

Summary
In a genetic screen for Kinesin heavy chain (Khc)-interacting proteins, we identified APLIP1, a neuronally expressed Drosophila homolog of JIP-1, a JNK scaffolding protein [1]. JIP-1 and its homologs have been proposed to act as physical linkers between kinesin-1, which is a plus-end-directed microtubule motor, and certain anterograde vesicles in the axons of cultured neurons [2]. Mutation of Aplip1 caused larval paralysis, axonal swellings, and reduced levels of both anterograde and retrograde vesicle transport, similar to the effects of kinesin-1 inhibition. In contrast, Aplip1 mutation caused a decrease only in retrograde transport of mitochondria, suggesting inhibition of the minus-end microtubule motor cytoplasmic dynein [3]. Consistent with dynein defects, combining heterozygous mutations in Aplip1 and Dynein heavy chain (Dhc64C) generated synthetic axonal transport phenotypes. Thus, APLIP1 may be an important part of motor-cargo linkage complexes for both kinesin-1 and dynein. However, it is also worth considering that APLIP1 and its associated JNK signaling proteins could serve as an important signaling module for regulating transport by the two opposing motors.
Results and Discussion
To identify proteins that influence kinesin-1-based axonal transport, genetic interaction tests were done to search for mutations that act as dominant enhancers of Kinesin heavy chain [4, 5]. A number of such E(Khc) mutations were found that caused synthetic axonal transport phenotypes (i.e., larval paralytic “tail flipping” and organelle-filled “axon swellings”) when combined with a Khc null (Khc27/+; E(Khc)/+). Tail flipping was not seen and swellings were rare in Khc27/+ or E(Khc)/+ single heterozygotes. A subset of E(Khc) loci caused tail flipping and swellings when homozygous mutant in a wild-type Khc background, suggesting that the products of those loci have direct roles in axonal transport. That subset includes Kinesin light chain (Klc), Dynein heavy chain 64C (Dhc64C), Glued [4, 6], and an unknown locus on chromosome 3 initially designated E(Khc)ek4 (abbreviated as ek4 below).
To gain more insight into the functions of ek4 products, a number of phenotypic tests were done. Homozygous ek4 mutant larvae showed classic posterior paralysis and axonal swelling phenotypes (Figure 1A) with severities similar to those caused by strong hypomorphic Khc genotypes [4, 7, 8]. However, in contrast to such Khc mutants, which die during larval and pupal stages of development, ek4 mutants survived to become active, fertile adults. Severity comparisons with a null (Df(3L)Fpa2) indicated that the ek4 mutation is a strong hypomorphic allele, causing nearly a complete loss of function (Table 1). These observations suggest that wild-type products of the ek4 locus have important axonal transport functions in larvae and that they have a positive functional relationship with kinesin-1. However, ek4 is not itself essential, suggesting that its products contribute to only a subset of kinesin-1 functions.
Figure 1.
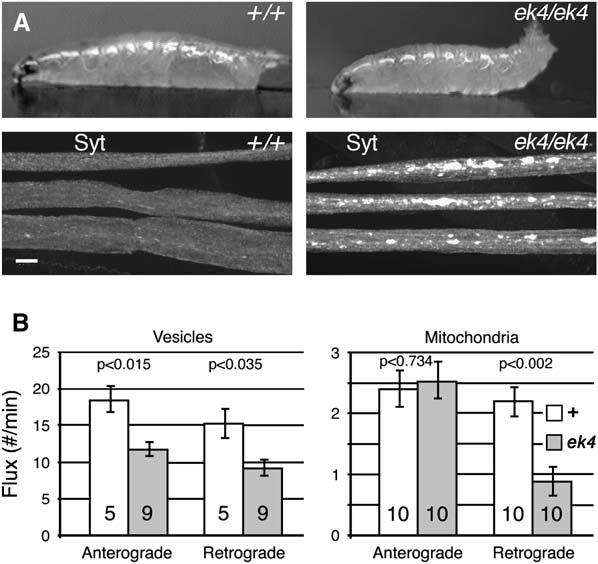
Axonal Transport Phenotypes in ek4 Mutants
(A) Top: single video frames of wild-type and ek4 mutant third instar larvae crawling across an agar block (anterior is to the left). Note the paralytic tail flipping of the mutant. Bottom: micrographs of segmental nerves from similar larvae that were dissected, fixed, and immunostained for the axonal vesicle protein synaptotagmin (Syt).
(B) The number of vesicles (GFP-nSyb) or mitochondria (mito-GFP) passing a given point in a nerve per minute (flux) was determined by time-lapse confocal microscopy of wild-type or mutant animals (see Movies S1–S4). Mean flux values (±SEM) are shown for anterograde and retrograde organelle transport. The number of different larvae tested is shown within each bar. P values, determined by a two-tailed t test (unequal variances), show the significance of differences between corresponding wild-type and ek4 mutant values.
Table 1.
Genetic Interaction Tests of ek4
| Genotype | % Flippersa | Swelling Severityb |
|---|---|---|
| +/+ | 0.0 (0, 0) | — |
| ek4/ek4 | 80.5 (82, 79) | +++ |
| Df(3L)Fpa2/ek4c | 90.0 (92, 88) | ++++ |
| Khc27+; ek4/+ | 86.0 (88, 84) | +++ |
| Khc27/+; ek4/P[Aplip1+]d | 0.0 (0, 0) | + |
| Df(3L)34ex5/ek4e | 66.5 (61, 72) | +++ |
| cDhc64Cek1/ek4f | 32.0 (29, 35) | ++ |
| Klp64Dk5/ek4g | 0.0 (0, 0) | — |
Third instar larvae were scored for posterior paralysis (tail flipping) and axonal swellings (synaptotagmin accumulations).
Two independent tests; Avg.% (Test 1%, Test 2%). n = 100 larvae per test.
The size and abundance of axonal swellings were scored according to the criteria of Martin et al. [4]. n = 5 larvae per test.
Df(3L)Fpa2 is a deletion that removes the ek4 locus, serving as an ek4 null.
P[Aplip1+] is a P element that caries a wild-type Aplip1 gene.
Df(3L)34ex5 is a deletion that removes Klc.
cDhc64Cek1 is slightly more severe than a null [4].
Klp64Dk5 is similar to a null when homozygous but is hypomorphic when hemizygous (K. Ray, personal communication).
To test the effects of ek4 mutations on kinesin-1-dependent fast axonal transport, we used time-lapse confocal microscopy. GFP-neuronal synaptobrevin (GFP-nSyb) [9] was used to image transport vesicles (see Movie S1 in the Supplemental Data available with this article online), while cytochrome c oxidase-GFP (mito-GFP) [3] was used to image mitochondria (Movie S3). They were expressed in motoneurons of larvae by virtue of Gal4-UAS promoters that were activated by P[GawB]D42-Gal4 (abbreviated D42), a motor neuron Gal4 driver [10]. With this system, it has been shown that hypomorphic Khc mutations cause anterograde and retrograde flux reductions for GFP-nSyb (60%–70%) and for mito-GFP (75% and 90%), supporting the hypothesis that normal dynein function in some processes depends on kinesin-1 [3]. Both anterograde and retrograde GFP-nSyb flux were reduced ∼35% in ek4 mutant axons (Figure 1B, Movie S2), supporting the idea that wild-type ek4 products facilitate some kinesin-1 functions. Surprisingly, ek4 mutant axons showed no change in anterograde mito-GFP flux and a 60% reduction in retrograde flux (Figure 1B, Movies S3 and S4). Currently, the only mutations known to cause a similar unidirectional inhibition of retrograde mitochondrial flux are in Dhc64C (∼80%) [3], which encodes the motor subunit of cytoplasmic dynein.
To further test the possibility that ek4 influences dynein, additional genetic interaction tests were done (Table 1). Consistent with the original genetic screen for dominant enhancers of Khc, ek4 acted as a dominant enhancer of Kinesin light chain (Klc), causing synthetic tail flipping and axonal swelling phenotypes. No such interaction was seen when ek4 was combined with a mutant allele of Klp64D, which encodes an anterograde axonal motor of the kinesin-2 family [11]. However, when ek4 was combined with a mutant allele of Dhc64C, synthetic tail flipping and axonal swelling phenotypes were seen. In summary, our results support the hypothesis that wild-type ek4 gene products facilitate vesicle transport by kinesin-1 and mitochondrial transport by cytoplasmic dynein.
To identify the ek4 locus, meiotic recombination and deletion mapping approaches were initially used. Our results indicated a position near the tip of the left arm of chromosome 3 within the 61F3-4 cytological region (Figure 2A). That interval included APP-like interacting protein 1 (Aplip1), a gene that encodes a neuronally expressed Drosophila homolog of c-Jun N-terminal kinase (JNK)-interacting protein 1 (JIP-1), a scaffolding protein that has been shown to bind Kinesin light chain (KLC), a reelin receptor (ApoER2), and Alzheimer's amyloid precursor protein (APP), as well as JNK pathway kinases [1, 2, 12]. It has been proposed that JIP-1 and its close relative JIP-2 link kinesin-1 with axon vesicles to facilitate anterograde vesicle transport. Similar kinesin-1 linker functions have been proposed for an unrelated JNK scaffolding protein, sunday driver (syd, JSAP, JIP-3), and for APP [13, 14], although the APP-kinesin relationship may be mediated by APLIP1/JIP-1 [1, 15-17]. A P element transgene that included Aplip1 and flanking sequences fully rescued the tail flipping and partially rescued the axonal swelling phenotypes of larvae that were doubly heterozygous for Khc27 and ek4 (Table 1). Finally, sequencing of the Aplip1 locus from ek4 mutant animals revealed a single base change that converts a conserved proline at position 483 to leucine (Figure 2B). This proline is within a conserved 11 amino acid C-terminal region (KBD) that has been shown to be important for binding of mammalian JIP-1 to KLC [2]. The transgenic rescue and sequencing results confirm that ek4 is a mutant allele of the Aplip1 gene, and hence it will be referred to as Aplip1ek4.
Figure 2.
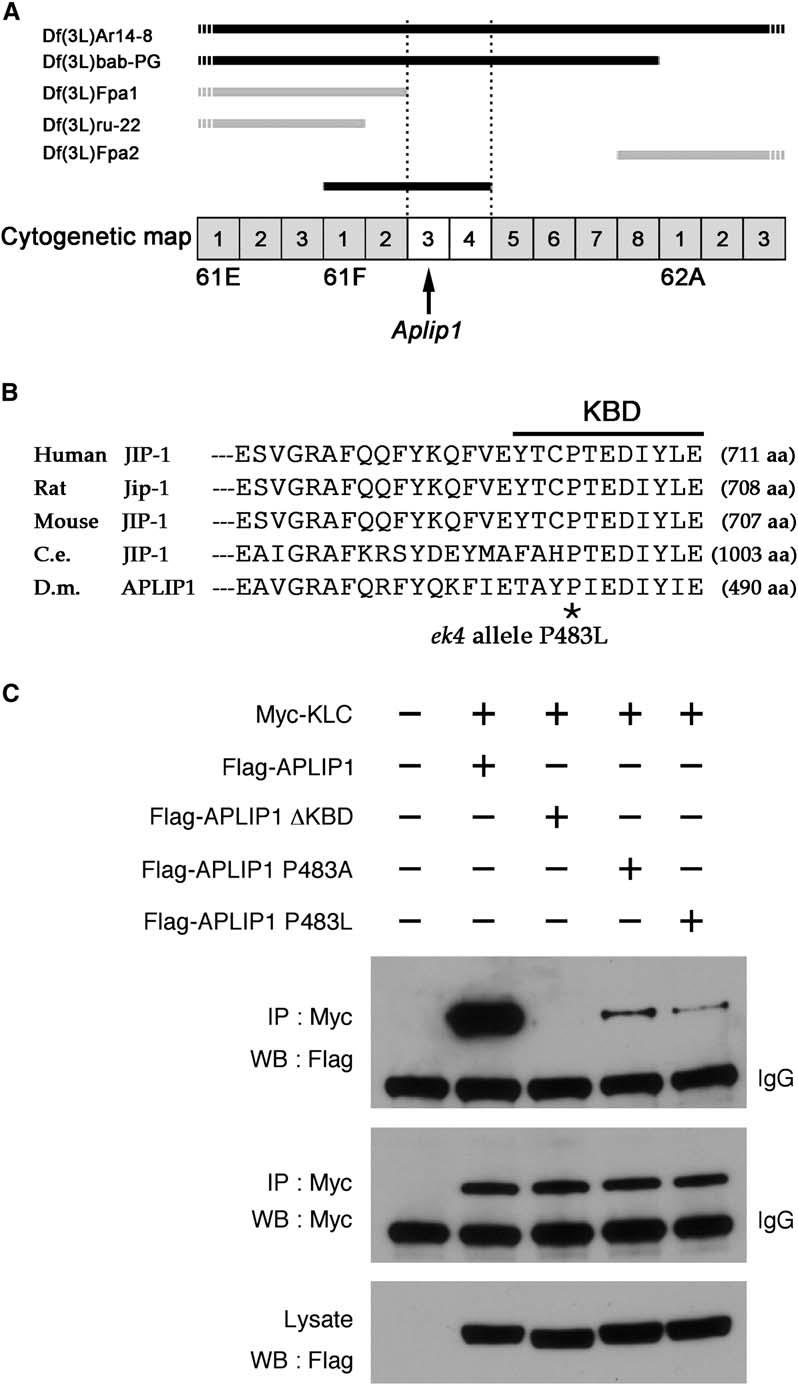
The ek4 Dominant Enhancer of Khc Mutation Is an Allele of Aplip1 that Inhibits Binding between APLIP1 and Kinesin Light Chain
(A) Chromosomal deficiencies (Df) on the left arm of chromosome 3 (3L) were used to map the ek4 locus to 61F3-4 by genetic complementation. The horizontal bars indicate chromatin that is absent from the listed deficiency chromosomes. Those that complemented ek4 are indicated by gray bars, and those that failed to complement are indicated by black bars.
(B) Sequence alignment of the C-terminal 27 residues of JIP-1 proteins from various species and of APLIP1 from Drosophila. Full sequence length for each protein is noted in parentheses. The 11 residues previously defined as a KLC binding domain [2] are noted by a black line (KBD). The location of a single amino acid change in the Aplip1 gene of ek4 mutants (P483L) is noted by an asterisk.
(C) The effects of P483L and other sequence changes on binding between KLC and APLIP1. S2 cells were transfected with the indicated epitope-tagged constructs. Cell lysates were incubated with anti-Myc antibody to immunoprecipitate Myc-KLC. The resulting pellets were then analyzed by Western blotting with anti-Myc or anti-Flag (for Flag-APLIP1). The bottom panel demonstrates that the wild-type and mutant Flag-APLIP1 proteins were present at similar levels in the starting cell lysates.
To determine whether the P483L mutation affected KLC-APLIP1 binding, we used epitope-tagged versions of KLC and APLIP1 for immunoprecipitation studies. After coexpression of Myc-KLC and wild-type Flag-APLIP1 in S2 cultured cells, anti-Myc antibody precipitated both proteins (Figure 2C, second lanes). Removal of the 11 amino acid KBD from Flag-APLIP1 eliminated detectable binding to Myc-KLC (Figure 2C, third lanes). Furthermore, changing proline 483 to either leucine or alanine substantially reduced KLC binding (Figure 2C, lanes 4 and 5). This shows that P483 is indeed important for KLC binding, which suggests that at least some of the Aplip1ek4 mutant phenotypes were due to poor association of APLIP1 and kinesin-1.
If APLIP1 links kinesin-1 to anterograde transport vesicles in Drosophila axons, as has been proposed for JIP-1 in vertebrates [2], it should localize in axons and such localization should depend on its ability to bind KLC. To test those predictions, we transformed flies with P elements that carried either full-length UAS-Flag-Aplip1 or UAS-Flag-Aplip1ΔKBD. When driven by D42-Gal4, the two constructs produced equivalent levels of mRNA, which were many times in excess relative to the endogenous gene in larvae (Figure S1). Western blots of larvae with anti-Flag were not successful, but both the full-length and the ΔKBD Flag-tagged proteins were seen at equivalent levels in Westerns of transfected S2 cells, suggesting that both were stable (Figure 2 and Figure S2, middle panel). Interestingly, D42-Gal4-driven expression of one copy of full-length UAS-Flag-Aplip1 in motoneurons caused dramatic tail flipping (Figure 3A) and nearly 100% lethality during late larval and pupal stages. In larval nerves, it caused axon swellings that stained intensely for vesicles (anti-Syt) and APLIP1 (anti-Flag) (Figure 3C). D42-Gal4-driven expression of the deletion construct caused no tail flipping (Figure 3B) or lethality. It did cause some axon swellings in larval nerves, and Flag-APLIP1ΔKBD staining was visible in those swellings (Figure 3D). However, the overall amount of staining in nerves was substantially reduced relative to the amount seen after expression of the full-length protein.
Figure 3.
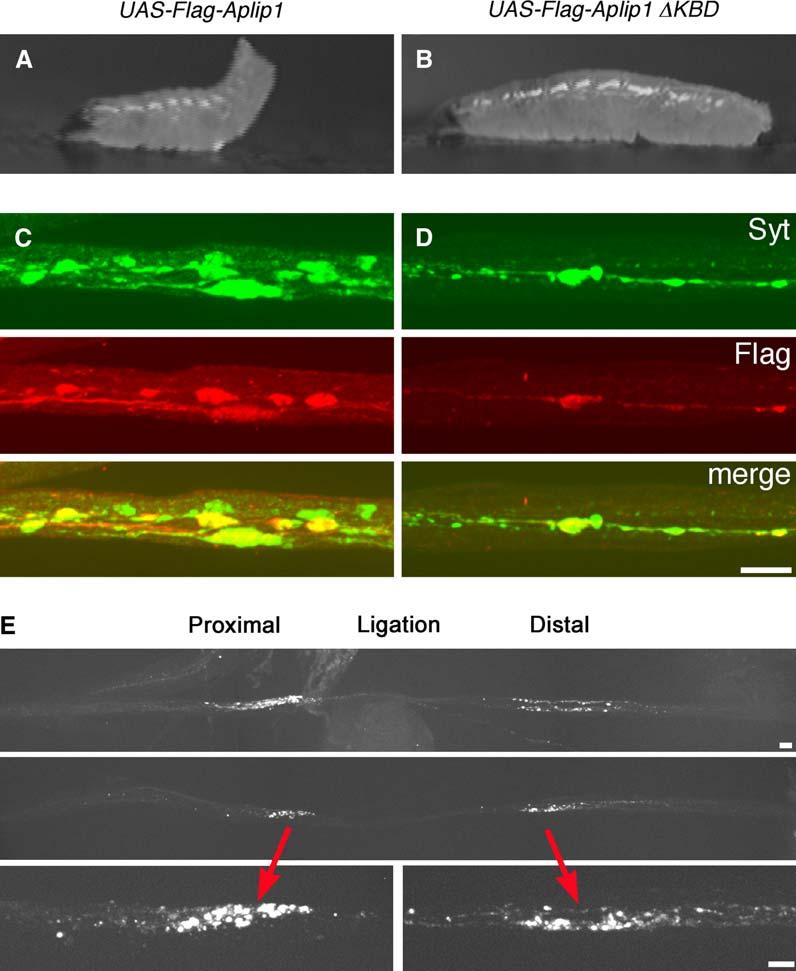
Bidirectional Axonal Transport of APLIP1 and Overexpression-Induced Axonal Transport Phenotypes
(A–D) Wandering third instar larvae (A and B) or segmental nerves (C and D) with overexpression of one copy of either full-length Flag-APLIP1 (left) or Flag-APLIP1 with the KBD deleted (right). Expression was driven in motor neurons by one copy of the D42-Gal4 driver. Expression rates for the two APLIP1 constructs were similar and many times greater than the endogenous protein (see Figure S1).
(A and B) Single video frames of live larvae crawling on agar blocks. The full-length construct caused severe tail flipping and slowed growth.
(C and D) Micrographs of larval segmental nerves costained for the vesicle protein Synaptotagmin (Syt) and for Flag-APLIP1 (Flag). These images were selected as representative from 40 different nerve locations in 10 larvae per genotype.
(E) Ligated segmental nerves of larvae expressing GFP-APLIP1 in motor neurons. Whole live larvae were constricted with synthetic filaments for 4 hr to generate focal compressions of segmental nerves that would block organelle transport. They were then dissected and fixed and the GFP was imaged. The top two panels show composite images of nerves from two different animals generated with a 40× objective lens. Note the accumulation of GFP-APLIP1 at the ligation site on both the cell body side (proximal) and the axon terminal side (distal). The bottom panels show single images (60× objective) of the accumulations in the middle panel. Scale bars equal 10 μm.
The presence of residual Flag-APLIP1ΔKBD in larval nerves indicates that some was transported into axons despite the fact that its binding to kinesin-1 was compromised. JIP-1 as well as APLIP1 is known to form multimers [1, 12]. Indeed, immunoprecipitation tests indicated that tagged APLIP1 and APLIP1ΔKBD can form stable multimers with one another (Figure S2). Thus, it is possible that in larval neurons, endogenous wild-type APLIP1 mediated linkage of some transgenic Flag-APLIP1ΔKBD to kinesin-1. Overall, our results suggest that binding between APLIP1 and KLC is an important factor in the presence of APLIP1 in axons, providing in vivo support for the hypothesis that APLIP1 is transported anterograde by kinesin-1.
To test the possibility that APLIP1 is associated with dynein-driven retrograde transport as well as with kinesin-1-driven anterograde transport, we developed transgenic flies carrying a UAS-GFP-Aplip1 transgene that expressed a stable fusion protein (Figure S2). When combined with the D42-Gal4 driver, some transformant lines showed paralysis and GFP-filled swellings, similar to the Flag-APLIP1 lines. Time-lapse imaging did not reveal obvious transport, suggesting that the GFP-APLIP1 was transported in a form too dispersed for imaging of discrete punctate signals. Turning to a classic axonal transport approach, we developed a method for nerve ligation [18, 19] in Drosophila larvae. A homozygous UAS-GFP-Aplip1 D42-Gal4 transformant line was used in which there were few axonal swellings and little visible axonal GFP fluorescence, presumably because of low expression. Intact live larvae were constricted with a fine synthetic fiber midway between head and tail to compress their segmental nerves. After 4 hr, they were partially dissected in fix, the ligation threads were cut, dissection was completed, and the nerves were imaged (Figure 3E). Distinct compressed regions were flanked by bright accumulations of GFP-APLIP1 on both the proximal and distal sides. This provides a strong indication that APLIP1 is carried not only by anterograde, but also by retrograde axonal transport.
Conclusions
By using an in vivo genetic approach to identify proteins that contribute to the mechanism of kinesin-1-driven anterograde axonal transport, we identified APLIP1, a Drosophila homolog of the JNK-interacting protein JIP-1. In vivo axonal transport analysis with intact nervous systems suggests roles for APLIP1 in anterograde and retrograde transport of nSyb-tagged vesicles and in retrograde transport of mitochondria. Similar neuronal phenotypes were seen with either Aplip1 inhibition or overexpression, suggesting that correct stoichiometry of APLIP1 and its interacting proteins is critical for normal organelle transport. The influence of APLIP1 on nSyb vesicle transport in both directions could be explained simply by its importance for kinesin-1 function. Khc is required for normal retrograde dynein activity as well as for anterograde kinesin-1 activity, probably because of a physical or regulatory relationship between the two motors [3, 4, 20, 21]. Alternatively, APLIP1 might make separate contributions to kinesin-1-driven anterograde and dynein-driven retrograde vesicle transport.
The selective influence of APLIP1 on retrograde, but not anterograde, transport of mitochondria, as well as Aplip1-Dhc64C genetic interactions, suggests that APLIP1 does have distinct, kinesin-independent functions in dynein-driven transport, at least for mitochondria. Considering how APLIP1 and other JIP-1-related proteins contribute to axonal transport mechanisms, binding studies suggest they may be structural components of kinesin-1-cargo linkage complexes (this report) [1, 2]. However, the APLIP1 influence on retrograde mitochondria, the well-known scaffolding role of APLIP1/JIP-1 in the JNK signaling pathway, and indications that JNK may influence motor linkage [17] must also be kept in mind. Mitochondrial transport and distribution in axons responds dramatically to extracellular signaling [22, 23] and may also respond to intracellular signaling stimulated by changes in mitochondrial membrane potential (P. Hollenbeck, personal communication) [24]. APLIP1 might be important in those or in other pathways that regulate dynein-cargo linkage and/or mechanochemistry. Future tests for a physical APLIP1-dynein association and for influences of JNK signaling on axonal transport may provide important insights into the microtubule-based transport mechanisms required to sustain neurons and other large asymmetric cells.
Supplementary Material
Acknowledgements
We thank Debra Rose, Jim Powers, Thom Kaufman, Joe Duffy, Susan Strome, and Beth Raff for advice throughout this project and Michelle Post and Olga Klyachko for technical assistance. This work was supported by NIH GM46295 (W.M.S.), an Established Investigatorship from the American Heart Association (W.M.S.), and predoctoral fellowships to D.H., R.V.B., and A.D.P. from the American Heart Association Midwest Affiliate.
Footnotes
Supplemental Data
Supplemental Data include two figures, four movies, and Supplemental Experimental Procedures and can be found with this article online at http://www.current-biology.com/cgi/content/full/15/23/2137/DC1/.
References
- 1.Taru H, Iijima K, Hase M, Kirino Y, Yagi Y, Suzuki T. Interaction of Alzheimer's beta-amyloid precursor family proteins with scaffold proteins of the JNK signaling cascade. J. Biol. Chem. 2002;277:20070–20078. doi: 10.1074/jbc.M108372200. [DOI] [PubMed] [Google Scholar]
- 2.Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 2001;152:959–970. doi: 10.1083/jcb.152.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pilling A. Analysis of the role of kinesin-1 and cytoplasmic dynein in axonal organelle transport in Drosophila melanogaster. Indiana University; Bloomington, Indiana: 2005. PhD thesis. [Google Scholar]
- 4.Martin M, Iyadurai SJ, Gassman A, Gindhart JG, Jr., Hays TS, Saxton WM. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol. Biol. Cell. 1999;10:3717–3728. doi: 10.1091/mbc.10.11.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin M, Ahern-Djamali SM, Hoffmann FM, Saxton WM. Abl tyrosine kinase and its substrate Ena/VASP have functional interactions with kinesin-1. Mol. Biol. Cell. 2005;16:4225–4230. doi: 10.1091/mbc.E05-02-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gindhart JG, Jr., Desai CJ, Beushausen S, Zinn K, Goldstein LSB. Kinesin light chains are essential for axonal transport in Drosophila. J. Cell Biol. 1998;141:443–454. doi: 10.1083/jcb.141.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saxton WM, Hicks J, Goldstein LSB, Raff EC. Kinesin heavy chain is essential for viability and neuromuscular functions in Drosophila, but mutants show no defects in mitosis. Cell. 1991;64:1093–1102. doi: 10.1016/0092-8674(91)90264-y. [DOI] [PubMed] [Google Scholar]
- 8.Hurd DD, Saxton WM. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics. 1996;144:1075–1085. doi: 10.1093/genetics/144.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estes PS, Ho GL, Narayanan R, Ramaswami M. Synaptic localization and restricted diffusion of a Drosophila neuronal synaptobrevin—green fluorescent protein chimera in vivo. J. Neurogenet. 2000;13:233–255. doi: 10.3109/01677060009084496. [DOI] [PubMed] [Google Scholar]
- 10.Yeh E, Gustafson K, Boulianne GL. Green fluorescent protein as a vital marker and reporter of gene expression in Drosophila. Proc. Natl. Acad. Sci. USA. 1995;92:7036–7040. doi: 10.1073/pnas.92.15.7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ray K, Perez SE, Yang Z, Xu J, Ritchings BW, Steller H, Goldstein LSB. Kinesin-II is required for axonal transport of choline acetyltransferase in Drosophila. J. Cell Biol. 1999;147:507–518. doi: 10.1083/jcb.147.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, Goldstein LSB. Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell. 2000;103:583–594. doi: 10.1016/s0092-8674(00)00162-8. [DOI] [PubMed] [Google Scholar]
- 14.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LSB. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 15.Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR, Koliatsos VE, Kins S, Lee VM, Wong PC, et al. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J. Neurosci. 2005;25:2386–2395. doi: 10.1523/JNEUROSCI.3089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsuda S, Matsuda Y, D'Adamio L. Amyloid beta protein precursor (AbetaPP), but not AbetaPP-like protein 2, is bridged to the kinesin light chain by the scaffold protein JNK-interacting protein 1. J. Biol. Chem. 2003;278:38601–38606. doi: 10.1074/jbc.M304379200. [DOI] [PubMed] [Google Scholar]
- 17.Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, Kitamura N. A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J. Biol. Chem. 2003;278:22946–22955. doi: 10.1074/jbc.M212160200. [DOI] [PubMed] [Google Scholar]
- 18.Hirokawa N, Sato-Yoshitake R, Kobayashi N, Pfister KK, Bloom GS, Brady ST. Kinesin associates with anterogradely transported membranous organelles in vivo. J. Cell Biol. 1991;114:295–302. doi: 10.1083/jcb.114.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirokawa N, Sato-Yoshitake R, Yoshida T, Kawashima T. Brain dynein (MAP1C) localizes on both anterogradely and retrogradely transported membranous organelles in vivo. J. Cell Biol. 1990;111:1027–1037. doi: 10.1083/jcb.111.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ligon LA, Tokito M, Finklestein JM, Grossman FE, Holzbaur EL. A direct interaction between cytoplasmic dynein and kinesin I may coordinate motor activity. J. Biol. Chem. 2004;279:19201–19208. doi: 10.1074/jbc.M313472200. [DOI] [PubMed] [Google Scholar]
- 21.Kural C, Kim H, Syed S, Goshima G, Gelfand VI, Selvin PR. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science. 2005;308:1469–1472. doi: 10.1126/science.1108408. [DOI] [PubMed] [Google Scholar]
- 22.Chada SR, Hollenbeck PJ. Mitochondrial movement and positioning in axons: the role of growth factor signaling. J. Exp. Biol. 2003;206:1985–1992. doi: 10.1242/jeb.00263. [DOI] [PubMed] [Google Scholar]
- 23.Chada SR, Hollenbeck PJ. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr. Biol. 2004;14:1272–1276. doi: 10.1016/j.cub.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 24.Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.