

Abstract
The existence of an in-frame deletion mutant correlates with the sensitivity of lung cancers to EGFR (epidermal growth factor receptor)-targeted tyrosine kinase inhibitors. We reported previously that the in-frame 15-bp deletional mutation (delE746–A750 type deletion) was constitutively active in cells. Kinetic parameters are important for characterizing an enzyme; however, it remains unclear whether the kinetic parameters of deletion mutant EGFR are similar to those of wild-type EGFR. We analysed autophosphorylation in response to ATP and inhibition of gefitinib for deletion mutant EGFR and wild-type EGFR. Kinetic studies, examining autophosphorylation, were carried out using EGFR fractions extracted from 293-pΔ15 and 293-pEGFR cells transfected with deletion mutant EGFR and wild-type EGFR respectively. We demonstrated the difference in activities between unstimulated wild-type (Km for ATP=4.0±0.3 μM) and mutant EGFR (Km for ATP=2.5±0.2 μM). There was no difference in Km values between EGF-stimulated wild-type EGFR (Km for ATP=1.9±0.1 μM) and deletion mutant EGFR (Km for ATP=2.2±0.2 μM). These results suggest that mutant EGFR is active without ligand stimulation. The Ki value for gefitinib of the deletion mutant EGFR was much lower than that of wild-type EGFR. These results suggest that the deletion mutant EGFR has a higher affinity for gefitinib than wild-type EGFR.
Keywords: autophosphorylation, epidermal growth factor receptor (EGFR), gefitinib, kinase inhibition, tyrosine kinase
Abbreviations: EGF, epidermal growth factor; EGFR, EGF receptor; HEK-293, human embryonic kidney; 293-pEGFR, HEK-293 cells transfected with wild-type EGFR; 293-pΔ15, HEK-293 cells transfected with deletion mutant EGFR; TBS-T, Tris-buffered saline with Tween 20; TGF-α, transforming growth factor-α
INTRODUCTION
EGFR [EGF (epidermal growth factor) receptor] is among the most important targets for lung cancer therapy, and many EGFR-targeted inhibitors have been developed [1]. These EGFR-targeted compounds inhibit the tyrosine kinase activity of EGFR by competing at the ATP-binding site [2]. Many EGFR-targeted tyrosine kinase inhibitors such as gefitinib and erlotinib have been assessed clinically [3,4]. Recently, an EGFR mutation was found in patients who responded to gefitinib, and mutant EGFR has been reported to be a determinant of the response to EGFR tyrosine kinase inhibitors [5,6]. To date, over 30 EGFR mutations including delE746–A750, L858R and delL747–P753insS, have been reported in lung cancer. These EGFR mutations, except for T790M, are considered to be of the ‘gain-of-function’ type. Differences exist among them; for example, constitutively active in delE746–A750 compared with hyperresponsive to ligand stimulation in L858R and delL747–P753insS, although these mutant EGFRs increase sensitivity to EGFR-targeted tyrosine kinase inhibitors [7–9]. In general, the observation of hyperresponsiveness to ligand stimulation, as in the case of L858R, raises the possibility of high affinity for ATP. We reported previously that deletion mutant EGFR was constitutively phosphorylated under unstimulated conditions, whereas wild-type EGFR was not phosphorylated until ligand stimulation [7]. The differences in cellular phenotype and sensitivity to gefitinib between deletion mutant EGFR and wild-type EGFR raise the possibility that the enzymatic properties of the deletion mutant EGFR may differ from those of wild-type EGFR. However, it remains unclear whether the kinetic parameters of deletion mutant EGFR are different from those of wild-type EGFR. In the present study, we focused on the autophosphorylation of deletion mutant EGFR, and investigated the inhibition constant of gefitinib. Technically, we used deletion mutant EGFR and wild-type EGFR extracted from ectopically expressed HEK-293 (human embryonic kidney) cells. The autophosphorylation assay reflects the native behaviour of EGFR in maintaining cellular functions.
MATERIALS AND METHODS
Reagents
Gefitinib (Iressa®, ZD1839) was provided by AstraZeneca.
Cell culture
The HEK-293 cell line was obtained from the A.T.C.C. (Manassas, VA, U.S.A.) and was cultured in RPMI 1640 medium (Sigma) supplemented with 10% heat-inactivated foetal bovine serum (Life Technologies).
Plasmid construction and transfection
Construction of the expression plasmid vector of wild-type EGFR and the 15-bp deletion mutant EGFR (delE746–A750 type deletion), which has the same deletion site as that observed in detail in PC-9 cells, has been described elsewhere [7,10,11]. The plasmids were transfected into HEK-293 cells and the transfectants were selected using Zeosin (Sigma). The stable transfectants (pooled cultures) of the wild-type EGFR and its deletion mutant were designated 293-pEGFR and 293-pΔ15 cells respectively.
Immunoblotting
The 293-pΔ15 and 293-pEGFR cells were treated with or without gefitinib for 3 h, stimulated with EGF (100 ng/ml) under serum-starvation conditions and then lysed for immunoblot analysis. Immunoblot analysis was performed as described previously [12]. Equivalent amounts of protein were separated by SDS/PAGE (2–15% gradient) and transferred to a PVDF membrane (Millipore). The membrane was probed with a mouse monoclonal antibody against EGFR (Transduction Laboratories), a phospho-EGFR antibody (specific for Tyr1068) (Cell Signaling Technology) as the first antibody, followed by a horseradish-peroxidase-conjugated secondary antibody. The bands were visualized with ECL® (enhanced chemiluminescence) (Amersham Biosciences).
Determination of ligand secretion by ELISA
The 293-pΔ15 and 293-pEGFR cells were cultured in 12-well plates under serum-starvation conditions. The cell culture supernatant was collected for each cell line and stored at −80 °C for further analysis. Amounts of EGF and TGF-α (transforming growth factor α) in the culture medium from each cell line were determined with a DuoSet ELISA development kit (R&D Systems). The assay was performed in triplicate according to the manufacturer's instructions.
Preparation of cell lysates for EGFR autophosphorylation
Cultivated cells, after reaching 70–80% confluence, were starved in serum-free medium for 24 h, with or without EGF (100 ng/ml) stimulation. The cells were washed twice with ice-cold PBS containing 0.33 mM MgCl2 and 0.9 mM CaCl2 [PBS(+)], then lysed with lysis buffer [50 mM Tris/HCl, pH 7.4, 50 mM NaCl, 0.25% Triton X-100, 5 mM EDTA, protease inhibitor (Roche Diagnostics) and phosphatase inhibitor (Sigma)]. For the preparation of gefitinib-treated cell lysates, cultivated cells were starved in serum-free medium for 24 h, and were then pre-incubated with 2 μM gefitinib for 3 h. Either with or without EGF stimulation (100 ng/ml), the cells were washed twice with ice-cold PBS(+) and lysed with lysis buffer. The cell lysate was centrifuged at 20000 g for 10 min, and the protein concentration of the supernatant was measured with a BCA (bicinchoninic acid) protein assay (Pierce).
Autophosphorylation assay
The amount of EGFR in 293-pΔ15 and 293-pEGFR cells was determined by quantitative immunoassay (R&D Systems) according to the manufacturer's instructions. The autophosphorylation assay was carried out with a quantitative immunoassay system. Wells in a 96-well immunomodule (Nalge Nunc International) were incubated with 0.8 μg/ml goat anti-(human EGFR) antibody in PBS (provided with the EGFR quantitative immunoassay system) and incubated at 4 °C overnight. The plates were washed three times with TBS-T (Tris-buffered saline with Tween 20; 20 mM Tris/HCl, pH 7.4, 150 mM NaCl and 0.05% Tween 20) and were then filled with blocking buffer (PBS containing 1% BSA and 5% sucrose) and incubated for 2 h at room temperature (25 °C). The wells were washed three times with TBS-T and incubated with cell lysates of 293-pEGFR or 293-pΔ15 including equal amounts of EGFR (130 ng of EGFR/well) diluted with lysis buffer. After a 2 h incubation at room temperature, the 96-well plate was washed with TBS-T. Autophosphorylation of EGFR was initiated by addition of ATP (0–32 μM in 50 mM Tris/HCl, pH 7.5, 20 mM MgCl2 and phosphatase inhibitor) followed by incubation for 5 min. In some experiments, various concentrations of gefitinib were added to the wells before the addition of ATP. Following the autophosphorylation reaction, the wells were washed with TBS-T. Next, horseradish-peroxidase-conjugated anti-phosphotyrosine antibody, PY-99-HRP (0.4 μg/ml in PBS containing 1% BSA and 0.1% Tween 20) (Santa Cruz Biotechnology) was added to the wells for 2 h at room temperature. The wells were washed three times with TBS-T. Bound phosphotyrosine antibody was detected colorimetrically after adding 100 μl of substrate (tetramethylbenzidine and H2O2) to each well. After a 10 min incubation, the colour reaction was quenched by the addition of 100 μl of 1M H2SO4. The absorbance readings for each well were determined at 450 nm with Delta-soft on an Apple Macintosh computer interfaced to a Bio-Tek Microplate Reader EL-340 (BioMetallics).
Data analysis
For kinetic analysis, an Eadie–Hofstee plot was applied for the calculation of Km (Michaelis constant) and Vmax (maximum velocity). The data obtained were plotted as velocity against velocity/substrate concentration (V/ATP). The slope of the line is equal to −Km and the x-intercept is Vmax. The Ki value was calculated as follows:
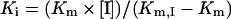 |
(1) |
in which Km is the Michaelis constant for ATP, Km,I is the Michaelis constant for ATP in the presence of gefitinib and [I] is the concentration of gefitinib. The statistical analysis was performed using KaleidaGraph (Synergy Software).
RESULTS
Autophosphorylation of deletion mutant EGFR and wild-type EGFR
We performed the autophosphorylation assay and immunoblot analysis using lysates extracted from 293-pΔ15 and 293-pEGFR cells under unstimulated and EGF-stimulated conditions (Figures 1A and 1B). Under unstimulated conditions, deletion mutant EGFR was highly phosphorylated in the absence of ATP. Addition of ATP did not affect the autophosphorylation of deletion mutant EGFR. On the other hand, autophosphorylation of wild-type EGFR was barely detectable without ATP, and proceeded in an ATP-dependent manner. In the EGF-stimulated case, wild-type EGFR was phosphorylated to a greater extent in the absence of ATP than unstimulated wild-type EGFR. The autophosphorylation of EGF-stimulated wild-type EGFR additively increased with the addition of ATP. These findings indicate that the deletion mutant retains the constitutive activity in our autophosphorylation assay. In the immunoblot analysis, phosphorylation of deletion mutant EGFR was detected in 293-pΔ15 cells without ligand stimulation. Addition of EGF increased phosphorylation of EGFR in the 293-pEGFR cells. Taken together, these results indicate that the deletion mutant has constitutive autophosphorylation activity.
Figure 1. Autophosphorylation reactions of deletion mutant EGFR and wild-type EGFR.

(A) The 293-pΔ15 and 293-pEGFR cells were treated with or without EGF (100 ng/ml) for 10 min after serum-starvation. EGFR was extracted from the cells and immobilized on wells with anti-EGFR antibody. Autophosphorylation reactions were initiated by the addition of ATP, and autophosphorylation was detected using horseradish-peroxidase-conjugated phosphotyrosine antibody, measuring the absorbance (‘optical density’) at 450 nm. Autophosphorylation was seen for unstimulated (○) and EGF-stimulated (●) deletion mutant EGFR, and unstimulated (△) and EGF-stimulated (▲) wild-type EGFR. Results are representative of at least three independent experiments. (B) The 293-pΔ15 and 293-pEGFR cells were treated with or without EGF (100 ng/ml) for the indicated times after serum-starvation. Phosphorylation of EGFR and total EGFR was determined by immunoblotting. (C) The 293-pΔ15 and 293-pEGFR cells were exposed to gefitinib (0.002–2 μM) for 3 h under serum-starvation conditions, and stimulated with EGF (100 ng/ml) for 10 min. The cells were then lysed and subjected to immunoblot analysis.
In addition, we examined the secretion of major ligands for EGFR such as EGF and TGF-α from transfected HEK-293 cells by ELISA. No detectable EGF and TGF-α secretion was observed in the cultivation medium used for HEK-293 transfectants (results not shown), indicating that these transfectants are not activated via EGF-mediated autocrine loops. We considered that autophosphorylation using unstimulated EGFR represents a low level of EGF-independent basal phosphorylation, whereas autophosphorylation using EGF-stimulated EGFR represents EGF-induced phosphorylation.
Kinetic parameters of autophosphorylation
The deletion mutant EGFR is constitutively phosphorylated under unstimulated conditions. Measuring the autophosphorylation activity of deletion mutant EGFR requires unphosphorylated tyrosine residues of EGFR. An autophosphorylation assay was reconstructed to determine the kinetic parameters of deletion mutant EGFR. The method is summarized in Figure 2. The concentrations of gefitinib used (2 μM) completely inhibited phosphorylation of both the deletion mutant and wild-type EGFR, as demonstrated by immunoblot analysis (Figure 1C). We performed autophosphorylation assays with various amounts of EGFR (results not shown). In our autophosphorylation assay, a constant amount of EGFR (130 ng/well) was adopted to measure its autophosphorylation, because this amount of EGFR was found to be appropriate for detecting changes in the absorbance of both wild-type and deletion mutant EGFR. The autophosphorylation of deletion mutant EGFR and wild-type EGFR was analysed by comparison with unstimulated and EGF-stimulated EGFR (Figure 3). The higher phosphorylation of deletion mutant EGFR shown in Figure 1(A) was lowered by using gefitinib-treated lysates, while the autophosphorylation reaction was initiated by addition of ATP. The ATP-dependent autophosphorylation reactions of deletion mutant EGFR and wild-type EGFR in crude cellular extracts were monitored (Figure 3, insets). The data were transformed into an Eadie–Hofstee plot, and the kinetic parameters were determined as apparent Km and Vmax values for ATP (Figure 3 and Table 1). Under unstimulated conditions, differences in activities were seen between unstimulated wild-type (Km for ATP=4.0±0.3 μM) and deletion mutant EGFR (Km for ATP=2.5±0.2 μM). Under EGF-stimulated conditions, there was no difference in Km values between EGF-stimulated wild-type EGFR (Km for ATP=1.9±0.1 μM) and deletion mutant EGFR (Km for ATP=2.2±0.2 μM). The Vmax values of wild-type EGFR and deletion mutant EGFR were equal under both conditions. These results suggest that the wild-type EGFR is conformationally activated by EGF stimulation, and that the mutant EGFR is active without ligand stimulation.
Figure 2. Schematic illustration of the cell-based autophosphorylation assay.

The 293-pΔ15 and the 293-pEGFR cells overexpressing deletion mutant EGFR and wild-type EGFR respectively were treated with 2 μM gefitinib for 3 h and stimulated with or without EGF (100 ng/ml) under serum-starvation conditions. EGFR was extracted from cells and immobilized on wells with anti-EGFR antibody. The autophosphorylation reaction was initiated by the addition of ATP with or without gefitinib, and horseradish-peroxidase-conjugated anti-phosphotyrosine antibody was used to detect the phosphorylation of EGFR. TKI, tyrosine kinase inhibitor.
Figure 3. Autophosphorylation activities of deletion mutant EGFR and wild-type EGFR.

Plots of absorbance (‘optical density’) against ATP concentration (inset) were fitted to an Eadie–Hofstee plot to calculate the values of kinetic parameters (Km and Vmax) for deletion mutant EGFR (A) and wild-type EGFR (B) under unstimulated (○) and EGF-stimulated conditions (●). Results are representative of at least three independent experiments with similar results.
Table 1. Kinetic parameters for ATP.
The autophosphorylation reaction was performed using the indicated enzyme and gefitinib (0.5–5 nM). The steady-state kinetic parameters for ATP were determined from the Eadie–Hofstee plot in Figure 5. Results are means±S.D. for three independent duplicate experiments.
| Km (μM) | Vmax (μM·min−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Deletion mutant | Wild-type | Deletion mutant | Wild-type | ||||||
| Gefitinib (nM) | EGF stimulation… | − | + | − | + | − | + | − | + |
| 0 | 2.5±0.2 | 2.2±0.2 | 4.0±0.3 | 1.9±0.1 | 1.9±0.1 | 2.1±0.1 | 2.0±0.0 | 1.9±0.0 | |
| 0.5 | 5.6±0.5 | 5.7±0.4 | 4.1±0.4 | 2.3±0.1 | 1.9±0.1 | 1.9±0.2 | 2.0±0.1 | 1.9±0.1 | |
| 1 | 9.8±2.8 | 10.9±3.0 | 4.6±1.2 | 2.5±0.1 | 2.0±0.1 | 1.9±0.0 | 2.0±0.2 | 1.8±0.1 | |
| 5 | 26.1±5.4 | 30.2±4.2 | 7.0±2.3 | 4.9±0.9 | 1.9±0.1 | 1.8±0.2 | 2.0±0.1 | 1.8±0.2 | |
Gefitinib inhibits autophosphorylation of deletion mutant EGFR
We examined the inhibitory effect of gefitinib (0.5, 1 and 5 nM) on the autophosphorylation of deletion mutant EGFR in comparison with wild-type EGFR under unstimulated and EGF-stimulated conditions. The data were transformed into a Lineweaver–Burk plot for estimation of the mode of inhibition (Figure 4). Lineweaver–Burk plot analysis showed that gefitinib competitively inhibited the autophosphorylation of deletion mutant EGFR as well as that of wild-type EGFR. The data were transformed into an Eadie–Hofstee plot for determination of kinetic parameters (Figure 5). Eadie–Hofstee plot analysis revealed the apparent Km and Vmax values for ATP in the presence of various gefitinib concentrations, and the kinetic parameters are summarized in Table 1. The Ki for deletion mutant EGFR and wild-type EGFR was calculated using eqn 1 (see the Materials and methods section). The Ki value of gefitinib for deletion mutant EGFR (Ki for gefitinib=0.5±0.1 nM) was 26-fold lower than that for wild-type EGFR (Ki for gefitinib=13.3±5.1 nM) under unstimulated conditions (Figure 5). Under EGF-stimulated conditions, the Ki value of gefitinib for deletion mutant EGFR (0.3±0.1 nM) was 10-fold lower than that for wild-type EGFR (3.0±0.6 nM) (Figure 5). Based on these comparative studies, we concluded that gefitinib binds deletion mutant EGFR more strongly than wild-type EGFR. In addition, we calculated the inhibitory effect of gefitinib for both types of EGFR in the presence of 2 μM ATP (Figure 6). Relatively strong inhibitory activity was detected for deletion mutant EGFR as compared with wild-type EGFR. These results suggest that gefitinib had a high affinity (low Ki value) for deletion mutant EGFR compared with wild-type EGFR.
Figure 4. Mechanism of inhibition of deletion mutant EGFR by gefitinib.

Autophosphorylation of unstimulated deletion mutant (A), unstimulated wild-type (B), EGF-stimulated deletion mutant (C) and EGF-stimulated wild-type (D) EGFR was measured with or without gefitinib at concentrations of 0 (○), 0.5 (●), 1 (△) and 5 (▲) nM. Reciprocal velocity against reciprocal ATP concentrations (0.5–32 μM) were plotted. Data are representative of at least three independent experiments.
Figure 5. Inhibition constant of gefitinib for autophosphorylation activity of deletion mutant EGFR.

The same dataset as shown in Figure 4 was fitted to an Eadie–Hofstee plot, and kinetic parameters from this fit are summarized in Table 1. Shown are the results for the unstimulated (A) and EGF-stimulated (C) deletion mutant EGFR and unstimulated (B) and EGF-stimulated (D) wild-type EGFR in response to ATP with or without gefitinib at concentrations of 0 (○), 0.5 (●), 1 (△) and 5 (▲) nM. Results are representative of at least three independent experiments.
Figure 6. Effects of gefitinib on autophosphorylation of deletion mutant EGFR.

The percentage of absorbance compared with the control under conditions of 2 μM ATP was calculated using the same dataset as shown in Figure 4 at a concentration of 2 μM ATP. The results shown are for unstimulated (A) and EGF-stimulated (C) deletion mutant EGFR and unstimulated (B) and EGF-stimulated (D) wild-type EGFR in response to ATP with or without gefitinib. Results are representative of at least three independent experiments.
DISCUSSION
Wild-type EGFR is unphosphorylated, being in an inactive form, under unstimulated conditions. The binding of ligands to the extracellular domain of EGFR induces dimerization and phosphorylation of the receptor into the active form [13]. The kinetic parameters of wild-type EGFR in our autophosphorylation assay are consistent with those of previous reports [14,15]. Crystallographic analysis has shown that the structure of the EGFR kinase domain after forming a complex with erlotinib exhibits a conformation consistent with the active form of protein kinases [16,17]. Previously, we reported that the deletion mutant EGFR was dimerized and phosphorylated constitutively without ligand stimulation, suggesting an active conformation [9]. We analysed the enzymatic properties of the deletion mutant EGFR, and determined the Ki value of gefitinib for deletion mutant EGFR. The inhibition constant of gefitinib for wild-type EGFR was similar to the value reported by Wakeling et al. [18]. We showed that the Ki value of gefitinib for deletion mutant EGFR was much lower than that for wild-type EGFR. The evidence of the decreased Ki value of gefitinib for deletion mutant EGFR means that gefitinib binds deletion mutant EGFR more strongly than wild-type EGFR. The high-affinity interaction between deletion mutant EGFR and gefitinib may be attributable to structural differences between deletion mutant EGFR and wild-type EGFR.
Our conclusion does not contradict the previous report by Stamos et al. [16] on a similar EGFR-targeted tyrosine kinase inhibitor, erlotinib, which binds to the active form of EGFR [14]. This result differs from that reported elsewhere: Fabian et al. [19] reported that there were no differences in the binding affinity of EGFR-targeted tyrosine kinase inhibitors between wild-type EGFR and mutant EGFR, including the deletion mutation. They constructed and expressed the kinase domain of EGFR on a bacteriophage surface, followed by interaction with immobilized inhibitors using biotin–avidin systems. Conversely, in our experiments, we performed autophosphorylation assays with EGFR extracted from 293-pΔ15 and the 293-pEGFR cells overexpressing deletion mutant and wild-type EGFR respectively. We consider our cell-based autophosphorylation assay results to reflect the native state of deletion mutant EGFR and to possibly explain the hypersensitivity of mutant-expressing cells to gefitinib.
We demonstrated that the deletion mutant actually binds gefitinib more strongly than wild-type EGFR. This is likely to be the mechanism of action of other tyrosine kinase inhibitors such as erlotinib, ZD6474 [dual inhibitor targeted to VEGFR2 (vascular endothelial growth factor receptor 2)/KDR (kinase insert domain-containing receptor) and EGFR] and other possible multi-targeted tyrosine kinase inhibitors. Indeed, EGFR-specific tyrosine kinase inhibitors AG1478 and erlotinib, as well as ZD6474, as described in our previous report [7] showed different growth-inhibitory activities against HEK-293 transfected with deletion mutant EGFR (results not shown). Thus it is likely that these (ATP competitive) tyrosine kinase inhibitors have different binding property effects on wild-type and deletion mutant EGFR to those of gefitinib.
In the present study, we focused on the enzymatic properties of in-frame deletion mutant EGFR (delE746–A750). The inhibition of receptor autophosphorylation in deletion mutant EGFR by gefitinib was much greater than that in wild-type EGFR. Next, it is necessary to examine the kinetic properties of other types of EGFR mutants, especially L858R, and these findings may pave the way for the discovery of different kinase inhibitors with different inhibition profiles for EGFR.
Acknowledgments
This work was supported by funds for the Third Term Comprehensive 10-Year Strategy for Cancer Control.
References
- 1.Arteaga C. L. ErbB-targeted therapeutic approaches in human cancer. Exp. Cell Res. 2003;284:122–130. doi: 10.1016/s0014-4827(02)00104-0. [DOI] [PubMed] [Google Scholar]
- 2.Traxler P., Furet P., Mett H., Buchdunger E., Meyer T., Lydon N. Design and synthesis of novel tyrosine kinase inhibitors using a pharmacophore model of the ATP-binding site of the EGF-R. J. Pharm. Belg. 1997;52:88–96. [PubMed] [Google Scholar]
- 3.Shepherd F. A., Rodrigues Pereira J., Ciuleanu T., Tan E. H., Hirsh V., Thongprasert S., Campos D., Maoleekoonpiroj S., Smylie M., Martins R., et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 4.Bell D. W., Lynch T. J., Haserlat S. M., Harris P. L., Okimoto R. A., Brannigan B. W., Sgroi D. C., Muir B., Riemenschneider M. J., Iacona R. B., et al. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J. Clin. Oncol. 2005;23:8081–8092. doi: 10.1200/JCO.2005.02.7078. [DOI] [PubMed] [Google Scholar]
- 5.Lynch T. J., Bell D. W., Sordella R., Gurubhagavatula S., Okimoto R. A., Brannigan B. W., Harris P. L., Haserlat S. M., Supko J. G., Haluska F. G., et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 6.Paez J. G., Janne P. A., Lee J. C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F. J., Lindeman N., Boggon T. J., et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 7.Arao T., Fukumoto H., Takeda M., Tamura T., Saijo N., Nishio K. Small in-frame deletion in the epidermal growth factor receptor as a target for ZD6474. Cancer Res. 2004;64:9101–9104. doi: 10.1158/0008-5472.CAN-04-2360. [DOI] [PubMed] [Google Scholar]
- 8.Tracy S., Mukohara T., Hansen M., Meyerson M., Johnson B. E., Janne P. A. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. 2004;64:7241–7244. doi: 10.1158/0008-5472.CAN-04-1905. [DOI] [PubMed] [Google Scholar]
- 9.Sakai K., Arao T., Shimoyama T., Murofushi K., Sekijima M., Kaji N., Tamura T., Saijo N., Nishio K. Dimerization and the signal transduction pathway of a small in-frame deletion in the epidermal growth factor receptor. FASEB J. 2005;20:311–313. doi: 10.1096/fj.05-4034fje. [DOI] [PubMed] [Google Scholar]
- 10.Nishio K., Arioka H., Ishida T., Fukumoto H., Kurokawa H., Sata M., Ohata M., Saijo N. Enhanced interaction between tubulin and microtubule-associated protein 2 via inhibition of MAP kinase and CDC2 kinase by paclitaxel. Int. J. Cancer. 1995;63:688–693. doi: 10.1002/ijc.2910630514. [DOI] [PubMed] [Google Scholar]
- 11.Kawamura-Akiyama Y., Kusaba H., Kanzawa F., Tamura T., Saijo N., Nishio K. Non-cross resistance of ZD0473 in acquired cisplatin-resistant lung cancer cell lines. Lung Cancer. 2002;38:43–50. doi: 10.1016/s0169-5002(02)00175-7. [DOI] [PubMed] [Google Scholar]
- 12.Koizumi F., Shimoyama T., Taguchi F., Saijo N., Nishio K. Establishment of a human non-small cell lung cancer cell line resistant to gefitinib. Int. J. Cancer. 2005;116:36–44. doi: 10.1002/ijc.20985. [DOI] [PubMed] [Google Scholar]
- 13.Tanner K. G., Kyte J. Dimerization of the extracellular domain of the receptor for epidermal growth factor containing the membrane-spanning segment in response to treatment with epidermal growth factor. J. Biol. Chem. 1999;274:35985–35990. doi: 10.1074/jbc.274.50.35985. [DOI] [PubMed] [Google Scholar]
- 14.Nair N., Davis R. J., Robinson H. L. Protein tyrosine kinase activities of the epidermal growth factor receptor and ErbB proteins: correlation of oncogenic activation with altered kinetics. Mol. Cell. Biol. 1992;12:2010–2016. doi: 10.1128/mcb.12.5.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wood E. R., Truesdale A. T., McDonald O. B., Yuan D., Hassell A., Dickerson S. H., Ellis B., Pennisi C., Horne E., Lackey K., et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 16.Stamos J., Sliwkowski M. X., Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 17.Noble M. E., Endicott J. A., Johnson L. N. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 18.Wakeling A. E., Guy S. P., Woodburn J. R., Ashton S. E., Curry B. J., Barker A. J., Gibson K. H. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62:5749–5754. [PubMed] [Google Scholar]
- 19.Fabian M. A., Biggs W. H., 3rd, Treiber D. K., Atteridge C. E., Azimioara M. D., Benedetti M. G., Carter T. A., Ciceri P., Edeen P. T., Floyd M., et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]