

Abstract
The atomic force microscope (AFM) was employed to investigate the extension and retraction dynamics of protruding and stable edges of motile 3T3 fibroblasts in culture. Such dynamics closely paralleled the results of earlier studies employing video microscopy that indicated that the AFM force-mapping technique does not appreciably perturb these dynamics. Force scans permitted height determinations of active and stable edges. Whereas the profiles of active edges are flat with average heights of 0.4–0.8 μm, stable edges smoothly ascend to 2–3 μm within about 6 μm of the edge. In the region of the leading edge, the height fluctuates up to 50% (SD) of the mean value, much more than the stable edge; this fluctuation presumably reflects differences in underlying cytoskeletal activity. In addition, force mapping yields an estimate of the local Young’s modulus or modulus of elasticity (E, the cortical stiffness). This stiffness will be related to “cortical tension,” can be accurately calculated for the stable edges, and is ≈12 kPa in this case. The thinness of the leading edge precludes accurate estimation of the E values, but within 4 μm of the margin it is considerably smaller than that for stable edges, which have an upper limit of 3–5 kPa. Although blebbing cannot absolutely be ruled out as a mechanism of extension, the data are consistent with an actin polymerization and/or myosin motor mechanism in which the average material properties of the extending margin would be nearly constant to the edge. Because the leading edge is softer than the stable edge, these data also are consistent with the notion that extension preferentially occurs in regions of lower cortical tension.
Cell locomotion consists of a complex set of integrated molecular events that are vital to many life processes. Recently, considerable progress has been made in uncovering various molecular mechanisms that must be involved in locomotion (for reviews, see refs. 1 and 2). Less is known about the physical properties of moving cells, including the mechanical properties of the cell surface and the actual forces involved in locomotion (3, 4). Local measurements of the surface stiffness and properties of the leading edge have been made by using “cell poking” (5) and microneedles (6). Recently, the laser trap has been used to probe connections between cell-surface integrins and the membrane-associated cytoskeleton to compare the leading and trailing edge of fibroblasts (7, 8). Such connections also have been probed by using magnetic methods (9). Advances in silicone rubber substratum technology have permitted the imaging of traction forces used by highly motile fish-scale keratocytes (10–12) and in cells undergoing cytokinesis (13). Fibroblast traction forces have been investigated by using silicon microlevers embedded in a silicon substrate (14), and the manifestations of these forces were studied by employing a deformable polyacrylamide substrate (15).
The atomic force microscope (AFM) (16) has evolved to become a very important tool in the family of scanning-probe microscopy techniques. Because it can easily be operated in a liquid environment, particularly under physiological conditions (17), biological processes at the cellular (18) and even molecular (19) level can be investigated. AFM combines in a unique way high spatial resolution (20) with very high force sensitivity (21), with the result that the elastic properties of biological samples can be measured (reviewed in ref. 22). The spatial resolution and force sensitivity of AFM are combined in the force-mapping mode (23), which can be used to map the viscoelastic properties of living cells in considerable spatial detail (24–26). The resulting images can be thought of as a pixel-by-pixel “cell poking” experiment (5) yielding a map of the local modulus of elasticity (E). E is the basic measure relating stress applied to the specimen to the resulting strain in the sample; values range from ≈200 GPa for very hard materials such as steel to between 1 and 100 kPa for cellular specimens (22).
In this paper, we demonstrate that the AFM force-mapping mode allows the protrusion of active cell edges to be interrogated in terms of the kinetics, thickness, and mechanical properties of the protrusion. The results are compared with the more stable edges of motile fibroblasts. The data permit calculation of the local value of E to a spatial resolution of about 100 nm. E is a measure of cortical stiffness, a function of the material properties of the cytoskeleton and, when it is present, cortical tension. Thus, for example, higher cortical stiffness could result from an intrinsically stiffer cytoskeletal matrix and/or from increased cortical tension in the specimen. The derived mechanical properties of the leading edge are discussed in terms of current models for protrusion.
MATERIALS AND METHODS
Cells.
3T3 fibroblasts were cultured in DMEM supplemented with 5% fetal calf serum and 1% penicillin/streptomycin (GIBCO) following standard procedures. Cells were plated in 35-mm plastic Petri dishes (Nunclon, Naperville, IL) and cultured at 37°C in a 5% CO2 atmosphere. Typically, cells were subcultured 1 or 2 days before the experiment.
Atomic Force Microscopy.
A commercial AFM (Bioscope; Digital Instruments, Santa Barbara, CA) combined with an Axiomat custom-modified inverted optical microscope (Zeiss) was employed for these studies. This combination allowed lateral positioning of the AFM tip on the sample to micrometer precision. Soft silicon nitride cantilevers (Microlever; Park Scientific, Sunnyvale, CA) were employed and have a force constant of 8 mN/m as determined by measuring the thermal noise of the free lever (27). The AFM was covered with a gastight Plexiglas box so that the environment of the specimen could be controlled. Resistance-heating of this specimen chamber was accomplished by gluing resistors to the stage (a 20-mm thick stainless steel plate) of the optical microscope on top of which the AFM and the sample were mounted. Specimen temperature was monitored with a PT100 sensor by using a commercial display unit (DPM 235; Schwille Elektronik, Kircheim, Germany) to measure temperature. CO2 was flushed slowly through the chamber to maintain a steady-state concentration of 5% as measured with a gas sensor (Model IR1580 diffusion probe; Servomex, Crowborough, U.K.).
Data Acquisition.
E can be calculated from force curves, in which the deflection of the AFM cantilever is monitored as it approaches the sample. The deflection will be zero as long as the AFM tip is off the surface and will increase monotonically beyond the contact point. This increase is linear on a stiff sample; however, it will be smaller and nonlinear on softer samples. To measure local, laterally resolved E, the AFM was operated in the force-mapping or force-volume mode. A force map is a two-dimensional array of force curves recorded while the tip is raster-scanned across the sample. Because it takes about 20 minutes to record a single map of 64 × 64 force curves, standard force mapping is not suitable for investigating processes as rapid as protrusive activity. Therefore, two-dimensional resolution had to be sacrificed by using the y-disable mode (where force curves are continuously recorded along the same line). The calculation then yields time sequences of height and elasticity profiles along a single, scanned line.
Data Analysis.
To calculate E from the force curves, we employed Sneddon’s modification of the Hertzian model for the elastic indentation of a flat, soft sample by a stiff cone (28, 29). The model relates the applied loading force F to the indentation depth δ:
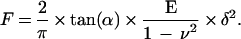 |
1 |
Here, E is the Young’s modulus, ν is the Poisson ratio of the sample, and α is the half-opening angle of the AFM tip. On a stiff sample, the cantilever deflection d(z) will be equal to the piezo movement z, whereas on a soft sample the deflection is decreased because of elastic indentation:
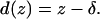 |
2 |
The force can be obtained by multiplying the measured cantilever deflection d(z) by the known spring constant k of the cantilever:
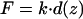 |
3 |
Thus, we can substitute F and δ in Eq. 1 to obtain E as a function of the measured quantities z and d(z). Fitting this function to the force-curve data will not only yield E but also the position z0, where the cantilever initially contacts the sample, which corresponds to the real height of the sample at the point where the force curve was recorded. This calculation is described elsewhere in more detail (30).
The Hertzian model has been derived for an infinitely thick soft sample. It also can be applied to thin films as long as the indentation into the sample by the tip is sufficiently small compared with its thickness. This is demonstrated in Fig. 1a, where a typical force curve obtained on a thicker stable edge is fitted by the Hertz model in four different ranges of cantilever deflection (arrows indicate zero deflection). Calculated contact points (arrowheads) do not differ significantly. At the leading margin, however, there are locations where cell heights are only 200–300 nm, and the indentation depth becomes significant compared with sample thickness. Thus, at higher loading forces, the tip will sense the elastic properties of the underlying stiff substrate and the calculated E must be considered an upper limit of the actual values. Different fits to a sample force curve obtained on a flat lamellipodium are shown in Fig. 1b. Only for the lowest range of analysis does the calculation yield a contact point at a position where the deflection does not differ appreciably from the zero deflection value. Thus, we conclude that the influence of the substratum is lowest if force curves from such thin regions of the cell are fit to the Hertz model in the lowest force regime (small cantilever deflections). This procedure gives the best fit of the data and allows a reasonable estimate of specimen height. It should be noted that the calculated cell heights will not necessarily reflect the undisturbed topography of the cell if, for example, the surface is covered with a layer that is too soft to deflect the cantilever, such as the glycocalyx.
Figure 1.

(a) Typical force curve on stable edge fitted in four different deflection ranges showing good fit of the Hertz model to the data, which allows extraction of the E value and contact point that does not appreciably depend on the force range used for the fit. (b) Typical force curve on a thin part of a protruding edge fitted in four different deflection ranges showing that the curve can only be fit by the Hertz model in the lowest applied force regime. For a and b, cantilever deflection range used for the fit is shown in nm at right margin, arrows indicate zero deflection, calculated contact points are indicated by arrowheads, and values of E are given in c. (c) Dependence of E on the mean force of the force range used for analysis for stable and active edges of the cell.
The influence of noise in the determination of zero deflection was reduced by averaging deflection values of various points in the off-surface portion of the force curve. The z height of the contact point is then calculated by fitting the force curve in a higher deflection regime to the Hertz model and extrapolating the fit curve to zero deflection. This procedure will reduce noise significantly.
The dependence of measured E values as a function of mean loading force (corresponding to the range of cantilever deflections employed for analysis) shows that the calculated values are independent of the range of analysis for stable edges but depend on this parameter for protruding edges (Fig. 1c). Consequently, analysis typically was carried out for small cantilever deflections in the range of 10–40 nm (corresponding to 80–320 pN) to minimize the influence of the underlying stiff substrate.
RESULTS
Kinetics of Edge Dynamics.
The AFM in the present configuration can be used to “visualize” various properties of live cells while allowing their motile state to be maintained. A typical AFM deflection image of a region of a motile NIH 3T3 fibroblast is seen in Fig. 2. (The deflection mode of imaging is sensitive to local slope and is somewhat analogous in this sense to a differential-interference contrast image.) Successive line scans of force curves across active and stable edges (compare encircled and rectangular regions in Fig. 2) were recorded to yield time sequences of height and elasticity profiles along the scan line for a protruding (Fig. 3 a and b) and a more stable (Fig. 3 c and d) edge. By thresholding the elasticity data, the intersections of the line scan with the edge were calculated, and thus the dynamics of both edge types could be tracked at the frequency of the line scan (Fig. 3 a and c Inset). Over a period of about 30 min, the leading edge extends in a fluctuating manner about 10 μm. We use the term “stable” edge somewhat loosely, as the edge does not move actively but rather undergoes a slow extension of about 4 μm during the observation period.
Figure 2.

Deflection image of a portion of a representative 3T3 cell showing leading (encircled region) and stable (rectangular region) edges. Both regions show, schematically, the position of a scan line interrogating the respective edge.
Figure 3.

Successive single line scans of height (a) and E (b) across the protruding edge of a motile 3T3 cell in which values are encoded in a gray scale. Inset in a shows position vs. time for a point on the margin, revealing fluctuations superimposed on a general protrusion. Successive single line scans of height (c) and E (d) of the stable edge of a 3T3 cell in which values are encoded in a gray scale. Inset in c shows position vs. time for a point on the margin. The band structure in the E plot presumably is caused by stress fibers crossing the scan line. Note the gradual extension.
The position of the edge, sampled at a frequency of 0.1 Hz, is shown for the leading and trailing edges in Fig. 3 a and c (Inset), respectively. It is clear that the leading edge is much more dynamic than the stable edge in terms of fluctuations in the position of the margin. By differentiating this data, instantaneous velocities of protrusion and retraction can be calculated. With the differentiation algorithm used, the minimum detectable instantaneous velocity (5.2 nm/s = 0.31 μm/min) represents edge movement of 1 pixel (110 nm) during the time needed to record two lines of force curves (21 s). Mean velocities can be calculated with a better accuracy of 0.055 nm/s (1 pixel in 2,000 s). The distribution of instantaneous velocities is shown for both extending (Fig. 4a) and stable (Fig. 4b) edges. For the extending edge, the mean velocity of extension is 5.5 nm/s (0.33 μm/min) and is composed of both protruding and retracting phases. Because the protruding phase dominates, the edge undergoes net extension. For both phases, maximum velocities are 50–60 nm/s (0.8–1.0 μm/min). The stable edge does not fluctuate appreciably, but rather extends slowly, with a mean velocity of 2.2 nm/s (0.13 μm/min). Here, 93% of the measured velocity values are less than or equal to the minimum detectable value (±5.2 nm/s).
Figure 4.

Histograms of instantaneous velocities for the protruding edge (a, calculated from the Inset in Fig. 3a) and for the stable edge (b, calculated from the Inset in Fig. 3c).
Force Mapping Permits Height Determinations of Cell Edges.
The data in Fig. 3 a and c constitute time series of height profiles permitting the thickness of the cell edges to be determined as described in Materials and Methods. Average height profiles of protruding and stable edges are shown in Fig. 5 Lower and were obtained by averaging the rows after aligning all of the lines in Fig. 3 so that the cell edge was at the same lateral position.
Figure 5.

(a, Upper) Average E as a function of position from the edge for protruding lamella. (a, Lower) Average cell thickness as a function of position from the edge for protruding lamella. (b, Upper) Average E as a function of position from the edge for stable margin. Arrows indicate distinct, local increases in apparent E for the stable edge that may correspond to stress fiber-like structures. (b, Lower) Average cell thickness as a function of position from the edge for the stable margin. Force curves analyzed in the cantilever-deflection range of 10–40 μm. Error bars represent the SD of the mean values of height or E at a given distance from the edge.
One issue that arises is whether the AFM cantilever is compressing ruffles. We argue that for these cells this is not the case. Ruffles are comparatively stiff structures (6); this stiffness prevents them from being entirely compressed by the AFM tip. To test this notion, we deliberately detached a leading edge from the substrate before force mapping. In the height and elasticity images calculated from the force map, these artificial ruffle-like structures were clearly visible (data not shown). Because similar structures were not observed in the data reported, we conclude that no large ruffles were present. It is possible, however, that fluctuations in height at a single position in the leading lamella reflected the retrograde movement of small ruffles (see below).
Spatially Resolved E Values for Leading and Stable Edges.
E can be calculated from the force-displacement curves by using the Hertz model (24). These data are shown in image format in Fig. 3 b and d. The band structure in the data set for the stable edge presumably arises from fibers perpendicular to the scan line having larger apparent E values. Average profiles of E are shown in Fig. 5 Upper. On the stable edges, E decreases from ≈12 kPa to ≈5 kPa 5 μm from the edge. Again, spatial variations are measured that may correspond to regions containing actin stress fibers (31). In such regions, E increases distinctly, but it should be noted that the values for E on stress fibers are approximations, because the Hertz model assumes homogeneity of the soft sample—an assumption that is violated when fibers are present. As indicated in Materials and Methods, for the stable edges, the values of E do not appreciably depend on the force range used for analysis (Fig. 1c). In the thin parts of the protruding lamellae, the E values depend on force, but at low force values there is a reasonable fit to the Hertz model (Fig. 1b). E values in these regions (Fig. 1c) are in the range of 3–5 kPa and are an upper limit because of the influence of the substratum.
Fluctuations in Height Are More Pronounced in Active than Stable Edges.
From the family of successive line scans taken at the same position, the height and E value at given distances behind the leading and stable edges can be recorded as functions of time. In Fig. 6, the time variations of height are given for a point 700 nm behind the leading and stable edges. At this point, where both the leading and stable edges have similar average heights, the fluctuations in height are considerably greater in the leading lamella. Time-dependent fluctuations also occur in E (data not shown), but there is a correlation between height and elasticity data that may reflect, in part, the influence of the underlying stiff substrate. These differences in height and elasticity fluctuations presumably reflect differences in underlying cytoskeletal dynamics in the two regions. In particular, height fluctuations in the leading lamella could reflect the passage of small (<1-μm high) ruffles.
Figure 6.

Variations of cell height with time 700 nm behind the leading and stable edges.
DISCUSSION
Although fibroblasts often have a mean velocity of less than 1 μm/min, it is well known that their leading edge is dynamic, often protruding at rates many times exceeding that of the mean cell velocity. Given the differences in cell types, our measurements are in general agreement with the earlier literature (6, 32) in terms of mean velocities of protrusion having an average of about 5 μm/min. At the leading margin, numerous brief retracting phases are superimposed on the general protrusive activity. These fluctuations and the similarities of the velocities of the forward and backward phases also are in agreement with the results of Ambercrombie et al. (32). The predominance of the protrusive phase results in an overall extension of the edge (32). On the other hand, for chicken heart fibroblasts, the work of Felder and Elson (6) showed that a nearly constant velocity of protrusion occurred in lamellar extension preceded by rapid acceleration and ending with a quick deceleration. In addition, the earlier work on chicken heart fibroblasts (6, 32) detected the centripetal movement of ruffles. Such movement was not observed in the study of NIH 3T3 cells, although modest thickenings of the lamella were seen (see, for example, Fig. 3a at 750 s and 1,300 s). However, the general agreement of the AFM results with those obtained by light microscopy suggests that interrogation of the edge by soft AFM cantilevers does not greatly perturb the system.
Mechanisms for protrusion can be divided into several classes (2, 33): protrusion driven by actin polymerization; protrusion driven by myosin I-type motors with obligate actin polymerization; osmotic gel swelling; or pressure-driven blebbing (toothpaste tube model). Regulation of protrusive and retractive phases, which would be responsible for fluctuations in edge position (as the lamellipod generally extends) is likely to be complex. For example, as suggested by Welch et al. (34), a protrusive phase could result from a burst of actin polymerization, a reduction of myosin-powered retraction, an increase in substratum coupling anchoring the actin-based cytoskeleton and preventing retrograde actin flow within the lamellipod, or a combination of such factors. On the other hand, an increase in myosin activity or a decrease in actin polymerization at the edge may produce the transient retractive phases observed in lamellar extension.
The elasticity data (Fig. 5a) are not consistent with a blebbing mechanism of protrusion because (to 100-nm spatial resolution) no pronounced softening of the leading edge is measured, which would be envisioned in such a mechanism (33, 35, 36). Such softening on a length scale detectable by force mapping would be expected for a blebbing mechanism in which the bleb is initially deficient in cytoskeletal structure (35) because blebs have micrometer dimensions (35, 37). Indeed, softening due to loss of f-actin structure caused by the cytochalasins can be observed in mechanical indentation measurements (5, 25, 31, 38). It should be noted that there are cells in which blebbing clearly appears to be the predominant mode of protrusion (35, 37). The lack of softening also argues against the osmotic gel-swelling mechanism postulated by Oster and Perelson (39) where f-actin crosslinks are severed, allowing the gel to swell to a new equilibrium position provided such softening is not confined to within approximately 100 nm of the leading edge.
Our measurements are consistent with a straight actin polymerization mechanism for protrusion (40) or one that combines obligatory actin polymerization with the action of myosin I-type motors (2) to extend the leading edge. In this regard, it is significant that actin polymerization models can account for this protrusion rate both in terms of actual growth (40) and in terms of diffusional delivery of actin monomer to the leading edge (41). The original Brownian ratchet mechanism requires excursions of the leading edge of ≈5 nm (diameter of actin) to allow insertion of an actin monomer. Such softening owing to the fluctuating leading edge would be undetectable at the resolution of current force mapping (100 nm). In the event that membrane fluctuations are damped, bending of the terminal actin filaments is hypothesized to permit insertion (42). It also is possible that hydrostatic pressure within the cell caused by contraction (43) provides just enough displacement at the edge to allow insertion of actin monomer. These latter mechanisms, too, will permit extension without softening of the leading edge on a length scale detectable by force mapping. The polymerization or motor-plus-polymerization mechanisms also are consistent with the material properties of the leading edge being constant to the edge, a view supported by electron microscopy of the dense lamellipodial actin network (44, 45).
Cortical tension is a term often invoked to explain properties of motile systems (46). Force mapping provides a measure of cortical stiffness to which cortical tension will contribute. For example, the fact that the leading edge is softer (lower E value) within 4 μm from the edge than the stable edge is consistent with the notion that lower cortical tension is associated with regions of the cell that are capable of extension (47).
Acknowledgments
This work was supported in part by National Institutes of Health Grant GM 35325 (K.J.) and in part by the Deutsche Forschungsgemeinschaft (C.R. and M.R.). We thank the Sonderforschungsbereich 266 for travel support (K.J.).
ABBREVIATIONS
- AFM
atomic force microscope
- E
Young’s modulus or modulus of elasticity
References
- 1.Lauffenburger D A, Horwitz A F. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 2.Mitchison T J, Cramer L P. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- 3.Oliver T, Lee J, Jacobson K. Semin Cell Biol. 1994;5:139–147. doi: 10.1006/scel.1994.1018. [DOI] [PubMed] [Google Scholar]
- 4.Elson E L, Felder S F, Jay P Y, Kolodney M S, Pasternak C. Motion Analysis of Living Cells. New York: Wiley–Liss; 1997. pp. 67–83. [Google Scholar]
- 5.Petersen N O, McConnaughey W B, Elson E L. Proc Natl Acad Sci USA. 1982;79:5327–5331. doi: 10.1073/pnas.79.17.5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Felder S, Elson E L. J Cell Biol. 1990;111:2513–2526. doi: 10.1083/jcb.111.6.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt C E, Horwitz A F, Lauffenburger D A, Sheetz M P. J Cell Biol. 1993;123:977–991. doi: 10.1083/jcb.123.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choquet D, Felsenfeld D P, Sheetz M P. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- 9.Wang N, Butler J P, Ingber D E. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 10.Lee J, Leonard M, Oliver T, Ishihara A, Jacobson K. J Cell Biol. 1994;127:1957–1964. doi: 10.1083/jcb.127.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliver T, Dembo M, Jacobson K. Cell Motil Cytoskeleton. 1995;31:225–240. doi: 10.1002/cm.970310306. [DOI] [PubMed] [Google Scholar]
- 12.Dembo M, Oliver T, Ishihara A, Jacobson K. Biophys J. 1996;70:2008–2022. doi: 10.1016/S0006-3495(96)79767-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burton K, Taylor D L. Nature (London) 1997;385:450–454. doi: 10.1038/385450a0. [DOI] [PubMed] [Google Scholar]
- 14.Galbraith C G, Sheetz M P. Proc Natl Acad Sci USA. 1997;94:9114–9118. doi: 10.1073/pnas.94.17.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelham R J, Wang Y-L. Proc Natl Acad Sci USA. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Binnig G, Quate C F, Gerber C. Phys Rev Lett. 1986;56:930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- 17.Drake B, Prater C B, Weisenhorn A L, Gould S A C, Albrecht T R, Quate C F, Cannell D S, Hansma H G, Hansma P K. Science. 1989;243:1586–1589. doi: 10.1126/science.2928794. [DOI] [PubMed] [Google Scholar]
- 18.Fritz M, Radmacher M, Gaub H E. Biophys J. 1994;66:1328–1334. doi: 10.1016/S0006-3495(94)80963-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radmacher M, Fritz M, Hansma H G, Hansma P K. Science. 1994;265:1577–1579. doi: 10.1126/science.8079171. [DOI] [PubMed] [Google Scholar]
- 20.Müller D J, Schabert F A, Büldt G, Engel A. Biophys J. 1995;68:1681–1686. doi: 10.1016/S0006-3495(95)80345-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rief M, Oesterheld F, Berthold M, Gaub H E. Science. 1997;275:1295–1297. doi: 10.1126/science.275.5304.1295. [DOI] [PubMed] [Google Scholar]
- 22.Radmacher, M. (1997) IEEE Eng. Med. Biol. 47–57. [DOI] [PubMed]
- 23.Radmacher M, Cleveland J P, Fritz M, Hansma H G, Hansma P K. Biophys J. 1994;66:2159–2165. doi: 10.1016/S0006-3495(94)81011-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radmacher M, Fritz M, Kacher C M, Cleveland J P, Hansma P K. Biophys J. 1996;70:556–567. doi: 10.1016/S0006-3495(96)79602-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hofmann U G, Rotsch C, Parak W J, Radmacher M. J Struct Biol. 1997;119:84–91. doi: 10.1006/jsbi.1997.3868. [DOI] [PubMed] [Google Scholar]
- 26.A-Hassan E, Heinz W F, Antonik M D, D’Costa N P, Nageswaran S, Schonenenberger C-A, Hoh J H. Biophys J. 1998;74:1564–1578. doi: 10.1016/S0006-3495(98)77868-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butt H-J, Jaschke M. Nanotechnology. 1995;6:1–7. [Google Scholar]
- 28.Hertz H. Reine Angew Mathematik. 1882;92:156–171. [Google Scholar]
- 29.Sneddon I N. Int J Eng Sci. 1965;3:47–57. [Google Scholar]
- 30.Domke J, Radmacher M. Langmuir. 1998;14:3320–3325. [Google Scholar]
- 31.Henderson E, Haydon P G, Sakaguchi D S. Science. 1992;257:1944–1946. doi: 10.1126/science.1411511. [DOI] [PubMed] [Google Scholar]
- 32.Abercrombie M J, Heaysman J E M, Pegrum S M. Exp Cell Res. 1970;59:393–398. doi: 10.1016/0014-4827(70)90646-4. [DOI] [PubMed] [Google Scholar]
- 33.Condeelis J. Annu Rev Cell Biol. 1993;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- 34.Welch M D, Mallavarapu A, Rosenblatt J, Mitchison T J. Curr Opin Cell Biol. 1997;9:54–61. doi: 10.1016/s0955-0674(97)80152-4. [DOI] [PubMed] [Google Scholar]
- 35.Cunningham C C. J Cell Biol. 1995;129:1589–1599. doi: 10.1083/jcb.129.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fedier A, Keller H U. Cell Motil Cytoskeleton. 1997;37:326–337. doi: 10.1002/(SICI)1097-0169(1997)37:4<326::AID-CM4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Keller H U, Bebie H. Cell Motil Cytoskeleton. 1996;33:241–251. doi: 10.1002/(SICI)1097-0169(1996)33:4<241::AID-CM1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 38.Rotsch C, Braet F, Wisse E, Radmacher M. Cell Biol Int. 1998;21:685–696. doi: 10.1006/cbir.1997.0213. [DOI] [PubMed] [Google Scholar]
- 39.Oster G, Perlson A. J Cell Sci Suppl. 1987;8:35–54. doi: 10.1242/jcs.1987.supplement_8.3. [DOI] [PubMed] [Google Scholar]
- 40.Peskin C H, Odell G M, Oster G F. Biophys J. 1993;65:316–324. doi: 10.1016/S0006-3495(93)81035-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olbris D J, Herzfeld J. Mut Res Soc Symp Proc. 1997;463:129–134. [Google Scholar]
- 42.Mogilner A, Oster G. Biophys J. 1996;71:3030–3045. doi: 10.1016/S0006-3495(96)79496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strohmeier R, Bereiter-Hahn J. J Cell Sci. 1987;88:631–640. doi: 10.1242/jcs.88.5.631. [DOI] [PubMed] [Google Scholar]
- 44.Small J V. J Cell Biol. 1981;91:695–705. doi: 10.1083/jcb.91.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Svitkina T M, Verkhovsky A B, Borisy G G. J Struct Biol. 1995;115:290–303. doi: 10.1006/jsbi.1995.1054. [DOI] [PubMed] [Google Scholar]
- 46.Bray D, White J G. Science. 1988;239:883–887. doi: 10.1126/science.3277283. [DOI] [PubMed] [Google Scholar]
- 47.Kolega J. J Cell Biol. 1986;102:1400–1411. doi: 10.1083/jcb.102.4.1400. [DOI] [PMC free article] [PubMed] [Google Scholar]