

Abstract
The frequency of resistance to β-lactams among nosocomial isolates has been increasing due to extended-spectrum β-lactamase (ESBL)-producing enteric bacilli. Although clinical outcome data are desirable, assessment of clinical efficacy has been limited due to the lack of a statistically meaningful number of well-documented cases. Since time above the MIC (T>MIC) is the pharmacokinetic-pharmacodynamic (PK-PD) measure that best correlates with in vivo activity of β-lactams, a stochastic model was used to predict the probability of PK-PD target attainment ranging from 30 (P30) to 70% (P70) T>MIC, for standard dosing regimens of both piperacillin-tazobactam and cefepime against Escherichia coli and Klebsiella pneumoniae ESBL phenotypes. The P70/30 T>MIC for cefepime at 2 g every 12 h against E. coli and K. pneumoniae was 0.99/1.0 and 0.96/1.0 and for a regimen of 1 g every 12 h was 0.96/1.0 and 0.93/0.99, respectively. For piperacillin-tazobactam at 3.375 g every 4 h against E. coli and K. pneumoniae, the P70/30 T>MIC was 0.77/0.96 and 0.48/0.77 and for a regimen of 3.375 g every 6 h was 0.28/0.91 and 0.16/0.69, respectively. These data suggest that the probability of achieving T>MIC target attainment rates is generally higher with cefepime than with piperacillin-tazobactam for present-day ESBL-producing strains when one uses contemporary dosing regimens.
Increasing antimicrobial resistance among extended-spectrum β-lactamase (ESBL)-producing Escherichia coli and Klebsiella pneumoniae is a growing concern (5, 32). Infection with ESBL-producing E. coli or K. pneumoniae has been associated with a significantly longer duration of hospital stay and greater hospital charges. Prior cumulative drug exposure (measured by the number of antimicrobial agents and total duration of treatment) has been demonstrated to be an independent predictor of ESBL-producing E. coli or K. pneumoniae infection (22). It is therefore important, especially when choosing empirical therapy, to identify agents with a relative high probability of in vivo efficacy against these pathogens.
Nonclinical pharmacodynamic models of infection (i.e., in vitro and animal) have been used to establish the conditions under which an anti-infective agent is effective (7, 10, 30). By manipulation of the pharmacokinetics of agents within these models, mean concentration-in-human-serum-time courses have been simulated for many agents. For penicillins and cephalosporins, experiments have shown that antibacterial effects best correlate with the duration of time that drug concentrations exceed the MIC of the microorganism (7, 8). For enteric gram-negative bacilli such as E. coli or K. pneumoniae, antibacterial effects were observed for cephalosporins when free-drug (f) concentrations in serum were above the MIC for as little as 35 to 40% of the dosing interval, and this effect appeared to plateau when concentrations were above the MIC for 60 to 70% of the dosing interval (9). For penicillin derivatives and enteric gram-negative bacilli, antibacterial effects were observed at somewhat lower time-above-the-MIC (T>MIC) targets than for cephalosporins, i.e., 30 to 35% of the dosing interval (W. A. Craig, S. Ebert, and Y. Watanabe, Abstr. 33th Intersci. Conf. Antimicrob. Agents Chemother., abstr. 89, 1993).
The impact of ESBL-producing E. coli and K. pneumoniae on the clinical efficacy of agents commonly used in the critical-care setting may be difficult to quantify due to the lack of a statistically meaningful number of well-documented cases in the literature. Monte Carlo simulation is a tool that may be used to estimate the probability of attaining optimal pharmacokinetic-pharmacodynamic (PK-PD) targets. This approach involves incorporating the variability in drug exposure observed in a population of patients and the variability in MICs encountered in clinical practice into a stochastic model (1, 11, 13, 14).
The Antimicrobial Resistance Rate Epidemiology Study Team was established in 2001 and represents an integration of microbiological surveillance data and statistical and analytic techniques. The goal of this ongoing program is to better interpret the clinical significance of antimicrobial resistance patterns. The objectives of the analyses described herein were twofold: first, to compare the resistance rates and patterns of ESBL-producing E. coli and K. pneumoniae phenotypes obtained during 2000 for piperacillin-tazobactam and cefepime; and second, to estimate the probability of achieving T>MIC of 30 to 70% of the dosing interval for two regimens of piperacillin-tazobactam and two regimens of cefepime against these two organisms.
(The material contained herein was presented at the 41st Annual Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, Ill., 2002).
MATERIALS AND METHODS
Microbiological data.
The SENTRY Antimicrobial Surveillance Program was established in 1997 to monitor the occurrence of prominent pathogens and antimicrobial resistance patterns of nosocomial and community-acquired infections via a broad network of sentinel hospitals distributed by geographic location and bed capacity. Susceptibility data for E. coli and K. pneumoniae bloodstream isolates recovered during 2000 in the North America region (United States and Canada) were included in this analysis. All isolates were saved on transport swabs and sent to the University of Iowa College of Medicine (Iowa City) for storage and further identification and/or susceptibility testing. Upon receipt, isolates were subcultured on blood agar to ensure viability and purity. Species identifications were confirmed with the Vitek System (bioMérieux Vitek) or API (bioMérieux Vitek) products and standard reference methods (15). Isolates were frozen at −70°C until they were processed.
Antimicrobial susceptibility testing of isolates was performed by reference broth microdilution methods as described by the National Committee for Clinical Laboratory Standards (NCCLS) (24, 25). Microdilution trays were purchased from MicroScan (West Sacramento, Calif.), TREK (West Lake, Ohio), and PML Microbiologicals (Wilsonville, Oreg.). Antimicrobial agents were obtained from their respective manufacturers. Quality control was performed by testing E. coli ATCC 25922. Interpretive criteria were those published by the NCCLS. K. pneumoniae and E. coli isolates expressing an ESBL phenotype, as defined by a ceftazidime, ceftriaxone, or aztreonam MIC of ≥2.0 mg/liter, were further characterized with ESBL Etest (AB Biodisk, Solna, Sweden) strips containing antimicrobial gradients ranging from 0.016 to 256 mg/liter and were paired with strips containing the same cephalosporin gradient in the presence of 2 mg of clavulanic acid/liter or with commercial ESBL Etest strips that contain a stable gradient of ceftazidime (1 to 32 mg/liter) on one half and ceftazidime plus clavulanic acid (2 mg/liter) on the other half. The Etest inoculum was adjusted to the turbidity of a 0.5 McFarland standard from a 24-h subculture and was swabbed onto the surface of a 150-mm plate of Mueller-Hinton agar. An eightfold-or-greater reduction in MIC with clavulanate acid in comparison with the MIC with the substrate oxyimino cephalosporin alone was considered evidence of a positive ESBL test (6).
Pharmacokinetic data.
Serum pharmacokinetic parameter estimates following intravenous (i.v.) dosing of piperacillin-tazobactam and cefepime were obtained from the medical literature (3, 18). In these studies, 3.375 g of piperacillin-tazobactam (3.0 and 0.375 g, respectively) or 1 g of cefepime was administered over a 30-min period in patients with estimated creatinine clearances between 60 and 91 ml/min. For cefepime, the mean (plus or minus standard deviation [SD]) elimination half-life was 3.33 ± 0.74 h and the peak concentration in serum was 70.5 ± 20.8 mg/liter. Mean (plus or minus SD) total body clearance was 75.5 ± 12.9 ml/min. Renal clearance accounted for 80.3% ± 10.6% of drug removal. The mean volume of distribution at steady state was 19.6 ±2.99 liters. For piperacillin, the elimination half-life was 1.1 ± 0.187 h and the peak concentration in serum was 228 ± 25 mg/liter. Mean (plus or minus SD) clearance in plasma was 159 ± 19 ml/min. Renal clearance accounted for 48.4% ± 5.8% of drug removal. The mean volume of distribution at steady state was 13.0 ± 1.4 liters (25). For tazobactam, the mean (plus or minus SD) elimination half-life was 1.2 ± 0.002 h. Mean (plus or minus SD) clearance of tazobactam in plasma was 141 ± 21 ml/min, and the mean volume of distribution at steady state was 14.7 ± 1.9 liters.
The fraction of unbound drug for piperacillin-tazobactam and cefepime assumed in these analyses was 70 and 84%, respectively (2, 20).
PK-PD target attainment analyses.
PK-PD target attainment analyses were carried out by using Monte Carlo simulation. Dosing regimens modeled included piperacillin-tazobactam (3.375 g i.v. administered every 4 and 6 h) and cefepime (1 and 2 g i.v. administered every 12 h). Five thousand patient simulations were carried out in order to estimate the probability of attaining T>MIC targets of 30, 40, 50, 60, and 70% of the dosing interval for each drug regimen-organism combination by using Crystal Ball 2000.1 by Decisioneering, Inc. (Denver, Colo.). The following PK-PD structural model was used in the simulations:
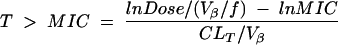 |
where Vβ is the volume that, when multiplied by β for a two-compartment model provides the true total clearance, CLT is total clearance, and f is the fraction of unbound drug.
PK-PD target attainment analyses evaluating β-lactam and β-lactamase inhibitor combination agents against β-lactamase-producing strains of bacteria require one to account for the exposure of both drug components. To this end we utilized a two-step algorithm, an approach that was first suggested by Dudley, in our PK-PD target attainment analyses of piperacillin-tazobactam (12, 19). The logic was as follows: first, the β-lactamase inhibitor (tazobactam) exposure must be sufficiently large enough to render the bacteria functionally β-lactamase negative; and second, the β-lactam (piperacillin) concentration must exceed the drug MIC for the pathogen for at least the duration of the desired PK-PD target. Bacterial killing is predicted only in those instances when both of these endpoints have been met.
Identification of the magnitude of exposure that would satisfy the first-step of the algorithm was based upon the concentration of tazobactam used in the MIC determination method described by the NCCLS. The broth microdilution MIC determination method described by the NCCLS utilizes a fixed tazobactam concentration of 4 mg/liter. Since the integral of the drug concentration-time profile is described by the area under the concentration-time curve (AUC), a fixed in vitro tazobactam concentration of 4 mg/liter over a 24-h incubation period can be thought of as a 24-h tazobactam AUC of 96 mg · h/liter. Thus, the first exposure target in the two-step algorithm was met in those instances where the 24-h tazobactam AUC was 96 mg · h/liter or greater. In those instances where this endpoint was met, the probability of meeting the second target simplified to a single probability, i.e., achieving an f piperacillin T>MIC of 30 to 70% of the dosing interval.
RESULTS
Microbiological data.
During 2000, the SENTRY Program evaluated 1,909 E. coli and 743 K. pneumoniae bloodstream isolates. Of these, 65 (3.4%) E. coli and 40 (5.4%) K. pneumoniae isolates manifested ESBL phenotypes (i.e., isolates for which ceftazidime and/or ceftriaxone and/or aztreonam manifested MICs that were ≥ 2 mg/liter). The in vitro activity of piperacillin-tazobactam and cefepime against these strains is summarized in Table 1. Piperacillin-tazobactam was significantly more active against E. coli isolates (MIC at which 50% of the isolates tested are inhibited [MIC50] of 4 mg/liter) than against K. pneumoniae strains (MIC50 = 16 mg/liter). Also at the NCCLS breakpoint for susceptibility (≤16 mg/ml), 89.1 and 62.5% of E. coli and K. pneumoniae strains were inhibited, respectively. When MIC50s were used as a basis for comparison, the ESBL phenotypes were 16-fold more susceptible to cefepime than to piperacillin-tazobactam. The potency of cefepime was evident by the high rates of inhibition of ESBL-producing K. pneumoniae and E. coli strains (92.5 and 98.5%, respectively), when the NCCLS breakpoint for susceptibility of Enterobacteriaceae was used.
TABLE 1.
In vitro activity of piperacillin-tazobactam and cefepime tested against 105 ESBL phenotype (NCCLS criteria) strains of E. coli and K. pneumoniae (SENTRY Antimicrobial Surveillance Program, 2000)
| Agent | Organism (no. of strains) | Cumulative % inhibited at MIC (mg/liter) of:
|
% Susceptiblea | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ≤0.12 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | 32 | 64 | |||
| Piperacillin-tazobactam | E. coli (65) | 3.1b | 6.3 | 26.6 | 62.5 | 81.3 | 89.1 | 89.1 | 92.2 | 89.1 | ||
| K. pneumoniae (40) | 0.0 | 2.5 | 12.5 | 35.0 | 47.5 | 62.5 | 72.5 | 80.0 | 62.5 | |||
| Cefepime | E. coli (65) | 49.2 | 63.1 | 78.6 | 84.6 | 89.2 | 93.8 | 98.5 | 100 | 98.5 | ||
| K. pneumoniae (40) | 7.5 | 12.5 | 37.5 | 62.5 | 82.5 | 92.5 | 92.5 | 95.0 | 92.5 | |||
PK-PD target attainment analyses.
Probabilities for attaining the selected range of the PK-PD target (T>MIC) for piperacillin-tazobactam and cefepime, stratified by microorganism and dosing regimen, are presented in Table 2. For the regimen of 3.375 g of piperacillin-tazobactam i.v. every 4 or 6 h, the P for attaining PK-PD target measures (e.g., 30 to 40% T>MIC) for ESBL-producing E. coli phenotypes exceeded 0.86.
TABLE 2.
Probability of attaining T>MIC target measures following standard dosing regimens of piperacillin-tazobactam and cefepime against E. coli and K. pneumoniae ESBL-producing phenotypes
| Regimen |
P of achieving PK-PD target measuresa,b
|
||||
|---|---|---|---|---|---|
| 30% | 40% | 50% | 60% | 70% | |
| E. coli | |||||
| Piperacillin-tazobactam, 3.375 g every 4 h | 0.96 | 0.92 | 0.90 | 0.86 | 0.77 |
| Piperacillin-tazobactam, 3.375 g every 6 h | 0.91 | 0.86 | 0.73 | 0.50 | 0.28 |
| Cefepime, 2 g every 12 h | 1.0 | 1.0 | 1.0 | 1.0 | 0.99 |
| Cefepime, 1 g every 12 h | 1.0 | 0.99 | 0.99 | 0.98 | 0.96 |
| K. pneumoniae | |||||
| Piperacillin-tazobactam, 3.375 g every 4 h | 0.77 | 0.72 | 0.65 | 0.57 | 0.48 |
| Piperacillin-tazobactam, 3.375 g every 6 h | 0.69 | 0.57 | 0.43 | 0.29 | 0.16 |
| Cefepime, 2 g every 12 h | 1.0 | 1.0 | 1.0 | 0.98 | 0.96 |
| Cefepime, 1 g every 12 h | 0.99 | 0.96 | 0.95 | 0.94 | 0.93 |
Percentage of the dosing interval when drug concentration in serum remained above the MIC is given.
Boldfaced values represent the PK-PD target ranges for each drug.
Similarly, against ESBL-producing K. pneumoniae phenotypes, the P of attaining PK-PD target measures for the regimen of 3.375 g of piperacillin-tazobactam i.v. every 4 h ranged from 0.72 to 0.77. As would be expected, P values were comparatively less for the regimen of 3.375 g i.v. every 6 h. For example, the P of achieving T>MIC of 40% or more of the dosing interval was 0.57.
In general, the probability of meeting PK-PD targets (i.e., 50 to 60% T>MIC) with cefepime was equal to or higher than that with piperacillin-tazobactam, regardless of the dosing regimen modeled or microorganism considered. The magnitude of difference between regimens in these probabilities appeared to be more dramatic between the dosing regimens of cefepime and piperacillin-tazobactam against K. pneumoniae ESBL-producing strains.
DISCUSSION
ESBLs hydrolyze oxyimino cephalosporins (e.g., ceftazidime and ceftriaxone) and monobactams (e.g., aztreonam) and have generally evolved from TEM-1, TEM-2, SHV, or related β-lactamases (5, 16, 27, 31). Microorganisms that elaborate ESBLs include K. pneumoniae, E. coli, Proteus mirabilis, and Salmonella species (5, 27). The emergence of ESBL-producing organisms coincided with the popularization of cephalosporins that occurred during the 1980s. After initial identification in Europe (21) and later in the United States (17), a number of epidemic outbreaks of infection involving these organisms were reported (4, 23, 29). The prevalence of these organisms appears to be increasing and has high geographic variability. For instance, a recent worldwide surveillance report found that, in Latin America, 45.4% of K. pneumoniae isolates expressed ESBL phenotypes compared with 24.6% in the Western Pacific region, 22.6% in Europe, 7.6% in the United States, and 4.9% in Canada. Similarly, within the United States there was considerable regional or institutional variability with the highest prevalence in the Northeastern and South-Central states and the lowest in the Western states (32).
Controversy has existed as to whether or not cephalosporins and β-lactam and β-lactamase inhibitor combination agents may be used clinically against ESBL-producing strains. Largely this controversy arises from the observation that many of these plasmid-mediated β-lactamases are capable of hydrolyzing late-generation cephalosporins, monobactams, and many penicillin derivatives and that these agents are subject to inoculum effects.
To this end, Andes and Craig studied the impact of ESBL production in E. coli and K. pneumoniae on the activity of cefepime in the neutropenic murine-thigh model of infection (D. Andes and W. A. Craig, Abstr. 41st Intersci. Conf. Antimicrob. Agents Chemother., abstr. A-1099, 2001). Mice were infected with high inocula, 107 to 108 CFU per thigh. Four E. coli (one non-ESBL and three ESBL) and three K. pneumoniae (one non-ESBL and two ESBL) strains were used. ESBL production in E. coli and K. pneumoniae strains (E. coli, 14714, TEM-10; 102-94090, unknown; and SC15243, SHV-2; and K. pneumoniae, MCV2, SHV-4; and UA-834, SHV-2) that were studied had no impact upon the T>MIC necessary for in vivo cefepime efficacy. This is an important observation, as it suggests that the presence of ESBL production in and of itself is not predictive of efficacy. Rather, the information needed to predict efficacy is captured by the MIC in relation to the magnitude of drug exposure in vivo.
One aim of these analyses was to examine the probability of achieving PK-PD targets associated with favorable in vivo outcomes of these commonly used agents against E. coli and K. pneumoniae ESBL-producing phenotypes. As the results clearly demonstrate, a higher proportion of patients are likely to achieve the given PK-PD targets when piperacillin-tazobactam is given every 4 h rather than every 6 h. Moreover, the probability of piperacillin-tazobactam achieving PK-PD targets was markedly greater for E. coli isolates than for K. pneumoniae ESBL phenotypes. Against K. pneumoniae ESBL phenotypes, the P of PK-PD target attainment was as low as 0.16. These results suggest that piperacillin-tazobactam may be a less effective empirical choice against contemporary K. pneumoniae than against E. coli ESBL-producing isolates.
In general, PK-PD targets were likelier to be achieved by cefepime regardless of the dosing regimen modeled, the PK-PD target, or the microorganism considered. Cefepime achieved desired PK-PD targets at a P of >0.90, suggesting that it may be a more appropriate empirical choice than piperacillin-tazobactam would be against many ESBL-producing isolates.
Many authors have recommended strict adherence to infection control procedures and the judicious use of antimicrobial agents as a remedy for ESBL outbreaks of infection or their prevention. The meaning of the term “judicious use,” however, lies in the eyes of the beholder. In the case of ESBL-producing organisms, judicious use may mean the replacement of all cephalosporin and β-lactam and β-lactamase inhibitor combination agents with more β-lactamase-stable carbapenems. While, at first this may seem a reasonable strategy, such efforts may only lead to modest success. In one report, such a strategy led to the further selection of other resistance problems more deleterious than the first (28). Moreover, outbreaks of infections due to ESBL-producing organisms have been successfully controlled without restricting an entire class(es) of agents (26). It is important that more than one-half of infections involving ESBL-producing organisms are isolated to the urinary tract, and since penicillins and cephalosporins concentrate in the urine to a high degree, the use of these agents for ESBL-producing organisms has resulted in positive clinical outcomes (22).
In conclusion, the increasing prevalence of ESBL-producing strains is of clinical concern. However, ESBL production in and of itself is not predictive of efficacy. Our analyses demonstrate that (i) most ESBL-producing E. coli strains isolated in North America have MICs for cefepime and piperacillin-tazobactam that are in the susceptible range, (ii) more ESBL-producing strains of K. pneumoniae are susceptible to cefepime than to piperacillin-tazobactam, and (iii) T>MIC targets that are associated with in vivo efficacy of ESBL-producing strains obtained during this surveillance study may be achieved in patients treated with cefepime and to a lesser extent in those treated with piperacillin-tazobactam.
REFERENCES
- 1.Ambrose, P. G., and D. M. Grasela. 2000. The use of Monte Carlo simulation to examine pharmacodynamic variance of drugs: fluoroquinolone pharmacodynamics against Streptococcus pneumoniae. Diagn. Microbiol. Infect. Dis. 38:151-157. [DOI] [PubMed] [Google Scholar]
- 2.Barbhaiya, R. H., S. T. Forgue, W. C. Shyu, et al. 1987. High-pressure liquid chromatographic analysis of BMY-28142 in plasma and urine. Antimicrob. Agents Chemother. 31:55-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbaiya, R. H., C. A. Knupp, S. T. Forgue, G. R. Matzke, D. R. P. Guay, and K. A. Pittman. 1990. Pharmacokinetics of cefepime in subjects with renal insufficiency. Clin. Pharmacol. Ther. 48:268-276. [DOI] [PubMed] [Google Scholar]
- 4.Brun-Buisson, C., P. Legrand, A. Phillippon, F. Muntravers, M. Ansquer, and J. Duval. 1987. Transferable enzymatic resistance to third-generation cephalosporins during nosocomial outbreak of multi-resistant Klebsiella pneumoniae. Lancet ii:302-306. [DOI] [PubMed]
- 5.Bush, K. 2001. New beta-lactamases in gram-negative bacteria: diversity and impact on the selection of antimicrobial therapy. Clin. Infect. Dis. 32:1085-1089. [DOI] [PubMed] [Google Scholar]
- 6.Cormican, M. G., S. A. Marshall, and R. N. Jones. 1996. Detection of extended-spectrum β-lactamase (ESBL)-producing strains by the Etest ESBL screen. J. Clin. Microbiol. 34:1880-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craig, W. A. 2001. Pharmacodynamics of antimicrobials: general concepts and applications, p. 1-22. In C. H. Nightingale, T. Murakawa, and P. G. Ambrose (ed.), Antimicrobial pharmacodynamics in theory and clinical practice. Marcel Dekker, New York, N.Y.
- 8.Craig, W. A. 1996. Antimicrobial resistance issues of the future. Diagn. Microbiol. Infect. Dis. 25:213-217. [DOI] [PubMed] [Google Scholar]
- 9.Craig, W. A. 1995. Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad-spectrum cephalosporins. Diagn. Microbiol. Infect. Dis. 22:89-96. [DOI] [PubMed] [Google Scholar]
- 10.Dudley, M. N., and D. Griffith. 2001. Animal models of infection for the study of antibiotic pharmacodynamics, p. 67-97. In C. H. Nightingale, T. Murakawa, and P. G. Ambrose (ed.), Antimicrobial pharmacodynamics in theory and clinical practice. Marcel Dekker, New York, N.Y.
- 11.Dudley, M. N., and P. G. Ambrose. 2000. Pharmacodynamics in the study of drug resistance and establishing in vitro susceptibility breakpoints: ready for prime time. Curr. Opin. Microbiol. 3:515-521. [DOI] [PubMed] [Google Scholar]
- 12.Dudley, M. N. 1995. Combination beta-lactamase and beta-lactamase-inhibitor therapy: pharmacokinetic and pharmacodynamic considerations. Am. J. Health Syst. Pharm. 52(Suppl. 2):S23-S27. [DOI] [PubMed] [Google Scholar]
- 13.Drusano, G. L., D. Z. D'Argenio, S. L. Preston, C. Barone, W. Symonds, S. LaFon, M. Rogers, W. Prince, A. Bye, and J. A. Bilello. 2000. Use of drug effect interaction modeling with Monte Carlo simulation to examine the impact of dosing interval on the projected antiviral activity of the combination of abacavir and amprenavir. Antimicrob. Agents Chemother. 44:1655-1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drusano, G. L., S. L. Preston, C. Hardalo, R. Hare, C. Banfield, D. Andes, O. Vega, and W. A. Craig. 2001. Use of preclinical data for selection of a phase II/III dose for evernimicin and identification of a preclinical MIC breakpoint. Antimicrob. Agents Chemother. 45:13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isenberg, H. D. (ed.) 1992. Clinical microbiology procedures handbook. American Society for Microbiology, Washington, D.C.
- 16.Jacoby, G. A., and A. A. Medeiros. 1991. More extended-spectrum β-lactamases. Antimicrob. Agents Chemother. 35:1697-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacoby, G. A., A. A. Medeiros, T. F. O'Brien, M. E. Pinto, and J. Jiang. 1989. Broad-spectrum transmissible beta-lactamases. N. Engl. J. Med. 33:1451-1460. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, C. A., C. E. Halstenson, J. S. Kelloway, B. E. Shapiro, S. W. Zimmerman, A. Tonelli, R. Faulkner, A. Dutta, J. Hayes, D. S. Greene, and O. Kuye. 1992. Single-dose pharmacokinetics of piperacillin and tazobactam in patients with renal disease. Clin. Pharmacol. Ther. 51:32-41. [DOI] [PubMed] [Google Scholar]
- 19.Jones, R. N., and M. N. Dudley. 1997. Microbiologic and pharmacodynamic principles applied to the antimicrobial susceptibility testing of ampicillin/sulbactam. Diagn. Microbiol. Infect. Dis. 28:1-14. [DOI] [PubMed] [Google Scholar]
- 20.Kessler, R. E., M. Bies, R. E. Buck, D. R. Chisholm, T. A. Pursiano, Y. H. Tsai, M. Misiek, K. E. Price, and F. Leitner. 1985. Comparison of a new cephalosporin, BMY 28142, with other broad-spectrum β-lactam antibiotics. Antimicrob. Agents Chemother. 27:207-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knothe, H., P. Shah, V. Kremery, M. Antal, and S. Mitshashi. 1983. Transferable resistance to cefotaxime, cefoxitin, cefamandole and cefuroxime in clinical isolates of Klebsiella pneumoniae and Serratia marcescens. Infection 11:315-317. [DOI] [PubMed] [Google Scholar]
- 22.Lautenbach, E., J. B. Patel, W. B. Bilker, P. H. Edelstein, and N. O. Fishman. 2001. Extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae: risk factors for infection and impact of resistance on outcomes. Clin. Infect. Dis. 32:1162-1171. [DOI] [PubMed] [Google Scholar]
- 23.Meyer, K. S., C. Urban, J. A. Eagan, B. J. Berger, and J. J. Rahal. 1993. Nosocomial outbreak of Klebsiella infection resistant to late-generation cephalosporins. Ann. Intern. Med. 119:353-358. [DOI] [PubMed] [Google Scholar]
- 24.National Committee for Clinical Laboratory Standards. 2000. Performance standards for antimicrobial susceptibility testing. Document M100-S10. Wayne, Pa.
- 25.National Committee for Clinical Laboratory Standards. 2000. Performance standards for antimicrobial susceptibility testing. Document M7-A5. Wayne, Pa.
- 26.Paterson, D. L., N. Singh, J. D. Rihs, C. Squier, B. L. Rihs, and R. R. Muder. 2001. Control of an outbreak of infection due to extended-spectrum β-lactamase-producing Escherichia coli in a liver transplant unit. Clin. Infect. Dis. 33:126-128. [DOI] [PubMed] [Google Scholar]
- 27.Philippon, A., G. Arlet, and P. H. Lagrange. 1994. Origin and impact of plasmid-mediated extended-spectrum beta-lactamases. Eur. J. Clin. Microbiol. Infect. Dis. 13:S17-S29. [DOI] [PubMed] [Google Scholar]
- 28.Rahal, J. J., C. Urban, D. Horn, et al. 1998. Class restriction of cephalosporins to control total cephalosporin resistance in nosocomial Klebsiella. JAMA 280:1233-1237. [DOI] [PubMed] [Google Scholar]
- 29.Rice, L. B., S. H. Willey, G. A. Papanicolaou, A. A. Medeiros, G. M. Eliopoulos, R. C. Moellering, Jr., and G. A. Jacoby. 1990. Outbreak of ceftazidime resistance caused by extended-spectrum β-lactamases at a Massachusetts chronic-care facility. Antimicrob. Agents Chemother. 34:2193-2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rybak, M. J., E. Hershberger, and G. P. Allen. 2001. In vitro models of infection for the study of antibiotic pharmacodynamics, p. 41-65. In C. H. Nightingale, T. Murakawa, and P. G. Ambrose (ed.), Antimicrobial pharmacodynamics in theory and clinical practice. Marcel Dekker, New York, N.Y.
- 31.Sirot, D. 1995. Extended-spectrum plasma-mediated beta-lactamases. J. Antimicrob. Chemother. 36(Suppl. A):19-34. [DOI] [PubMed] [Google Scholar]
- 32.Winokur, P. L., R. Canton, J. M. Casellas, and N. Legakis. 2001. Variations in the prevalence of strains expressing an extended-spectrum beta-lactamase phenotype and characterization of isolates from Europe, the Americas, and the Western Pacific region. Clin. Infect. Dis. 32(Suppl. 2):S94-S103. [DOI] [PubMed] [Google Scholar]