Is and Ip signals, whereas rapidly rotating fluorophores generate anticorrelated signals (12, 13). Data collected and analyzed for over 100 TMR-labeled SNase molecules indicate that Cys28-linked TMR rotates rapidly, with little restriction, on a time scale that is much faster than the integration time (5 ms) and slower than or comparable to the radiational lifetime of the fluorophore (Fig. 3 b and c). The rotation of Cy5, on the other hand, displays significant fluctuations on the 10 msec–1 sec time scale (Fig. 3d). Calculations and simulations described below demonstrate that these are clearly insufficient to account for the large fluctuations in FRET efficiency observed (Fig. 2e). The combination of free TMR rotation and hindered Cy5 rotation restricts the orientational factor κ2 in Ro to the range 1/3 to 4/3 (the worst-case scenario involves static dipole orientation for Cy5 that changes direction on the millisecond time scale). Assuming no variations in distance between donor and acceptor, the reduced uncertainty in κ2 predicts fluctuations in E(t) that are far smaller than those observed, according to the equation ΔE = E(1 − E)Δκ2/κ2. This conclusion holds even for the case of nonspecifically Cy5-labeled protein, where some or all of the Cy5 molecules might be in the worst-case scenario discussed above. In addition, the distribution of Cy5 rotational fluctuation time scales differs substantially from that of E(t) fluctuation time scales (Figs. 2c and 3d).
Figure 3.

(a) A typical smFPA time trace of Cys28 TMR-labeled SNase immobilized by means of histidine tag in buffer A. The two orthogonally polarized emissions, Is (dotted) and Ip (solid), are displayed as a function of time. Because correlated emissions correspond to a fixed fluorophore dipole and anticorrelated emissions correspond to a rapidly rotating fluorophore dipole, the SNase-conjugated TMR molecule shown here is rotating rapidly (much faster than the data integration time of 5 msec). (b) The angle parameter θem(t) calculated from a. The value of θem(t) is close to 45° when the fluorophore is rotating rapidly with little restriction (14). The break in graph represents a dark-state transition. In the inset is the autocorrelation of the angle parameter (in circles); there are clearly no significant temporal fluctuations in TMR rotation on the millisecond time scale. Also in the inset is the angle parameter autocorrelation for pTp-bound Cys28 TMR-labeled SNase (squares) together with an exponential fit. Here the rotational fluctuations are substantial, with a characteristic time constant τR of 96 msec. (c) Histograms of θ̄ for immobilized TMR-labeled SNase molecules with and without inhibitor pTp. The distribution is narrowly centered at 45° for uninhibited SNase, indicative of free and rapid rotation of the attached TMR fluorophore. The TMR of inhibitor-bound SNase, on the other hand, displays hindered and fluctuating rotational behavior, indicated by the broader mean angle parameter histogram; (d) Histogram of rotational fluctuation time constants τR for Cy5-labeled SNase. Only those molecules that showed single-step photobleaching were included to screen out multiply Cy5-labeled cases. The average value of τR is 220 msec, considerably longer than the majority of E(t) fluctuation time constants (average τE = 41 msec, Fig. 2c) .
Observations of spectral shifts by using SNase labeled singly with either TMR or Cy5 (estimated by using known dye and optical filter spectra) were infrequent; they were less than 5 nm in amplitude with time scales >300 msec, indicating that spectral shifts make negligible contributions to E(t) fluctuations. Thus, the above experiments show that, even in the case of nonspecific Cy5 labeling, orientational effects and spectral shifts cannot account for the magnitude and time scale of the observed fluctuations in FRET efficiency, i.e., these fluctuations must involve distance fluctuations and likely reflect conformational dynamics of the protein itself, possibly amplified by interactions with the dyes and their tethers.
spFRET and smFPA can also be used to probe the effects of ligand binding on protein dynamics. For example, TMR rotation, while rapid and unrestricted when conjugated to ligand-free SNase, displays hindered rotational dynamics and temporal fluctuations in the presence of the SNase active-site inhibitor deoxythymidine diphosphate (pTp) (Fig. 3 b and c) (16–21). Because Cys28 is not in the immediate vicinity of the active site, these changes in smFPA likely reflect changes in protein dynamics that result from inhibitor binding, such as decreases in backbone or side-chain flexibility. E(t) fluctuation time scales are also affected by the inhibitor binding; addition of the inhibitor increases the time scales of the FRET efficiency fluctuations (Fig. 2d). Average values of τE and τR were determined by fitting the histograms (bin size 30 msec) to a single exponent. Simulations indicate that the probability of statistical aberration alone giving rise to the differences in time constants observed (220 msec vs. 41 msec for Figs. 3d and 2c, and 133 msec vs. 41 msec for Figs. 2d and 2c) is less than 0.1%, given the number of proteins studied. The distributions in τE and τR must also include the effects of nonspecificity of Cy5 labeling. However, the data do not show any dominant peaks in the fluctuation time-scale distribution; hence it is unlikely that biased sampling of certain Cy5 labeling sites within the 100 molecules studied is responsible for the differences among the cases compared. These changes in spFRET could reflect changes in protein dynamics that result from inhibitor binding, but orientational effects of nonspecifically bound Cy5 labels could make significant contributions.
Intramolecular FRET is a powerful technique that has the potential to advance our understanding of the conformational states and dynamics of biological macromolecules in equilibrium, on ligand binding, during folding and denaturation, and during catalysis, in ways that ensemble methods cannot. We note that intramolecular spFRET can be a more powerful technique than smFPA because the former probes the internal conformational states in the center-of-mass frame of the system and hence is less prone to complications due to the overall motion of the biological molecule.
Intermolecular spFRET can be used to investigate interactions between a pair of biomolecules, such as TMR-labeled SNase and Cy5-labeled DNA substrate (10). Intermolecular spFRET measurements were made using the SNase mutant D40G (kcat = 7s−1, Km = 55 μM), whose turnover rate is better matched to the 5-msec resolution of our current methodology than wild-type SNase (kcat = 100 s−1, Km = 55 μM) (16–21).
TMR-labeled protein was immobilized on derivatized glass plates by means of histidine tag in buffer A while a constant flow (0.1 ml/min) of a 10-nM solution of Cy5-labeled 40-nt ssDNA was flowed over the protein. Under these conditions, Cy5-labeled DNA adhered to the derivatized glass at a density of approximately 10 molecules per 1 μm2 and remained fairly constant throughout the experiment (the Cy5-DNA molecules were visualized by using direct 632 nm HeNe laser excitation of the Cy5 fluorophores). Therefore, the probability of FRET arising from random proximity between SNase and DNA is at most 1/1,000.
Emission time traces were collected by finding an enzyme using the donor emission and waiting for the arrival of an acceptor-labeled DNA molecule. Anticorrelated donor and acceptor emissions are again indicative of spFRET (Fig. 4a). For each measured FRET event, the duration of the FRET signal (time for which there is continuous acceptor emission), or τassoc, was extracted. τassoc represents the duration of one of several possible FRET-generating events: single-instance cleavage of DNA substrate by SNase, multiple and successive cleavages of DNA by SNase, binding and unbinding of DNA by SNase without cleavage, or nonspecific interaction between SNase and DNA. The value of τassoc may not necessarily represent the full enzyme–DNA association time because of fluorophore rotation and/or photobleaching. When the integrated acceptor emission is scatter plotted against τassoc for 45 spFRET time traces, the points fall under a 1/x-shaped curve, indicating that Cy5 photobleaching plays a role in shifting the values of τassoc to lower times.
Figure 4.

(a) Emission time trace of a single TMR-labeled wild-type SNase molecule interacting with single Cy5-labeled 40-nt ssDNA molecule(s) (donor emission in squares, acceptor emission in circles). The donor signal decreases every time the acceptor signal increases (signifying the initiation of energy transfer) and vice versa (signifying the termination of energy transfer); this is direct evidence for spFRET. (b) Histograms of τassoc for D40G SNase with 5′-Cy5-ssDNA and 3′-Cy5-ssDNA. The average duration of the FRET signals measured from interactions between donor-labeled SNase and 5′-end acceptor-labeled substrate are longer (average = 257 msec) than those measured with 3′-end acceptor-labeled substrate (average = 110 msec). This difference is consistent with a SNase cleavage mechanism in which the 5′ cleavage product is released more slowly than the 3′ cleavage product, or the enzyme catalyzes cleavage processively in the 3′ to 5′ direction. (c) Histograms of DNA-SNase association times, τassoc, for cleavage-impaired D21Y SNase with 5′-Cy5-ssDNA (left; average τassoc = 169 msec) and 3′-Cy5-ssDNA (right; average τassoc = 162 msec). This control experiment demonstrates that fluorophore photophysics and statistical aberrations do not account for the differences in association times observed when using 5′- and 3′-end-labeled substrates with the cleavage-competent D40G SNase mutant.
To investigate the SNase-catalyzed DNA cleavage reaction, τassoc values were extracted from approximately 200 spFRET events recorded between D40G SNase and 40-nt ssDNA labeled at either the 3′ or 5′ terminus with Cy5. As a control, values of τassoc were also measured using the SNase mutant D21Y, which is approximately 105-fold decreased in activity relative to wild-type SNase. Histograms of τassoc values for D40G and D21Y SNase are shown in Fig. 4 b and c. Average values of τassoc were determined by fitting the histograms (bin size 200 msec) to a single exponent, assuming underlying Poisson statistics.
D21Y SNase, with a kcat of 1.5 × 10−3 sec−1, is virtually incapable of catalyzing DNA hydrolysis, although it can bind reversibly to DNA with approximately the same affinity as wild-type and D40G SNase (Km ≈ 55 μM (16–21). Because the interactions between D21Y and 3′- or 5′-end-labeled DNA are expected to have identical lifetimes in the absence of hydrolysis, this D21Y system can be regarded as a control for the intermolecular FRET experiment with D40G SNase. Histograms of the compiled τassoc values are shown in Fig. 4c, and the average values of τassoc obtained by using the 5′- and 3′-end-labeled ssDNAs in the latter case (with D21Y SNase) are very similar. It is highly unlikely that differences in photophysics when using 3′- and 5′-end-labeled DNA are so closely offset by differences between D21Y interaction times with these DNAs. Hence, this result indicates that there are no significant differences in the photophysics between cases where the fluorophore is attached to the 3′ and 5′ termini of ssDNA.
In contrast, different average values of τassoc were obtained for the cleavage of 3′- and 5′-end-labeled DNAs by D40G SNase (110 msec and 257 msec, respectively) as shown in Fig. 4b. Simulation showed that the probability of statistical aberration alone generating this observed difference in τassoc values is less than 0.05%, given the number of proteins studied. Therefore, these differences in τassoc may reflect differential off-rates of bound 3′- and 5′-end-labeled cleavage products. The results are also consistent with a 3′ to 5′ processive cleavage reaction in which the enzyme sequentially hydrolyzes the DNA without dissociation. In such a mechanism, a 3′ to 5′ processive enzyme would remain bound to the 5′-end-labeled cleavage product and retain a FRET signal during strand degradation, whereas the 3′-end labeled fragment would dissociate after a single cleavage event. Indeed, earlier results from our laboratory showed that a 5′-32P end-labeled 64-nt oligodeoxynucleotide was either degraded into small fragments or left fully intact by wild-type SNase, suggesting a processive cleavage mechanism (24).
In summary, observations of energy transfer and fluorophore rotation in single-molecule and single-pair systems are capable of yielding important insights into protein structure and function. The experimental methodology used here is general and may be applied to the study of many biological processes on a single-molecule level. Further developments, including improvements in our ability to label multiple sites selectively in a protein, should make it possible to follow a variety of biochemical processes with greater precision.
Acknowledgments
Financial support for this work was provided by the National Institutes of Health (Grant No. GM49220) and by the Director, Office of Energy Research, Office of Basic Energy Sciences, Division of Materials Sciences of the Department of Energy (Contract No. DE-AC03-76SF00098). P.G.S. is a Howard Hughes Medical Institute Investigator. A.Y.T. is supported by a National Science Foundation predoctoral fellowship. W.B.C. is supported by an Alexander Hollaender Postdoctoral Fellowship, sponsored by the Department of Energy and administered by Oak Ridge Institute for Science and Education. We are grateful to David King for high-resolution protein mass spectral analysis and to Chris Gandhi, Michael Gelman, Ted Laurence, and Andy Martin for their contributions.
ABBREVIATIONS
- FRET
fluorescence resonance energy transfer
- spFRET
single-pair FRET
- smFPA
single-molecule fluorescence polarization anisotropy
- SNase
staphylococcal nuclease
- TMR
tetramethylrhodamine
- pTp
deoxythymidine diphosphate
- ssDNA
single-stranded DNA
References
- 1.Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T. Nature (London) 1995;374:555–559. doi: 10.1038/374555a0. [DOI] [PubMed] [Google Scholar]
- 2.Sase I, Miyata H, Corrie J E, Craik J S, Kinosita K. Biophys J. 1995;69:323–328. doi: 10.1016/S0006-3495(95)79937-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vale R D, Funatsu T, Pierce D W, Romberg L, Harada Y, Yanagida T. Nature (London) 1996;380:451–453. doi: 10.1038/380451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt T, Schutz G J, Baumgartner W, Gruber H J, Schindler H. Proc Natl Acad Sci USA. 1996;93:2926–2929. doi: 10.1073/pnas.93.7.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sase I, Miyata H, Ishiwata S, Kinosita K. Proc Natl Acad Sci USA. 1997;94:5646–5650. doi: 10.1073/pnas.94.11.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu H P, Xun L, Xie X S. Science. 1998;282:1877–1882. doi: 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- 7.Stryer L. Science. 1968;162:526. doi: 10.1126/science.162.3853.526. [DOI] [PubMed] [Google Scholar]
- 8.Meer B. Resonance Energy Transfer: Theory & Data. New York: VCH; 1994. [Google Scholar]
- 9.Ha T, Enderle T, Ogletree D F, Chemla D S, Selvin P R, Weiss S. Proc Natl Acad Sci USA. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schütz G J, Trabesinger W, Schmidt T. Biophys J. 1998;74:2223–2226. doi: 10.1016/S0006-3495(98)77931-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belford G C, Belford R C, Weber G. Proc Natl Acad Sci USA. 1972;69:1392–1393. doi: 10.1073/pnas.69.6.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ha T, Enderle T, Chemla D S, Selvin P R, Weiss S. Phys Rev Lett. 1996;77:3979–3982. doi: 10.1103/PhysRevLett.77.3979. [DOI] [PubMed] [Google Scholar]
- 13.Ha T, Glass J, Enderle T, Chemla D S, Weiss S. Phys Rev Lett. 1998;80:2093–2096. [Google Scholar]
- 14.Warshaw D M, Hayes E, Gaffney D, Lauzon A M, Wu J, Kennedy G, Trybus K, Lowey S, Berger C. Proc Natl Acad Sci USA. 1998;95:8034–8039. doi: 10.1073/pnas.95.14.8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blackburn S. Enzyme Structure and Function. New York: Dekker; 1976. [Google Scholar]
- 16.Hynes T R, Fox R O. Proteins Struct Funct Genet. 1991;10:92–105. doi: 10.1002/prot.340100203. [DOI] [PubMed] [Google Scholar]
- 17.Cotton F A, Hazen E E, Legg M J. Proc Natl Acad Sci USA. 1979;76:2551–2555. doi: 10.1073/pnas.76.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serpersu E, Shortle D, Mildvan A S. Biochemistry. 1987;26:1289–1300. doi: 10.1021/bi00379a014. [DOI] [PubMed] [Google Scholar]
- 19.Evans P A, Kautz R A, Fox R O, Dobson C M. Biochemistry. 1989;28:362–370. doi: 10.1021/bi00427a050. [DOI] [PubMed] [Google Scholar]
- 20.Hale S P, Poole L B, Gerlt J A. Biochemistry. 1993;32:7479–7487. doi: 10.1021/bi00080a020. [DOI] [PubMed] [Google Scholar]
- 21.Wu P, Brand L. Biochemistry. 1994;33:10457–10462. doi: 10.1021/bi00200a029. [DOI] [PubMed] [Google Scholar]
- 22.Chapman E, Thorson J S, Schultz P G. J Am Chem Soc. 1997;119:7151–7152. [Google Scholar]
- 23.Corey D R, Pei D, Schultz P G. Biochemistry. 1989;28:8277–8286. doi: 10.1021/bi00447a004. [DOI] [PubMed] [Google Scholar]
- 24.Corey D. Ph.D. thesis. Berkeley: Univ. of California; 1990. [Google Scholar]