

Abstract
The nucleus is spatially ordered by attachments to a nonchromatin nuclear structure, the nuclear matrix. The nuclear matrix and chromatin are intimately connected and integrated structures, and so a major technical challenge in nuclear matrix research has been to remove chromatin while retaining a native nuclear matrix. Most methods for removing chromatin require first a nuclease digestion and then a salt extraction to remove cut chromatin. We have hypothesized that cut chromatin is held in place by charge interactions involving nucleosomal amino groups. We have tested this hypothesis by chemically modifying amino groups after nuclease digestion. By using this protocol, chromatin could be effectively removed at physiological ionic strength. We compared the ultrastructure and composition of this nuclear matrix preparation with the traditional high-salt nuclear matrix and with the third nuclear matrix preparation that we have developed from which chromatin is removed after extensive crosslinking. All three matrix preparations reveal internal nuclear matrix structures that are built on a network of branched filaments of about 10 nm diameter. That such different chromatin-removal protocols reveal similar principles of nuclear matrix construction increases our confidence that we are observing important architectural elements of the native structure in the living cell.
Nucleic acid metabolism is spatially ordered in the nucleus (1, 2). This order is achieved by association with the nonchromatin nuclear structure or nuclear matrix (3). The nuclear scaffolding juxtaposes factors, enzymes, and other macromolecules in appropriate spatial domains of the nucleus, thereby effecting an efficient assembly of the complexes involved in replication, transcription, and RNA processing. The nuclear matrix binds most nuclear RNA and organizes chromatin attachment sites responsible for chromatin loops.
Despite its apparent importance, visualizing and characterizing the nuclear matrix remains an ongoing task. Recent studies show the innermost component of the matrix to be a network of highly branched “core” filaments that have an average diameter of 10 nm (4–6). Many of the intermediates of nucleic acid metabolism, together with regulatory and catalytic factors, remain complexed to the nuclear matrix when separated from chromatin. This and other evidence suggest that nucleic acid synthesis and processing are both nuclear matrix-bound processes (reviewed in refs. 3, 7, 8).
A major technical challenge in nuclear matrix research has been to remove the tenaciously bound, dense, and obscuring chromatin while retaining the underlying structure in nearly native form and composition. Different strategies have been reported. All methods cleave chromatin with nuclease, but a major portion of the DNA remains bound to the matrix, probably in the form of nucleosomes. The methods then diverge in the means used to elute the chromatin fragments.
Various combinations of very high- and extremely low-salt extractions have been used by most matrix isolation protocols (0.25 M NaCl, 0.4 M KCl, or 2 M NaCl) to remove nuclease-cleaved chromatin from the nucleus (4, 9–12). This step is probably responsible for the less than complete preservation of fine structure seen with these older techniques. In addition, proteins that are associated with nonchromatin nuclear structures may be stripped from the nuclear matrix by salt extraction steps (discussed in ref. 3). Several efforts have been made to develop matrix preparation procedures conducted entirely at physiological ionic strength, with varying results (5, 13–15). The best characterized morphologically is the technique of Cook and colleagues, who used electrical fields to remove digested chromatin from the nucleus (5, 14, 15).
We have reported a new technique for separating the nuclear matrix from chromatin. This method begins with detergent-extracted cells and stabilizes nuclear ultrastructure by extensive crosslinking with formaldehyde before digesting chromatin (6). Surprisingly, the formaldehyde treatment does not crosslink nucleosomes to the nuclear matrix. Apparently the amino groups on nucleosomes are not juxtaposed to those on the matrix. Instead, the nuclease-digested chromatin is released directly and there is no need for elution at elevated salt concentrations. The matrix prepared by this technique appears to have much improved preservation of fine structure and closely resembles the ribonucleoprotein network of the unextracted nucleus (6). Most importantly, the stabilized nuclear matrix is built on an underlying network of 10-nm filaments that can be seen beneath and emerging from the more complicated fibrogranular structure of the internal nuclear matrix. This finding confirmed previous models of nuclear matrix organization (4, 5).
We hypothesized that residual chromatin remained bound to the nuclear matrix after chromatin digestion principally through electrostatic forces and might be released at physiological salt concentrations by formaldehyde modification of amino groups on histones and nuclear matrix proteins (6). We have now tested this hypothesis by replacing formaldehyde with N-hydroxysulfosuccinimide acetate (sulfo-NHS), a reagent that modifies protein amino groups without crosslinking. Digested chromatin—both DNA and histones—readily eluted under physiological salt conditions after sulfo-NHS treatment. This result supports the hypothesis that cut chromatin is bound through amino group and is of considerable practical value, allowing isolation of the nuclear matrix under nearly physiological salt conditions. The resulting nuclear matrix appears to have more complete preservation of ultrastructure than is possible with traditional salt-using methods (9, 11, 12) and may be more suitable for some biochemical studies than the formaldehyde-crosslinked structure.
MATERIALS AND METHODS
Detergent Extraction of Cells.
Caski (CRL 1550) or SAOS-2 cells (HTB 85) were fractionated by following previously published procedures (4, 11, 17) but with some modifications. The cells were washed two times with cold PBS and extracted in cytoskeletal buffer (10 mM Pipes, pH 6.8/100 mM NaCl/300 mM sucrose/3 mM MgCl2/1 mM EGTA/1 μg/ml leupeptin/1 μg/ml pepstatin/2 μg/ml aprotinin/1 μg/ml antipain/1 mM aminoethyl benzenesulfonyl fluoride/10 units/ml prime RNase inhibitor) containing 0.5% Triton X-100 for 7 min at 4°C to remove the soluble proteins.
High-Salt Elution of Chromatin.
After this extraction, cells were treated with 600 units/ml each of the restriction enzymes (PstI and HaeIII) in cytoskeletal buffer at 32°C for 1 hr. Chromatin was then removed by a two-step salt extraction. First, 1 M ammonium sulfate was added to make a final concentration of 0.25 M. The cells were then washed and placed in cytoskeletal buffer to which a stock of 4 M NaCl was added to a final concentration of 2 M and incubated at room temperature for 10 min. For suspension-grown cells, washing was by centrifugation. Cells grown on solid substrates [glass coverslips or electron microscopy (EM) grids] were washed by buffer changes.
Chromatin Removal by Amine Modification.
This technique removes chromatin after digestion by modifying protein amine groups with the reagent sulfo-NHS (Pierce). After digestion with restriction enzymes, extracted cells were exposed to 2 mg/ml freshly prepared sulfo-NHS-acetate (in cytoskeletal buffer, pH 7.0) for 20 min at room temperature. After being washed, cells were again treated with 2 mg/ml of sulfo-NHS for 20 min at room temperature. Cells were then washed with 10 mM glycine to quench the excess blocking reagent. The final pellet was resuspended in cytoskeletal buffer.
DNA Measurement.
The amount of DNA remaining in the nuclear matrix was measured either by labeling cells with [3H]thymidine and measuring radioactivity release or by staining with 4′,6-diamidino-2-phenylindole (DAPI). Cells were labeled with 10 uCi/ml of Methyl [3H]thymidine for 20 hr. Each fraction of cell extraction was collected, and the radioactivity of the trichloroacetic acid precipitable samples was measured. In some experiments, cells grown on cover glass were extracted, fixed in 3.7% formaldehyde, and stained in 20 μg/ml of DAPI for 10 min.
Analysis of Proteins.
To measure the histone content of cell fractions, cells were labeled with 10 μC/ml [3H]lysine overnight. After extraction, the cells treated with 2 M NaCl and/or with sulfo-NHS-acetate were analyzed by gel electrophoresis on an 18% polyacrylamide gel (4). Nonhistone protein content was measured by labeling cells with 50 μCi/ml of [35S]methionine for 2 hr. The nuclear matrix proteins in the high-salt or the blocking reagent treatment samples were analyzed by SDS/PAGE on 10% gels.
EM.
We used two methods of embedment-free microscopy (16, 17): resinless sections for detailed morphology and cell whole mounts for immunolocalization and global morphology. Cells for resinless section EM were grown on coverslips, extracted, and then processed as described (16, 17). For the whole-mount EM, cells were grown on EM grids covered with formvar and coated with carbon. Cells were extracted as above and fixed with 3.7% formaldehyde (16, 17). High-resolution immunolocalization of antigens used whole mounts. The antibody-staining procedure has been published (17).
Immunofluorescence Microscopy.
Cells were grown on coverslips. After extraction, cells were fixed in 3.7% formaldehyde for 30 min at 4°C and blocked in 10% normal goat serum for 30 min at room temperature. Antibody incubations were in PBS containing 0.1% Tween 20.
RESULTS
All nuclear matrix isolation protocols remove chromatin in two steps. First, DNA is partially digested by suitable nucleases. The resulting chromatin fragments remain bound to the nuclear matrix and, in most protocols, are then extracted at elevated ionic strength. These ionic conditions can effect changes in nuclear matrix composition and form. We report here a method for removing digested chromatin from detergent-extracted nuclei without a salt-extraction step. DNA is first cleaved with a nuclease, either DNase I or a combination of the restriction enzymes PstI and HaeIII. The nucleus is then treated with the reagent sulfo-NHS, which modifies accessible amino groups. The resulting modification of protein amino groups results in the release of chromatin fragments.
Residual DNA in the Nuclear Matrix Prepared with Sulfo-NHS.
CaSki cells were extracted in 0.5% Triton X-100 (Calbiochem) to remove membrane lipids, allowing soluble proteins to diffuse away. The extracted cells were then digested with the restriction enzymes HaeIII and PstI to partially digest chromatin. Chromatin fragments were then eluted by treatment with the primary amine-blocking reagent sulfo-NHS. The removal of DNA by this procedure was estimated in CaSki cells by staining with the DNA-specific dye DAPI (Fig. 1 a and c). The images suggest that approximately 95% of total DNA was removed by this procedure, with the remaining DNA distributed throughout the nucleus. A more precise measurement used cells grown in [3H]thymidine and treated by the same protocol. The results of many experiments show that 7–13% of trichloroacetic acid-precipitable DNA remained in the nuclear matrix. A comparison of nuclear matrix preparations analyzed by DAPI staining showed that almost as much DNA was removed by the restriction enzyme/sulfo-NHS as by a more traditional high-salt method (Fig. 1b). Measurements of histone removal gave similar results (Fig. 2).
Figure 1.

DNA release from CaSki cells by salt extraction or amine modification after nuclease digestion. (a) Control cells extracted only with Triton X-100. Cells were extracted with 0.5% Triton in CSK buffer, fixed with 3.7% formaldehyde, and treated with 20 μg/ml DAPI to selectively stain DNA. (b) Standard nuclear matrix prepared with high-salt extraction. The 0.5% Triton X-100-extracted cells were digested with HaeIII/PstI, and the cleaved chromatin was eluted with 0.25 M ammonium sulfate followed by 2 M NaCl. The cells were then fixed and stained with DAPI. (c) Sulfo-NHS-extracted nuclear matrix preparation. The 0.5% Triton X-100-extracted cells were digested with HaeIII/PstI, and cleaved chromatin was removed by treatment with sulfo-NHS. The cells were then fixed and stained with DAPI.
Figure 2.

Histone release by salt extraction or amine modification after nuclease digestion. CaSki cells were labeled with [3H]lysine and nuclear matrices prepared by either the high-salt method (lanes 1 and 2) or the sulfo-NHS procedure (lanes 3 and 4). The histones from nuclear fractions were labeled heavily with [3H]lysine and were analyzed by SDS/PAGE and fluorography. Most of each histone was released from the nucleus by HaeIII/PstI digestion and sulfo-NHS treatment (lane 3), whereas less than 10% remained in the nuclear matrix fraction (lane 4). By using the more traditional high-salt method, the histones were released after HaeIII/PstI digestion by application of 0.25M ammonium sulfate (lane 1), leaving little histone protein with the nuclear matrix (lane 2).
Nuclear Matrix Protein Composition.
Nuclear matrix proteins both from the traditional high-salt elution and by the amine blocking reagent were analyzed by 10% SDS/PAGE (Fig. 3). Only one-dimensional electrophoresis was used, because the amine modification by sulfo-NHS changes isoelectric points and hence makes two-dimensional gel patterns impossible to interpret. Most nuclear matrix proteins were present in both preparations, although there were some differences. These common proteins were usually proteins present in the amine-blocking protocol but removed by high-salt extraction. These are very likely authentic nuclear matrix proteins stripped by the more disruptive high-salt elution procedure.
Figure 3.
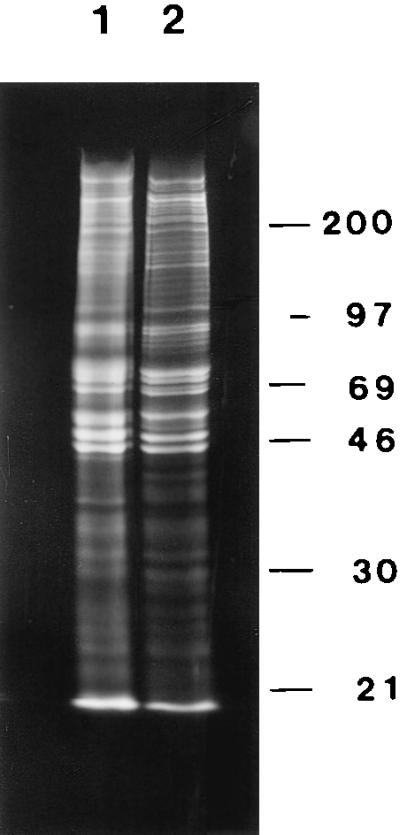
Protein composition of the high-salt and amine-modified nuclear matrices. CaSki cell proteins were labeled with [35S]methionine, extracted with 0.5% Triton X-100, and the chromatin digested with HaeIII/PstI. Chromatin was subsequently removed either by sequential 0.25 M ammonium sulfate/2 M NaCl extraction or by sulfo-NHS treatment. The proteins remaining with the nuclear matrix were analyzed by SDS/PAGE and fluorography. The protein composition of the amine-modified nuclear matrix (lane 1) was similar, but not identical, to the protein complement of the high-salt nuclear matrix (lane 2). There was also a greater total retention of proteins in the amine-modified nuclear matrix.
Morphology of Amine-Modified Nuclear Matrix.
Fig. 4 shows a nuclear matrix seen in whole mount, prepared from a SAOS cell by using sulfo-NHS. The nuclear matrix, seen here in its entirety, consists of the nuclear lamina enclosing a network of core filaments enmeshing residual nucleoli and smaller dense bodies.
Figure 4.

Amine-modified nuclear matrix seen in a whole-mount electron micrograph. SAOS-2 cells were extracted with 0.5% Triton X-100 in CSK buffer, digested with PstI and HaeIII, treated with sulfo-NHS, and processed for whole-mount EM. (a) Nuclear matrices, each bounded by a nuclear lamina (L), were enmeshed in and connected to the intermediate filaments (IF) of the cytoskeleton. Within each nucleus, residual nucleoli (Nu) were connected to nuclear matrix filaments, including 10-nm branched core filaments. (b) These connections between nuclear matrix filaments and nucleoli can be seen most clearly at higher magnification and in stereo.
The stereoscopic micrograph in Fig. 4b shows an important feature of the sulfo-NHS procedure: the preservation of detailed nucleolar ultrastructure. Although major nucleolar components such as ribosomal precursor RNA remain in a high-salt matrix, these are largely dispersed during high-salt elution. In contrast, the sulfo-NHS procedure affords, for the first time, an intact nucleolus visible in its entirety in an embedment-free EM preparation. Some of the granular substructure is visible at the nucleolar periphery. Numerous filaments anastomose with the nucleolus and correspond in size to the core filaments. Other filaments are larger and denser.
The unembedded whole mount affords a unique global view of the amine-modified nuclear matrix. However, for some purposes the images are too cluttered because of the dense array of matrix filaments. High-resolution studies require samples of the matrix volume that are thin enough to avoid optical confusion but still preserve structure. Such samples are obtained with resinless sections that allow very detailed images of the three-dimensional architecture of the nuclear interior.
The resinless section electron micrographs in Fig. 5 compare the matrix morphology of three nuclear matrix preparations that differ principally in their means of removing post-nuclease chromatin fragments. Fig. 5a shows the matrix prepared by the more conventional elution with 2 M NaCl. The electron micrograph exhibits the characteristic network of branched core filaments distributed throughout the nuclear interior and terminating on the nuclear lamina. Several dense bodies are enmeshed in the core filament network. Some these correspond to the interchromatin granule clusters seen in conventional embedded EM sections and to the splicing speckles seen in immunofluorescence microscopy (6, 18–20).
Figure 5.

Ultrastructural comparison of nuclear matrices prepared by three methods. CaSki cell nuclear matrices were prepared by three different methods and then visualized by resinless section EM. Higher magnification views of the nuclear interior are presented. (a) The high-salt nuclear matrix prepared by extracting cells with 0.5% Triton X-100 and then removing chromatin by digestion with PstI/HaeIII and sequential extractions with 0.25 M ammonium sulfate and 2 M NaCl. This procedure uncovered a network of branched filaments with an average diameter of 10 nm (arrowheads). The network of filaments connects to the inside of the nuclear lamina (not shown). (b) The amine-modified nuclear matrix was prepared in a similar way, except that the salt extractions were replaced by treatment with sulfo-NHS. The fundamental structural element revealed was a network of branched core filaments (arrowheads). More material coating the core filaments was retained. (c) The crosslink-stabilized nuclear matrix was prepared (6). After a 0.5% Triton X-100 extraction to remove soluble proteins, the structural networks were extensively crosslinked with formaldehyde. Chromatin was then removed by DNase I digestion. The more intricate structure of thicker fibers was built on an underlying network of core filaments (arrowheads). All three nuclear matrix methods reveal similar principles of nuclear construction with the nuclear matrix built on a network of branched core filaments connected to the nuclear lamina.
The resinless section micrograph of an amine-modified nuclear matrix in Fig. 5b shows the filament morphology is closely similar to the high-salt matrix in 5a. Enmeshed dense bodies are also seen here, and these often appear somewhat more extended and hence closer to what would be expected from images afforded by conventional embedded section micrographs.
The most accurate morphology is very likely that offered by the preparations fixed with formaldehyde before chromatin extraction (6). These preparations should retain the components that might be removed by either high concentrations of salt or sulfo-NHS. Fig. 5c shows a resinless section micrograph of a preextraction fixed preparation. This micrograph also shows the network of branched core filaments although, as expected, these show more filament-bound material.
Localization of SRm300 in the Amine-Modified Nuclear Matrix.
The identification of many of the dense bodies as interchromatin granule clusters was confirmed by immunogold staining with an antibody against a protein highly enriched in those structures. The whole-mount micrograph shown in Fig. 6 shows the distribution of the nuclear matrix-associated RNA splicing factor SRm300 in an amine-modified nuclear matrix. Detergent-extracted SAOS-2 cells were digested with HaeIII/PstI and treated with sulfo-NHS. They were then fixed, immunostained with monoclonal antibody-B4A11, which recognizes SRm300 (19, 20), and with a gold bead-conjugated second antibody. Gold beads heavily decorated the dense bodies retained in the matrix that must, therefore, correspond to interchromatin granule clusters. No staining was found either on the nucleoli or the filaments themselves. The inset of Fig. 6a shows the corresponding immunofluorescence staining pattern for B4A11. This antibody stained the splicing speckles, whereas nucleoli were unstained. The staining pattern was similar to that of unextracted chromatin-containing cells and therefore was consistent with a good preservation of the nuclear architecture by this new technique.
Figure 6.

Distribution of the RNA-splicing protein SRm300 in the amine-modified nuclear matrix. SAOS-2 cells, grown on gold grids, were processed by the sulfo-NHS nuclear matrix preparation method for whole-mount EM. The location of a nuclear matrix protein involved in RNA splicing was determined by staining with mAb B4A11 and a 10-nm gold-conjugated second antibody. (a) Low-magnification view of a B4A11-stained cell. The square marks the field selected for the higher magnification whole-mount stereo pair of b. The inset shows the location of SRm300 determined by immunofluorescent staining with mAb B4A11. (b) The dense bodies, corresponding to interchromatin granule clusters (splicing speckles) were heavily stained by mAb B4A11. In contrast, the larger dense bodies corresponding to nucleoli were completely unstained.
DISCUSSION
The intricate and precise nuclear architecture is ample evidence of an underlying organizing scaffold or nuclear matrix. Oddly, although the fact of nuclear organization is widely recognized, the nature of the nuclear matrix has been difficult to elucidate. Indeed, much controversy has surrounded this subject with some voices insisting, despite ample evidence to the contrary, that there was no nuclear matrix (21). Even amongst the convinced, there was vigorous contestation over the nature of the structure.
The clamor of opposing views came from the failure of many to consider ultrastructural evidence. Embedment-free EM techniques have improved the imaging of chromatin-depleted nuclear structure and allowed the use of ultrastructural criteria in the development of nuclear matrix isolation techniques (16, 17). This report compares three techniques for removing chromatin by those improved imaging methods. These three matrix preparations yield complementary results, revealing important principles of nuclear construction.
Chromatin is spatially organized by attachments to a nuclear scaffolding, the nuclear matrix. These attachments form chromatin into loop domains with an average size of 86 kb (15, 22). Nuclear matrix preparation protocols remove the majority of each loop, leaving with the matrix those DNA sequences nearest the loop base. This removal is usually achieved by a two-step protocol. In one step, a nuclease digestion cuts the DNA, presumably between nucleosomes.
A second step is required, the removal of chromatin fragments that remain bound. The older techniques use high-salt extraction. One alternative approach, yielding a nuclear matrix structure similar in basic construction to that shown in this report, removes DNA after nuclease digestion by electroelution under isotonic conditions (5). Digested chromatin may normally be held in place by electrostatic interactions that are broken down by salt treatment or overcome by electrical fields.
We previously reported that chromatin can be effectively removed from nuclei at physiological ionic strength by formaldehyde crosslinking and nuclease digestion (6). No salt elution or electroelution was required. Our hypothesis was that formaldehyde was modifying histone amino groups, allowing cut chromatin release. In this paper, we have tested that hypothesis and shown that chromatin—both DNA and histones — can be removed efficiently from the nucleus by a nuclease digestion and the modification of primary amino groups with sulfo-NHS. Both steps can be performed under isotonic solution conditions, avoiding any possibility of salt-induced rearrangements in the structure (15). Like formaldehyde, sulfo-NHS modifies amino groups and may alter their charge at physiological pH or sterically hinder their interactions. Treatment of chromatin with sulfo-NHS may, by this mechanism, reduce electrostatic interactions, holding cut chromatin in place.
The nuclear matrix uncovered in this way is built on a network of highly branched 10-nm filaments connected to the interior of the nuclear lamina and enmeshing nuclear structures including chromatin-depleted nucleoli and interchomatin granule clusters. These branched 10-nm filaments can be uncovered by many different nuclear matrix protocols (4–6) and can even be seen in whole nuclei when chromatin condenses (23). Their molecular characterization remains an important goal for future research.
Acknowledgments
This work was supported by National Institutes of Health Grant CA067628 to S.P. and American Cancer Society Grant IRG-93-033-05 to J.A.N.
ABBREVIATIONS
- sulfo-NHS
N-hydroxysulfosuccinimide acetate
- DAPI
4′,6-diamidino-2-phenylindole
- EM
electron microscopy
References
- 1.Spector D L. Annu Rev Cell Biol. 1993;9:265–315. doi: 10.1146/annurev.cb.09.110193.001405. [DOI] [PubMed] [Google Scholar]
- 2.Lamond A I, Earnshaw W C. Science. 1998;280:547–553. doi: 10.1126/science.280.5363.547. [DOI] [PubMed] [Google Scholar]
- 3.Nickerson J A, Blencowe B J, Penman S. Int Rev Cytol. 1995;162A:67–123. doi: 10.1016/s0074-7696(08)61229-2. [DOI] [PubMed] [Google Scholar]
- 4.He D C, Nickerson J A, Penman S. J Cell Biol. 1990;110:569–580. doi: 10.1083/jcb.110.3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson D A, Cook P R. EMBO J. 1988;7:3667–3677. doi: 10.1002/j.1460-2075.1988.tb03248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nickerson J A, Krockmalnic G, Wan K M, Penman S. Proc Natl Acad Sci USA. 1997;94:4446–4450. doi: 10.1073/pnas.94.9.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Driel R, Wansink D G, van Steensel B, Grande M A, Schul W, DeJong L. Int Rev Cytol. 1995;162A:151–189. doi: 10.1016/s0074-7696(08)61231-0. [DOI] [PubMed] [Google Scholar]
- 8.Berezney R, Mortillaro M J, Ma H, Wei X, Samarabandu J. Int Rev Cytol. 1995;162A:1–65. doi: 10.1016/s0074-7696(08)61228-0. [DOI] [PubMed] [Google Scholar]
- 9.Berezney R, Coffey D S. Biochem Biophys Res Commun. 1974;60:1410–1417. doi: 10.1016/0006-291x(74)90355-6. [DOI] [PubMed] [Google Scholar]
- 10.Kaufmann S H, Shaper J H. Exp Cell Res. 1991;192:511–523. doi: 10.1016/0014-4827(91)90071-2. [DOI] [PubMed] [Google Scholar]
- 11.Capco D G, Wan K M, Penman S. Cell. 1982;29:847–858. doi: 10.1016/0092-8674(82)90446-9. [DOI] [PubMed] [Google Scholar]
- 12.Fey E G, Krochmalnic G, Penman S. J Cell Biol. 1986;102:1654–1665. doi: 10.1083/jcb.102.5.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaughn J P, Dijkwel P A, Mullenders L H, Hamlin J L. Nucleic Acids Res. 1990;18:1965–1969. doi: 10.1093/nar/18.8.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson D A, Cook P R. EMBO J. 1985;4:919–925. doi: 10.1002/j.1460-2075.1985.tb03719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson D A, Dickinson P, Cook P R. EMBO J. 1990;9:567–571. doi: 10.1002/j.1460-2075.1990.tb08144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capco D G, Krochmalnic G, Penman S. J Cell Biol. 1984;98:1878–1885. doi: 10.1083/jcb.98.5.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nickerson J A, Krockmalnic G, Penman S. In: Cell Biology: A Laboratory Handbook. Celis J, editor. Vol. 1. Orlando: Academic; 1998. pp. 184–192. [Google Scholar]
- 18.Monneron A, Bernhard W. J Ultrastruct Res. 1969;27:266–288. doi: 10.1016/s0022-5320(69)80017-1. [DOI] [PubMed] [Google Scholar]
- 19.Blencowe B J, Nickerson J A, Issner R, Penman S, Sharp P A. J Cell Biol. 1994;127:593–607. doi: 10.1083/jcb.127.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blencowe B J, Issner R, Nickerson J A, Sharp P A. Genes Dev. 1998;12:996–1009. doi: 10.1101/gad.12.7.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singer R H, Green M R. Cell. 1997;91:291–294. doi: 10.1016/s0092-8674(00)80411-0. [DOI] [PubMed] [Google Scholar]
- 22.Vogelstein B, Pardoll D M, Coffey D S. Cell. 1980;22:79–85. doi: 10.1016/0092-8674(80)90156-7. [DOI] [PubMed] [Google Scholar]
- 23.Zhai Z H, Nickerson J A, Krochmalnic G, Penman S. J Virol. 1987;61:1007–1018. doi: 10.1128/jvi.61.4.1007-1018.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]