

Abstract
The recent expression of an azurin mutant where the blue type 1 copper site is replaced by the purple CuA site of Paracoccus denitrificans cytochrome c oxidase has yielded an optimal system for examining the unique electron mediation properties of the binuclear CuA center, because both type 1 and CuA centers are placed in the same location in the protein while all other structural elements remain the same. Long-range electron transfer is induced between the disulfide radical anion, produced pulse radiolytically, and the oxidized binuclear CuA center in the purple azurin mutant. The rate constant of this intramolecular process, kET = 650 ± 60 s−1 at 298 K and pH 5.1, is almost 3-fold faster than for the same process in the wild-type single blue copper azurin from Pseudomonas aeruginosa (250 ± 20 s−1), in spite of a smaller driving force (0.69 eV for purple CuA azurin vs. 0.76 eV for blue copper azurin). The reorganization energy of the CuA center is calculated to be 0.4 eV, which is only 50% of that found for the wild-type azurin. These results represent a direct comparison of electron transfer properties of the blue and purple CuA sites in the same protein framework and provide support for the notion that the binuclear purple CuA center is a more efficient electron transfer agent than the blue single copper center because reactivity of the former involves a lower reorganization energy.
The CuA centers (1) serve as the electron uptake site in the terminal respiratory enzyme cytochrome c oxidase (2) and also as a redox center in nitrous oxide reductase (N2OR) (3). A combination of x-ray structural characterization (4–7) and spectroscopic studies (for example, see refs. 8–17) on native enzymes, water-soluble fragments containing the CuA center (18–20), engineered CuA centers (21–23), and inorganic model compounds (24, 25) has established CuA as a mixed valence [Cu(1.5) − Cu(1.5)] (S = 1/2) center with two copper ions in a Cu2S2 diamond core, and these studies have provided a firm basis for understanding the structure and function of this class of biological copper centers. An immediate question that this unusual structure raised was what functional advantage has led to its selection, in particular compared with the type 1 (T1) blue copper centers. At least two distinct, though not mutually exclusive, rationales have been brought up so far. One is that the delocalized mixed-valence structure of the CuA site would facilitate the unidirectional long-range electron transfer (ET) to the cytochrome a site of the enzyme (11, 26). The other suggested that the CuA structure would yield a lower reorganization energy, as the metal-ligand bond length changes upon ET would amount to only half of those occurring in a mononuclear site (11, 14, 27).
To address the above question, ET studies on both the blue copper (28–31) and the purple CuA proteins (26, 32–36) have been carried out. An ideal system to directly answer the above question will be a well-characterized protein where either the blue copper or the purple CuA center is placed at the same location and ET is known to occur through the same protein framework. In this way many parameters affecting the direct comparison of ET efficiency of the two copper centers (such as different protein sequences and pathways) can be minimized and the difference between the two distinct copper centers can be highlighted.
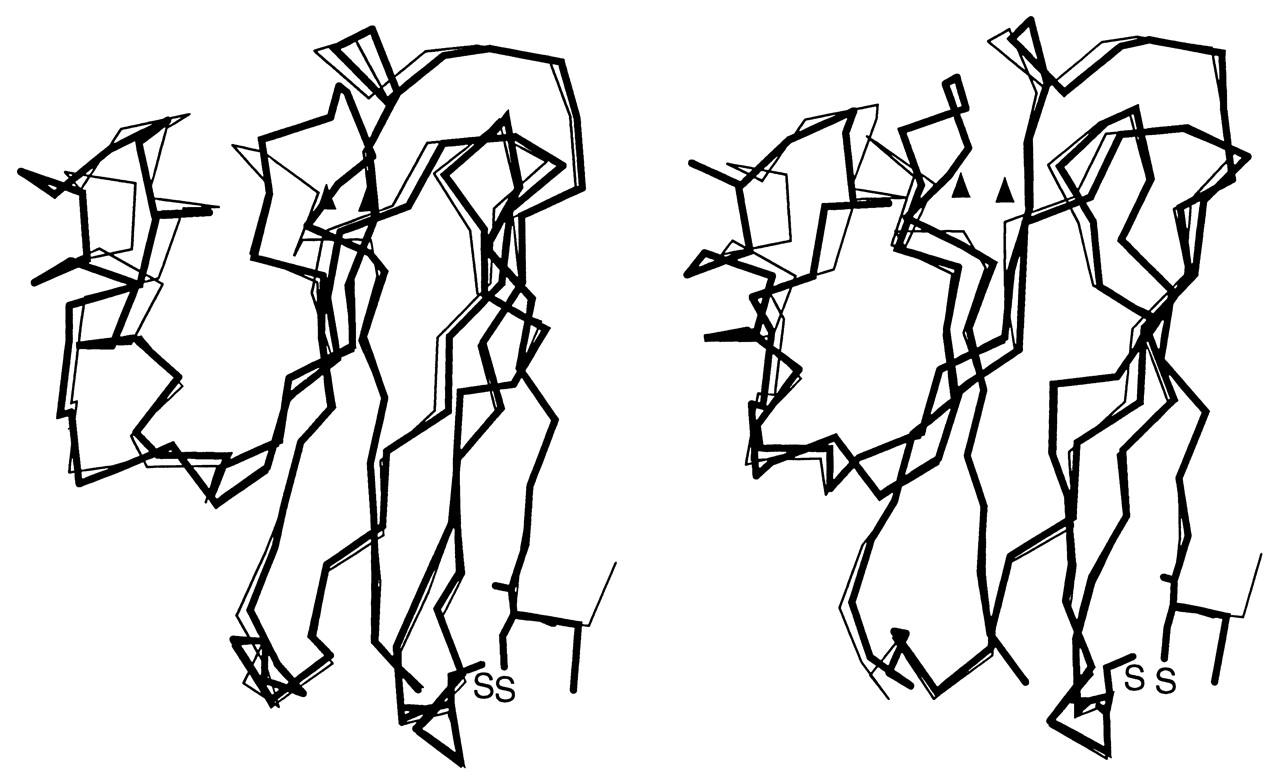
As part of our ongoing efforts to understand long-range ET processes in proteins (30) and to redesign metalloproteins (37), we have used the blue copper protein azurin as one of our model systems. Azurin consists of rigid β-pleated sheets and contains two potential redox centers: the T1 blue copper ion coordinated directly to amino acid residues, and a disulfide bridge (RSSR) present at the opposite end of the molecule, separated by a direct distance of 2.65 nm. Intramolecular ET between these sites was investigated in a large number of wild-type and single site-directed mutant azurins (38, 39), and the effect of specific changes in the protein structure on electronic couplings, reorganization energies, and the nature of the medium separating donor and acceptor were examined (30). We have succeeded in engineering an azurin variant (called purple CuA azurin hereafter) where the blue copper site has been replaced by the purple CuA center (23). Comprehensive spectroscopic characterization of the engineered azurin from Pseudomonas aeruginosa has demonstrated the striking similarity between the purple CuA azurin and the native purple CuA centers (11, 23, 40–43). The three-dimensional structure of the engineered purple CuA azurin at 1.65 Å (see supplemental data on the PNAS web site, www.pnas.org) shows close overall structural similarity between the native blue copper azurin and the engineered purple CuA azurin with an rms deviation of 1.07 Å for the α-carbon backbone when the ligand loop (corresponding to Cys-112 to Met-121 in the native blue copper azurin) is not considered in the calculation. Therefore, placing either a blue copper or a purple CuA center into the same protein framework permits a critical comparison of the efficiency of the two centers as electron mediators. The results of kinetic studies presented here suggest that the CuA center is a relatively more efficient ET agent because of its lower reorganization energy.
MATERIALS AND METHODS
Sample Preparation.
The purple CuA azurin was prepared and purified to homogeneity as described (23, 43). [Ru(NH3)5Py](ClO4)2 (Py = pyridine) was synthesized according to the procedure outlined by Cummins and Gray (44).
Redox Potential Measurements on Ru[(NH3)5Py[ClO4]2.
Redox potential measurements were made by using a Bioanalytical Systems (West Lafayette, IN) CV-50W potentiostat with a platinum working electrode, platinum auxiliary electrode, and a Ag/AgCl reference electrode, all supplied by Bioanalytical Systems.
Redox Titrations.
Titrations were performed by using 0.3–0.4 mM purple CuA azurin in 50 mM ammonium acetate, pH 5.1 and [Ru(NH3)5Py](ClO4)2 under a flow of Ar. The reduction of purple CuA azurin was monitored by CuA absorption at 774 nm where the [Ru(NH3)5Py]3+ and [Ru(NH3)5Py]2+ have no contribution to absorption. Each titration consisted of 8–9 aliquots of freshly prepared [Ru(NH3)5Py](ClO4)2 solution under Ar. Corrections were made for dilution. For the reaction,
 |
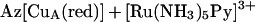 |
the equilibrium constants were obtained according to Eq. 1 (45),
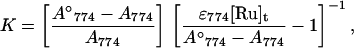 |
1 |
where A774, A774°, ɛ774, and [Ru]t are absorbance of purple CuA azurin, absorbance of fully oxidized purple CuA azurin, its extinction coefficient at 774 nm, and total concentration of ruthenium ions.
Kinetic Measurements.
The pulse radiolysis system using the Varian V-7715 linear accelerator at the Hebrew University in Jerusalem was used for carrying out the kinetic experiments (46). By using 5-MeV accelerated electrons and pulse lengths ranging from 0.1 to 1.5 μs, we produced 0.6–10 μM of CO2− radical ions. All optical measurements were carried out anaerobically under purified N2O in a 4 × 2 × 1 cm Spectrosil cuvette. Three light passes were used, which result in an overall optical path length of 12.3 cm. A 150-W xenon lamp produced the analyzing light beam, and an appropriate optical filter with cut-off at 385 nm was used to avoid photochemistry and light scattering. The data acquisition system consisted of a Tektronix 390 A/D digitizer connected to a personal computer. The temperature of the reaction solutions was controlled by a thermostating system and continuously monitored by a thermocouple attached to the cuvette (46). Practically all reactions were performed under pseudo-first order conditions, with typically a 10-fold excess of oxidized protein over reductant. The concentration of oxidized CuA was monitored at 485 and 510 nm, while formation and decay of the RSSR− radical was followed at 410 nm (ɛ410 ≅ 10,000 M−1⋅cm−1) (47). Kinetic runs at each temperature were repeated at least three times.
Aqueous solutions, 0.1 M in sodium formate (pH 5.1), were deaerated and saturated with N2O in glass syringes. Afterward the concentrated protein stock solution was added and N2O bubbling was continued for another 5 min. The solutions then were transferred into the pulse radiolysis cuvette under anaerobic conditions.
RESULTS
Fig. 1 shows a typical example of the time-dependent reduction of oxidized purple CuA azurin monitored at 485 nm and RSSR− reoxidation at 410 nm. The pulse radiolytically produced CO2− radicals led to reduction of purple azurin (k = 8 × 108 M−1⋅s−1 at 25°C, pH 5.1). The concentration of CO2− radicals produced was controlled so that less than 5% of the protein electron acceptor sites were reduced during a single pulse. Hence the probability for any azurin molecule being reduced by more than one electron during one pulse is quite low. A similar fast reduction can be monitored at 410 nm where the produced RSSR− radical anion absorbs. These fast (diffusion controlled) bimolecular reactions (not shown) are followed by a slower process (Fig. 1), which is the result of ET from the disulfide radical ion (Fig. 1B) to the oxidized CuA center (Fig. 1A). The observed rate constants of these processes were the same (kET = 650 ± 60 s−1, pH 5.1, 25°C) and were independent of both protein and radical concentrations, demonstrating that it proceeds intramolecularly. The temperature dependence of this process was examined for both blue copper (i.e., wild-type P. Aeruginosa azurin) and the engineered purple CuA azurin ranging from 0.5°C to 40°C for the former and 3.5°C to 42.8°C for the latter. The first-order rate constant, kET, can be written as:
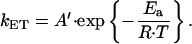 |
2 |
Accordingly, a plot of ln(kET) vs. 1/T for both proteins is shown in Fig. 2. From these data the activation enthalpy was determined to be: ΔH≠ = Ea − RT = 33.7 ± 3.1 kJ⋅mol−1 for purple CuA azurin, and ΔH≠ = 36.5 ± 3.0 kJ⋅mol−1 for the native blue copper azurin. The activation entropy ΔS≠, including the contribution from the electronic factor κ(r)ν for a nonadiabatic ET (compare with Eq. 5 below) can be calculated from:
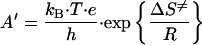 |
3 |
and was found to be ΔS≠ = −78 ± 5 J⋅K−1⋅mol−1 for purple CuA azurin, and ΔS≠ = −74 ± 6 J⋅K−1⋅mol−1 for the native blue copper azurin at pH 5.1 and 298 K.
Figure 1.

Time-resolved absorbance changes of a 4.3 μM purple CuA azurin solution after a pulse of accelerated electrons at (A) 485 nm, the intramolecular reduction of mixed valence [Cu(1.5)-Cu(1.5)] CuA azurin and (B) 410 nm, the decay of the RSSR− radical. Conditions were: N2O saturated aqueous solution, 100 mM sodium formate, pH 5.1; temperature, 25.0°C; pulse width, 0.4 μs. (Lower) The residuals of the fitting procedure.
Figure 2.

Temperature dependence of intramolecular RSSR− to Cu ET in purple CuA (■) and wild-type blue copper (•) azurin from P. aeruginosa shown as a plot of ln(kET vs. 1/T. The activation parameters were determined from this plot according to Eqs. 2 and 3.
Redox titrations of the purple CuA azurin were carried out at pH 5.1 and 298 K by using [Ru(NH3)5Py]2+. For the reaction
 |
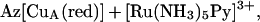 |
the equilibrium constants were found to vary slightly with different concentrations of [Ru(NH3)5Py]2+ between 0.1 and 2.0 mM. The final equilibrium constant (K = 0.48) was obtained by extrapolation to zero concentration of [Ru(NH3)5Py]2+. From the above K and the measured E° = 302 mV for [Ru(NH3)5Py]3+/2+ couple in 50 mM ammonium acetate (pH 5.1), a reduction potential of 283 mV is obtained for the purple CuA azurin. This value is lower than the potential of the T1 center in P. aeruginosa azurin (350 mV at pH 5) (48) and higher than the value of 240 mV reported for the CuA site in cytochrome c oxidase fragment (49). Hence, the driving force for the internal ET in purple azurin is lower than that operating in the wild-type protein. The reversibility of the above reaction was ascertained by using Ce(IV) as oxidant, which led to practically full reoxidation of the CuA center.
DISCUSSION
As the only change introduced into wild-type azurin is the replacement of the T1 site by that of CuA, comparing long-range ET between RSSR− and the respective copper centers in wild-type azurin and the purple azurin is expected to yield a better understanding of the ET properties of the CuA site. The intramolecular ET has been studied earlier in a large number of wild-type and site-directed mutant azurins (38, 39). The pH dependence of intramolecular ET exhibited a dramatic increase in the rate with decreasing pH. Thus, at pH 5.1 the rate constant kET becomes 250 ± 20 s−1 (50). The rate constant for the intramolecular ET in purple azurin is almost 3-fold larger than that of blue azurin at low pH, kET = 650 ± 60 s−1.
According to the semiclassical Marcus theory (51) the ET rate constant depends on the electronic coupling constant, HDA between electron donor (D) and acceptor (A), on the reorganization energy, λ and on the driving force, ΔG0:
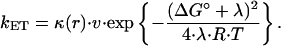 |
4 |
In the nonadiabatic regime, (κ ≪ 1),
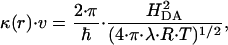 |
5 |
where the electronic coupling decreases exponentially with the distance:
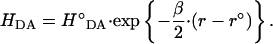 |
6 |
The question is then what causes the enhancement of the intramolecular ET rate in purple CuA azurin compared with the native blue copper protein.
The reduction potential for wild-type P. aeruginosa azurin at pH 5 is 350 mV (48). With a potential of −410 mV for the RSSR/RSSR− couple (47), the driving force for intramolecular RSSR− to Cu(II), −ΔG0 is 73.3 kJ⋅mol−1 (0.76 eV). The standard potential of the binuclear CuA center in the engineered azurin was determined to be 283 mV at pH 5.1. The driving force −ΔG0 = 66.9 kJ⋅mol−1 (0.69 eV) is now calculated. In spite of this smaller driving force in the latter, the rate of intramolecular ET in the purple CuA protein is nevertheless faster than in the blue copper protein.
An ET pathway in P. aeruginosa azurin from Cys-3 (i.e., part of the disulfide bond) to Cys-112 (a copper ligand in both blue and purple azurin) has been proposed (30). It links Cys-3 via a hydrogen bond to Thr-30 and further from Val-31 to Trp-48, by a 0.40 nm through-space jump. Then, Val-49 and Phe-111 are connected through another H-bond, followed by a backbone connection to the Cys-112 copper ligand. The close overall structural similarity between the native blue copper azurin and the engineered purple CuA azurin has been demonstrated recently by x-ray crystallography (see Fig. 3, which is published as supplemental data on the PNAS web site, www.pnas.org). An rms deviation taken over the residues separating electron donor and acceptor was calculated to be 0.24 Å (see Fig. 4, which is published as supplemental data on the PNAS web site, www.pnas.org). The same number of covalent bonds, the same two H-bonds and the through-space jump all are found in the purple CuA azurin structure, too. The hydrogen bonds are slightly longer (≈0.1 Å) in the mutant protein while the van der Waals contact distances are essentially identical (3.83 Å in wild-type azurin vs. 3.79 Å in the purple CuA azurin). Therefore, the same pathway is most probably also operative in the engineered purple azurin. Finally, it should be noted that the Cu-S covalency of the blue copper center is ≈38%, but it is slightly lower in the CuA center, where the total Cu-S covalency has been calculated to be 26% (11, 15). Thus, there is no structural indication for an improved overlap of donor and acceptor wave functions.
We previously have determined the total reorganization energy (including both electron donor [RSSR−] and acceptor [Cu(II)]) of wild-type P. aeruginosa azurin to be 1.0 eV (28). By using kET of 650 s−1 for the intramolecular ET in purple azurin we calculate a λTOT = 0.81 eV. Here λTOT is related to the reorganization energies of the individual redox centers (51):
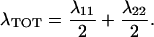 |
7 |
Di Bilio et al. (31) recently have analyzed the intramolecular long-range ET in Ru(II) modified azurin and calculated reorganization energies of both electron donor and acceptor centers from the temperature dependence of the observed rate constants (31). For the blue copper center λ = 0.82 eV was reported. Now, if λ11 in Eq. 7 denotes reorganization of the copper center whereas λ22 is that of the disulfide group, we can calculate the latter from the previously determined values of λTOT = 1.0 eV and λ11 = 0.82 eV in wild-type P. aeruginosa azurin to be λ22 = 1.2 eV. Assuming that this energy is unaffected by the substitution of the blue copper with the purple CuA center and by using the presently determined λTOT = 0.81 eV for purple azurin we now calculate λ11 = 0.4 eV for reorganization of the CuA center. Hence the reorganization energy of this site is only 50% that of the blue copper site. This finding constitutes strong experimental support for the notion that CuA is indeed a redox center with a more facile ET ability.¶
ET from CuA to heme a in cytochrome c oxidase is remarkably faster (kET ≈104 s−1) than what we observe here in the purple CuA azurin, particularly considering the much smaller driving force (0.05 eV) (35). However, an ET pathway has been identified by Ramirez et al. (26, 36), which links the two redox centers in cytochrome c oxidase. It consists of 14 covalent bonds and two hydrogen bonds. The coupling via this pathway would be much more efficient than the one in the purple azurin (19 covalent bonds, two hydrogen bonds, and one through-space-jump), which may explain the faster ET rate.
Theoretical calculations performed by Larsson et al. (27) and Gamelin et al. (11) have supported the idea that the advantage of the binuclear structure of the CuA center is in lowering both the inner and outer sphere reorganization energy compared with the T1 copper site. Their findings have been corroborated by the x-ray absorption study of Blackburn et al. (14), who observed minimal structural changes of the mixed valence binuclear center upon reduction.
In conclusion, by converting the single blue copper center into a binuclear CuA site and studying their ET reactivities in the same protein framework, we have demonstrated that the purple CuA centers are more efficient ET mediators than the blue copper center, mainly because of the low reorganization energy of the mixed-valence [Cu(1.5)-Cu(1.5)] site.
Supplementary Material
Acknowledgments
We thank Professor John H. Richards (Caltech) for providing the azurin gene, Mr. Alan Gengenbach (University of Illinois at Urbana-Champaign) for the synthesis and characterization of [Ru(NH3)5Py](ClO4)2, Mr. Michael R. Rosenblatt (University of Illinois at Urbana-Champaign) for measuring the reduction potential of [Ru(NH3)5Py]3+/2+ couple, and Dr. Jay Winkler (Caltech), Dr. Claire E. Slutter (The Weizmann Institute of Science), and Professor Harry B. Gray (Caltech) for helpful discussions during the preparation of the manuscript. O.F. thanks the Danish Natural Science Research Foundation for financial support (the Bioinorganic Research Program). I.P. acknowledges generous support from the German Israel Foundation and the Volkswagen Stifftung. This material is based on work supported by the National Science Foundation under Award No. CHE 95–02421 to Y.L. (CAREER Award and Special Creativity Extension). Y.L. is an Alfred P. Sloan Research Fellow, a Beckman Young Investigator of the Arnold and Mabel Beckman Foundation, and a Cottrell Scholar of the Research Corporation.
ABBREVIATIONS
- ET
electron transfer
- T1
type 1
- Py
pyridine
- RSSR
the disulfide bridge between Cys-3 and Cys-26 in azurin
Footnotes
We have assumed that the reduction potential of RSSR/RSSR− in the blue and purple azurins is similar to that of 5,5′-dithiobis(2-nitrobenzoic acid) modified hemoglobin (47). Under this assumption, intramolecular ET in both wild-type blue copper and purple CuA azurin should, according to Eq. 4, exhibit a near-zero activation enthalpy (because −ΔG° ≈ λ). The relatively high activation enthalpy observed (see above) must mean that −ΔG° ≠ λ, and one possibility is that the potential of the RSSR/RSSR− couple is more negative in the azurins. Our comparison of ET properties of blue and purple copper centers should be valid, however, because the same RSSR− center serves as the electron donor in both systems.
References
- 1.Beinert H. Eur J Biochem. 1997;245:521–532. doi: 10.1111/j.1432-1033.1997.t01-1-00521.x. [DOI] [PubMed] [Google Scholar]
- 2.Wikström M, editor. Minireview Series: Cytochrome Oxidase: Structure and Mechanism, Journal of Bioenergetics and Biomembranes. Vol. 30. 1998. [DOI] [PubMed] [Google Scholar]
- 3.Zumft W G, Kroneck P M H. Adv Inorg Biochem. 1996;11:193–221. [Google Scholar]
- 4.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 5.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 6.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature (London) 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 7.Wilmanns M, Lappalainen P, Kelly M, Sauer-Eriksson E, Saraste M. Proc Natl Acad Sci USA. 1995;92:11955–11959. doi: 10.1073/pnas.92.26.11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kroneck P M H, Antholine W E, Kastrau D H W, Buse G, Steffens G C M, Zumft W G. FEBS Lett. 1990;268:274–276. doi: 10.1016/0014-5793(90)81026-k. [DOI] [PubMed] [Google Scholar]
- 9.Neese F, Zumft W G, Antholine W E, Kroneck P M H. J Am Chem Soc. 1996;118:8692–8699. [Google Scholar]
- 10.Farrar J A, Neese F, Lappalainen P, Kroneck P M H, Saraste M, Zumft W G, Thomson A J. J Am Chem Soc. 1996;1996:11501–11514. [Google Scholar]
- 11.Gamelin D R, Randall D W, Hay M T, Houser R P, Mulder T C, Canters G W, de Vries S, Tolman W B, Lu Y, Solomon E I. J Am Chem Soc. 1998;120:5246–5263. [Google Scholar]
- 12.Andrew C R, Fraczkiewicz R, Czernuszewicz R S, Lappalainen P, Saraste M, Sanders-Loehr J. J Am Chem Soc. 1996;118:10436–10445. [Google Scholar]
- 13.Wallace-Williams S E, James C A, de Vries S, Saraste M, Lappalainen P, van der Oost J, Fabian M, Palmer G, Woodruff W H. J Am Chem Soc. 1996;118:3986–3987. [Google Scholar]
- 14.Blackburn N J, de Vries S, Barr M E, Houser R P, Tolman W B, Sanders D, Fee J A. J Am Chem Soc. 1997;119:6135–6143. [Google Scholar]
- 15.Williams K R, Gamelin D R, LaCroix L B, Houser R P, Tolman W B, Mulder T C, de Vries S, Hedman B, Hodgson K O, Solomon E I. J Am Chem Soc. 1997;119:613–614. [Google Scholar]
- 16.Dennison C, Berg A, Canters G W. Biochemistry. 1997;36:3262–3269. doi: 10.1021/bi961960u. [DOI] [PubMed] [Google Scholar]
- 17.Luchinat C, Soriano A, Djinovic-Carugo K, Saraste M, Malmstroem B G, Bertini I. J Am Chem Soc. 1997;119:11023–11027. [Google Scholar]
- 18.Lappalainen P, Aasa R, Malmström B G, Saraste M. J Biol Chem. 1993;268:26416–26421. [PubMed] [Google Scholar]
- 19.von Wachenfeldt C, de Vries S, van der Oost J. FEBS Lett. 1994;340:109–113. doi: 10.1016/0014-5793(94)80182-7. [DOI] [PubMed] [Google Scholar]
- 20.Slutter C E, Sanders D, Wittung P, Malmström B G, Aasa R, Richards J H, Gray H B, Fee J A. Biochemistry. 1996;35:3387–3395. doi: 10.1021/bi9525839. [DOI] [PubMed] [Google Scholar]
- 21.van der Oost J, Lappalainen P, Musacchio A, Warne A, Lemieux L, Rumbley J, Gennis R B, Aasa R, Pascher T, Malmström B G, Saraste M. EMBO J. 1992;11:3209–3217. doi: 10.1002/j.1460-2075.1992.tb05398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dennison C, Vijgenboom E, de Vries S, van der Oost J, Canters G W. FEBS Lett. 1995;365:92–94. doi: 10.1016/0014-5793(95)00429-d. [DOI] [PubMed] [Google Scholar]
- 23.Hay M, Richards J H, Lu Y. Proc Natl Acad Sci USA. 1996;93:461–464. doi: 10.1073/pnas.93.1.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houser R P, Young V G, Jr, Tolman W B. J Am Chem Soc. 1996;1996:2101–2102. [Google Scholar]
- 25.Barr, M. E., Smith, P. H., Antholine, W. E. & Spencer, B. (1993) J. Chem. Soc. Chem. Commun. 1649–1652.
- 26.Ramirez B E, Malmström B G, Winkler J R, Gray H B. Proc Natl Acad Sci USA. 1995;92:11949–11951. doi: 10.1073/pnas.92.26.11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larsson S, Källebring B, Wittung P, Malmström B G. Proc Natl Acad Sci USA. 1995;92:7167–7171. doi: 10.1073/pnas.92.16.7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farver O, Pecht I. In: Copper Proteins. Spiro T G, editor. New York: Wiley; 1981. pp. 151–192. [Google Scholar]
- 29.Gray H B. Chem Soc Rev. 1986;15:17–30. [Google Scholar]
- 30.Farver O, Pecht I. J Bioinorg Chem. 1997;2:387–392. [Google Scholar]
- 31.Di Bilio A J, Hill M G, Bonander N, Karlsson B G, Villahermosa R M, Malmström B G, Winkler J R, Gray H B. J Am Chem Soc. 1997;119:9921–9922. [Google Scholar]
- 32.Brzezinski P. Biochemistry. 1996;35:5611–5615. doi: 10.1021/bi960260m. [DOI] [PubMed] [Google Scholar]
- 33.Morgan J E, Li P M, Jang D J, El-Sayed M A, Chan S I. Biochemistry. 1989;28:6975–6983. doi: 10.1021/bi00443a030. [DOI] [PubMed] [Google Scholar]
- 34.Oliveberg M, Malmström B G. Biochemistry. 1991;30:7053–7057. doi: 10.1021/bi00243a003. [DOI] [PubMed] [Google Scholar]
- 35.Winkler J R, Malmström B G, Gray H B. Biophys Chem. 1995;54:199–209. doi: 10.1016/0301-4622(94)00156-e. [DOI] [PubMed] [Google Scholar]
- 36.Regan J J, Ramirez B E, Winkler J R, Gray H B, Malmström B G. J Bioenerg Biomembr. 1998;30:35–39. doi: 10.1023/a:1020551326307. [DOI] [PubMed] [Google Scholar]
- 37.Lu Y, Valentine J S. Curr Opin Struct Biol. 1997;7:495–500. doi: 10.1016/s0959-440x(97)80112-1. [DOI] [PubMed] [Google Scholar]
- 38.Farver O, Pecht I. Proc Natl Acad Sci USA. 1989;86:6968–6972. doi: 10.1073/pnas.86.18.6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farver O, Pecht I. J Am Chem Soc. 1992;114:5764–5767. [Google Scholar]
- 40.Hay M T, Milberg R M, Lu Y. J Am Chem Soc. 1996;118:11976–11977. [Google Scholar]
- 41.Andrew C R, Lappalainen P, Saraste M, Hay M T, Lu Y, Dennison C, Canters G W, Fee J A, Slutter C E, Nakamura N, Sanders-Loehr J. J Am Chem Soc. 1995;117:10759–10760. [Google Scholar]
- 42.Blackburn N J, Ralle M, Sanders D, Fee J A, De Vries S, Houser R P, Tolman W B, Hay M T, Lu Y. Am Chem Soc Symp Ser. 1998;692:241–259. [Google Scholar]
- 43.Hay M T, Ang M C, Gamelin D R, Solomon E I, Antholine W E, Ralle M, Blackburn N J, Massey P D, Wang X, Kwon A H, Lu Y. Inorg Chem. 1998;37:191–198. [Google Scholar]
- 44.Cummins D, Gray H. J Am Chem Soc. 1977;99:5158–5167. doi: 10.1021/ja00457a042. [DOI] [PubMed] [Google Scholar]
- 45.Goldberg M, Pecht I. Biochemistry. 1976;19:4197–4208. doi: 10.1021/bi00664a011. [DOI] [PubMed] [Google Scholar]
- 46.Pecht I, Farver O. In: Photochemistry and Radiation Chemistry: Complementary Methods for the Study of Electron Transfer. Wishart J, Nocera D, editors. Washington, DC: Am. Chem. Soc.; 1998. pp. 65–79. [Google Scholar]
- 47.Faraggi M, Klapper M H. J Am Chem Soc. 1988;110:5753–5756. [Google Scholar]
- 48.Pascher T, Karlsson B G, Nordling M, Malmström B G, Vänngård T. Eur J Biochem. 1993;212:289–296. doi: 10.1111/j.1432-1033.1993.tb17661.x. [DOI] [PubMed] [Google Scholar]
- 49.Immoos C, Hill M G, Sanders D, Fee J A, Slutter C E, Richards J H, Gray H B. J Biol Inorg Chem. 1996;1:529–531. [Google Scholar]
- 50.Farver O, Bonander N, Skov L K, Pecht I. Inorg Chim Acta. 1996;243:127–133. [Google Scholar]
- 51.Marcus R A, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}