

Abstract
Proinflammatory (TNF-α, IL-1β, and NO) and antiinflammatory (IL-10, CO) levels were assayed in serum, liver, and small bowel in order to verify a hypothetic inflammatory etiopathogeny of portal hypertension that could be the cause of its evolutive heterogeneity. Male Wistar rats were divided into one control group (n = 11) and one group with a triple stenosing ligation of the portal vein (n = 23) after 28 days of evolution. In one subgroup of portal hypertensive rats, portal pressure, collateral venous circulation, mesenteric vasculopathy, and liver and spleen weights were determined. In the remaining rats with portal hypertension TNF-α, IL-1β, and IL-10 were quantified in liver and ileum by enzyme-linked immunosorbent assay. NO synthase activity was studied in liver and ileum. CO and NO were measured in portal and systemic blood by spectrophotometry and Griess reaction, respectively. Portal hypertensive rats with mayor spleen weight show hepatomegaly and mayor development of collateral circulation. Ileum release of IL-10 (0.30 ± 0.12 versus 0.14 ± 0.02 pmol/mg protein; P < .01) is associated with a liver production of both proinflammatory mediators (TNF-α: 2 ± 0.21 versus 1.32 ± 0.60 pmol/mg protein; P < .05, IL-1β: 19.17 ± 2.87 versus 5.96 ± 1.84 pmol/mg protein; P = .005, and NO: 132.10 ± 34.72 versus 61.05 ± 8.30 nmol/mL; P = .005) and an antiinflammatory mediator (CO: 6.49 ± 2.99 versus 3.03 ± 1.59 pmol/mL; P = .005). In short-term prehepatic portal hypertension a gut-liver inflammatory loop, which could be fundamental in the regulation both of the portal pressure and of its complications, could be proposed.
INTRODUCTION
Portal hypertension (PHI) is a clinical syndrome which is usually secondary to intrahepatic or extrahepatic obstruction of portal flow. It is characterized by a pathological increase in portal pressure, associated with splenomegaly, and by the development of portosystemic collateral circulation which diverts portal flow to the systemic circulation bypassing the liver [1, 2, 3]. Moreover, PHT is the main complication of cirrhosis and is responsible for most of its common complications: variceal hemorrhage, ascites, and portosystemic encephalopathy [4]. Making an effort to create new PHT models or to improve previously existing ones is justified by the need to study the physiopathological mechanisms of this frequent and severe clinical syndrome for which several different types of medical [5, 6] and surgical [7] treatments have been proposed.
Partial portal vein ligation (PVL) in the rat is the most frequently used experimental model to study in the short term the pathophysiology of prehepatic PHT [8, 9]. Constriction of the portal vein (PV) is immediately followed by increased portal resistance and portal pressure as well as decreased portal venous inflow [10]. Partial development of portosystemic collaterals is found after 4 days of PVL and, after two weeks of evolution most (95%) of the increased portal blood is diverted from the liver by an extensive portocollateral vascular bed [9, 10]. Then, portal resistance decreases to control values and the splanchnic blood flow, secondary to a decrease in the splanchnic arteriolar resistance, increases, all of which contributes to the maintenance of increased portal pressure [10, 11].
Therefore, until now PHT in the rat has always been considered to be a hemodynamic impairment with much more homogeneous alterations than those described in human PHT because of a narrow range of PHT, grade of portosystemic shunts, and hepatic atrophy [12]. However, this evolutive uniformity could not be verified from our previous studies using a modified technique of PV calibrated stenosis in the rat since the degree of hepatic atrophy, splenomegaly, and portosystemic collateral circulation that developed were variable [13]. A possible inflammatory etiopathogeny of PHT could be one cause of this evolutive heterogeneity. Thus, an increased infiltration of the intestinal mucosa and submucosa by mast cells has been described in PHT rats [14]. These inflammatory cells could be considered to be primed to a noxious stimulus and this mechanism could be responsible for the increased susceptibility of the PHT mucosa. If so, the anaphylactic degranulation of the mast cells may cause inflammatory episodes that vary in number and duration in each individual. This difference could explain the great variability observed in prehepatic PHT in the rat.
In order to verify a hypothetic inflammatory etiopathogeny of PHT we have studied some mediators involved in an inflammatory response. These included tumor necrosis factor-α (TNF-α), Interleukin-1β (IL-1β), interleukin-10 (IL-10), nitric oxide synthase (NOS) isoforms in tissue (liver and/or intestine), and nitric oxide (NO) and carbon monoxide (CO) in serum in rats with short-term prehepatic PHT.
MATERIAL AND METHODS
Animals
Male Wistar rats from the Complutense University Vivarium in Madrid with body weights from 230 to 250 g were used. The experimental procedures employed in this study are in accordance with the Guidelines for the care and use of Laboratory Animals (1986) published in Spain (Royal Decree 223/1988).
Experimental design
The animals were divided for their study into three groups: one control group (I), in which the animals did not undergo any intervention, and two-subgroups (IIa and IIb), in which prehepatic PHT by triple stenosing ligation of the portal vein (TSLP) was carried out. All the animals were sacrificed by ether overdose after 28 days of evolution. In Group IIa portal pressure, collateral venous circulation, mesenteric vasculopathy, and hepatic and spleen weights were studied. In Group IIb TNF-α, IL-1β, and IL-10 levels were assayed in ileum, endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) activity were measured in liver and in intestine (duodenum, jejunum, and ileum). Finally, NO and CO concentrations were quantified in PV, suprahepatic inferior vena cava (SH-IVC), and infrahepatic inferior vena cava (IH-IVC) blood.
Portal vein stenosis technique
The surgical technique used to produce PHT by TSLP has been described previously [15]. In brief, while rats were under ketamine hydrochloride (80 mg/kg) and Xylacin (12 mg/kg) im anesthesia, the PV was isolated and three stenosing ligations were performed in its superior, medial, and inferior portions. The stenoses were calibrated by a simultaneous ligation (4-0 silk) around the PV and a 20-gauge blunt-tipped needle. The midline incision was closed on two layers with catgut and 3-0 silk.
Portal vein pressure measurement
The portal pressure is measured by an indirect technique of intrasplenic punction [16], inserting a 20 G needle in the splenic parenchyma which is, in turn, connected to a PE-50 tube. After verifying that free back blood flow was obtained, the catheter was then connected to a pressure recorder (PowerLab 200 ML 201) and to a transducer (Sensonor SN-844) with a Chart V 4.0 computer program (ADI Instruments). The system is calibrated before each experiment. The zero reference point was established at 1 cm above the operating table. Previous studies have demonstrated an excellent correlation between the indirect measurement of the portal pressure by intrasplenic punction and the direct measurement by canulation of the superior mesenteric vein [17].
Portosystemic collateral circulation study method
Portal hypertension was confirmed by the presence of splenomegaly and portosystemic collateral circulation. First, a midline abdominal incision with a large bilateral subcostal extension was made and then the areas in which the collateral venous circulation was developed, that is, the splenorenal (SR), gastroesophageal (paraesophageal collaterals (PE)), colorectal (hemorrhoidal or pararectal collateral (PR)) and hepatic hilum (portoportal (PP) and accessory hepatic vein (AHV)) [4], were carefully studied for the presence of increased collateral veins. The latter (AHV) reached the hepatic hilum following a pathway between the left lateral and caudate hepatic lobes [18].
Gross superior mesenteric vein study
Existence of dilation and tortuosity of the superior mesenteric vein branches has been named mesenteric venous vasculopathy (MVV). Three grade s of MMV were considered: (i) Grade 0: normal appearance of the superior mesenteric vein branches; (ii) Grade I: dilation and tortuosity of the mentioned branches secondary to the Pringle maneuver, and (iii) Grade II in which the dilation and tortuosity of the superior mesenteric vein branches were spontaneous.
Blood extraction method
Blood samples (1 mL) were drawn by puncture of the IH-IVC and SH-IVC. After 15 minutes of centrifugation at 1500 g they were separated and transferred to sterile polypropylene tubes. The serum was then frozen at −80°C until NO and CO were assayed.
Preparation of organ homogenates
Liver and intestine (duodenum, jejunum, ileum) were quickly dissected and frozen in dry ice. Frozen organ samples were weighed on a Mettler balance (model AE 100; Mettler Instrument Corp, Hightstown, NJ) and transferred to 50 mL polypropylene tubes (Falcon; Becton Dickinson, Lincoln Park, NJ) containing lysis buffer (4°C) at a ratio of 10 mL buffer/1 g of wet tissue. Lysis buffer consisted of 1 mM phenylmethylsulfonyl fluoride (PMSF; Sigma Chemical Company) and 1 μg/mL pepstatin A (Sigma Chemical Company), aprotinin (Sigma Chemical Company), antipain (Sigma Chemical Company), and leupeptin (Sigma Chemical Company) in 1x phosphate-buffered saline solution of pH 7.2 (Biofluids, Rockville, Md) containing 0.05% sodium azide (Sigma Chemical Company). Samples were homogenized for 30 seconds with an electrical homogenizer (Polytron; Brinkmann Instruments, Westminster, NY) at maximum speed, and the tubes were immediately frozen in liquid nitrogen. The samples were homogenized three times for optimal processing. The supernatants were frozen at −80°C to allow the formation of macromolecular aggregates. After thawing at 4°C, the aggregates were pelleted at 3000 g (4°C), and the final organ homogenate volume was measured with a graduated pipette [19]. The homogenates were stored at −80°C until assayed for the quantitative presence of rat TNF-α, IL-1β, and IL-10 with a rat enzyme-linked immunosorbent assay (ELISA).
Ileal and liver TNF-α, IL-1β, and IL-10 ELISA
Cytokine levels were measured using commercially available (ELISA) specific kits (BioNOVA Cientifica Ltd, Madrid, Spain). The minimum detectable level of TNF-α was 0.5 pg/mL (n = 10). The intraassay variation ranged from 3.1% (lower part of standard curve) to 4.2% (upper) and the interassay variation oscillated between 5.2% and 5.6%. The minimum detectable level of IL-1β was 1.5 pg/mL (n = 10). The intraassay variation ranged from 3.5% (lower part of standard curve) to 4.3% (upper) and the interassay variation osciled between 6.1% and 7.4%. The minimum detectable level of IL-10 was 2.5 pg/mL (n = 10). The intraassay variation ranged from 4.2% (lower part of standard curve) to 7.5% (upper) and the interassay variation osciled between 5.8% and 9.3%.
NOS activity
NOS activity was calculated as the rate of conversion of 14C-arginine in citrulline. Tissue was homogenized in 1 mL of a buffer containing 10−2 M HEPES, 0.32 M sucrose, 10−4 M EDTA, 10−3 M dithiothreitol, soybean trypsin inhibitor, 10 μg/mL leupeptin, 2 μg/mL aprotinin, and 1 mg/mL phenylmethanesulfonylfluoride and centrifuged for 20 minutes at 100 000 g at 4°C. An aliquot of the final supernatant was mixed with PBB containing calcium chloride, 14C-arginine, valine (to inhibit arginase activity) and NADPH, and incubated for 30 minutes at 37°C. After purification in a Dowex (50 W) column, radioactivity was measured. Constitutive NOS is calcium dependent while the inducible isoform is not. In contrast, the arginine analog N-methyl-L-arginine predominantly inhibits iNOS. To identify the activity of both NOS isoforms we used either the calcium chelator EGTA (1.4 mM) to isolate the activity of the iNOS or N-methyl-L-arginine (140 μM) to isolate the activity of the constitutive NOS [20].
Serum NO assay
The serum NO content was measured by the Griess reaction as nitrite concentration after nitrate reduction to nitrite [21]. Briefly, the samples were deproteinized by the addition of sulfosalicylic acid. They were then incubated for 30 minutes at 4°C and subsequently centrifuged for 20 minutes at 12 000 g. After incubation of the supernatants with E colischerichial nitrate reductase (37°C, 30 minutes), 1 mL of Griess reagent (0.5% naphthylethylenediamine dihydrochloride, 5% sulfonilamide, 25% phosphoric acid) (Merck, Darmstad, Germany) was added. The reaction was performed at 22°C for 20 minutes and the absorbance was measured at 546 nm, using sodium nitrite solution as a standard. The minimum detectable level of NO was 0.1 nmol/mL (n = 10). The intraassay variation was 1.6% and the interassay variation 2.7%.
Serum CO assay
To quantify the amount of CO, hemoglobin was added to bind CO as carboxyhemoglobin and the proportion of carboxyhemoglobin was estimated [22]. For this purpose, hemoglobin (4 μM) was gently mixed with the sample and 1 minute was allowed to ensure maximum CO binding. Then, samples were diluted with phosphate buffer (0.01 M monobasic potassium phosphate/dibasic potassium phosphate, pH 6.85) containing sodium hydrosulfite, allowed to stand at room temperature for 10 minutes, and absorbance was read at 420 and 432 nm against a matched cuvette only containing buffer. The minimum detectable level of CO was 1 pmol/mL (n = 10). The intraassay variation was 2.9% and the interassay variation 6.8%.
Statistical analysis
The results are expressed as the mean ± the standard deviation. Analysis of variance (ANOVA), the Duncan test and Student t for unpaired data were used for the statistical comparison of the quantitative variables between the different groups. Chi-square test was used for comparison of the qualitative variables. The linear correlation between two mutually dependent variables was determined and expressed as the correlation coefficient (r). The results are considered as statistically significant if P < .05.
RESULTS
Splenomegaly is always present in portal hypertensive rats. Since in rats with portacaval anastomosis (PCA) spleen weights correlated with portal pressure and preservation of portal pressure after PCA provides metabolic and nutritional benefits [23], two subgroups were defined within the TSLP group: one group in which spleen/body weight ratio was less than 0.29 (IIa; n = 9) and one group with a spleen/body weight ratio higher than or equal to 0.30 (IIb; n = 14) (Figure 1). The spleen weight increase is significant (P < .001) in all TSLP rats, and this increase is greater (P < .001) in Group IIb compared to Group IIa (Table 1).
Figure 1.
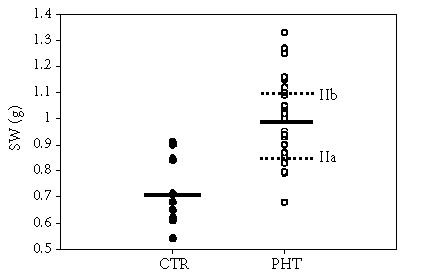
Residual plot showing the distribution of spleen weights in all control and TSLP rats. The solid lines represent the mean values for the overall group. The broken lines represent the mean value for the TSLP rats with lower spleen weights (IIa) and the TSLP rats with higher spleen weights (IIb).
Table 1.
Body weight increase (BWI), liver weight (LW), liver weight to body weight (BW), spleen weight (SW), and spleen weight to body weight in the control group (Group I) and in rats with triple stenosing ligation of portal vein (TSLP) (Group II) which, in turn, were divided into Groups IIa (SW/BW < 0.29) and IIb (SW/BW ≥ 0.30). The results are expressed as mean ± SD. ∗ denotes that P < .05 in relation to control group, ∗∗ denotes that P < .01 in relation to control group, and ∗∗∗ denotes that P < .001 which is a statistically significant value in relation to the control group. ••• denotes that P < .001 which is a statistically significant value of Group IIb in relation to Group IIa.
| Group | BWI (g) | LW (g) | LW/100 g BW | SW (g) | SW/100 g BW |
| I (cotrol) | 140 ± 30 | 13.4 ± 1.1 | 3.4 ± 0.15 | 0.7 ± 0.13 | 0.18 ± 0.04 |
| n = 11 | |||||
| II (TSLP) | 62 ± 38*** | 11 ± 2.5*** | 3.5 ± 0.8 | 1.0 ± 0.2*** | 0.3 ± 0.06*** |
| n = 23 | |||||
| IIa (TSLP) | 78 ± 40*** | 10.1 ± 1.5** | 3.1 ± 0.25 | 0.9 ± 0.12* | 0.3 ± 0.02*** |
| n = 9 | |||||
| IIb (TSLP) | 52 ± 34*** | 11.1 ± 2.9* | 3.7 ± 1 | 1.1 ± 0.13*** | 0.4 ± 0.04***,••• |
| n = 14 | |||||
The body weight increase after 28 days of evolution was lower (P < .001) in all portal hypertensive rats (Table 1). The liver weight to body weight ratio does not change when all the animals with PHT are considered (Group II), but it is smaller in comparison to control animals in the subgroup of lower spleen weight (IIa) and increases in the subgroup with greater spleen weight (IIb) (Table 1). As shown in Table 2, portal pressure increases (P < .001) in rats with PHT, although we did not find differences between the two subgroups of animals with different spleen weights. Portal pressure correlates with body weight increase (r = 0.71; P = .03) in Group IIa, with lower spleen weight. In Group IIb, with higher spleen weight, portal pressure is correlated with body weight increase (r = 0.56; P = .03) and there is an inverse correlation between portal pressure and liver/body weight ratio (r = −0.75; P = .002) and between portal pressure and spleen/body weight ratio (r = −0.68; P = .008) (Table 3).
Table 2.
Portal pressure (PP), mesenteric vasculopathy (MV), and portosystemic collateral circulation (CC) in control rats (Group I) and in rats with triple stenosing ligation of portal vein (TSLP) (Group II) which, in turn, were divided into Group IIa (spleen weight/body weight < 0.29) and Group IIb (spleen weight/body weight ≥ 0.30). The results are expressed as mean ± SD. ∗∗∗ denotes that P < .001 which is a statistically significant value relative to the control group. •• denotes that P = .005 which is a statistically significant value of Group IIb relative to Group IIa.
| Group | PP | MV | CC | ||
| mmHg | Grade I | Grade II | n ≤ 3 | n = 4 | |
| I (control) | 7 ± 0.6 | — | — | — | — |
| (n = 11) | |||||
| II (TSLP) | 13 ± 3.5*** | 11 (48%) | 12 (53%) | 12 (53%) | 11 (48%) |
| (n = 23) | |||||
| IIa (TSLP) | 13 ± 4*** | 5 (55.5%) | 4 (44.4%) | 8 (89%) | 1 (11%) |
| n = 9 | |||||
| IIb (TSLP) | 13 ± 3.4*** | 6 (43%) | 8 (57%) | 4••(28.6%) | 10•• (71.4%) |
| n = 14 | |||||
Table 3.
Correlations between portal pressure and body weight increase and liver and spleen weights in rats with triple stenosing ligation of portal vein (TSLP) (Group II) which, in turn, were divided into Group IIa (spleen weight/body weight < 0.29) and Group IIb (spleen weight/body weight ≥ 0.30).
| Correlations | Correlation (r) | ||
| TSLP (IIa + IIb) | TSLP (IIa) | TSLP (IIb) | |
| PP versus BWI | 0.57 (P = .004) | 0.71 (P = .03) | 0.56 (P = .03) |
| PP versus LW/BW | −0.46 (P = .02) | — | −0.75 (P = .002) |
| PP versus SW/BW | — | — | −0.68 (P = .008) |
Mesenteric vasculopathy, of either Grade I or II, is observed in all the animals with PV (Table 2). Portosystemic collateral circulation of superior and inferior SR, PE, and PR types is developed in rats with PHT. Two categories were established: one consisting in the development of 1 to 3 types of collateral circulation and another that consists in the development of 4 types of collateral circulation. When 3 or less types of collateral circulation are developed, superior and inferior SR types are the most frequent, and when four types exist PR and PE are the types associated.
Differences were observed between subgroups IIa and IIb in the development of collateral circulation. In the group with lower spleen weight (IIa) most rats (88.88%) had 3 or less types of collateral vessels, while in the group with greater spleen weight (IIb) 71.43% developed four types of collateral vessels, and this difference was statistically significant (P = .005) (Table 2). There is, therefore, a significant correlation (P = .003) between splenic weight and the number of newly formed collateral vessels.
The concentrations of proinflammatory cytokines TNF-α and IL-1β in ileum do not increase in rats with PHT (Figure 2). In contrast, IL-10 levels increase (P < .01) in these animals. TNF-α and IL-1β levels increase in the liver (P < .05 and P < .001, resp) (Figure 3).
Figure 2.
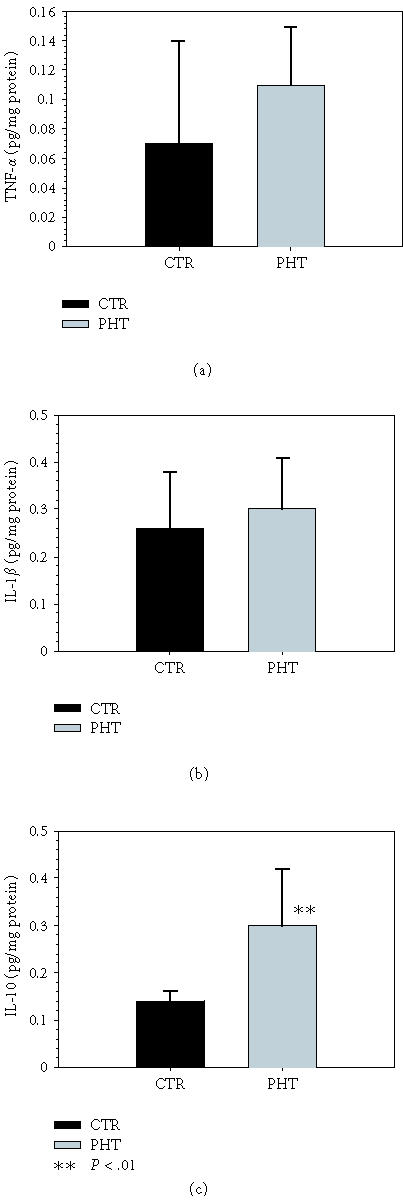
Concentrations of (a) TNF-α, (b) IL-1β, and (c) IL-10 in the ileum of control rats (Group I) and of rats with triple stenosing ligation of portal vein (TSLP) (Group IIb) after 28 days of evolution.
Figure 3.
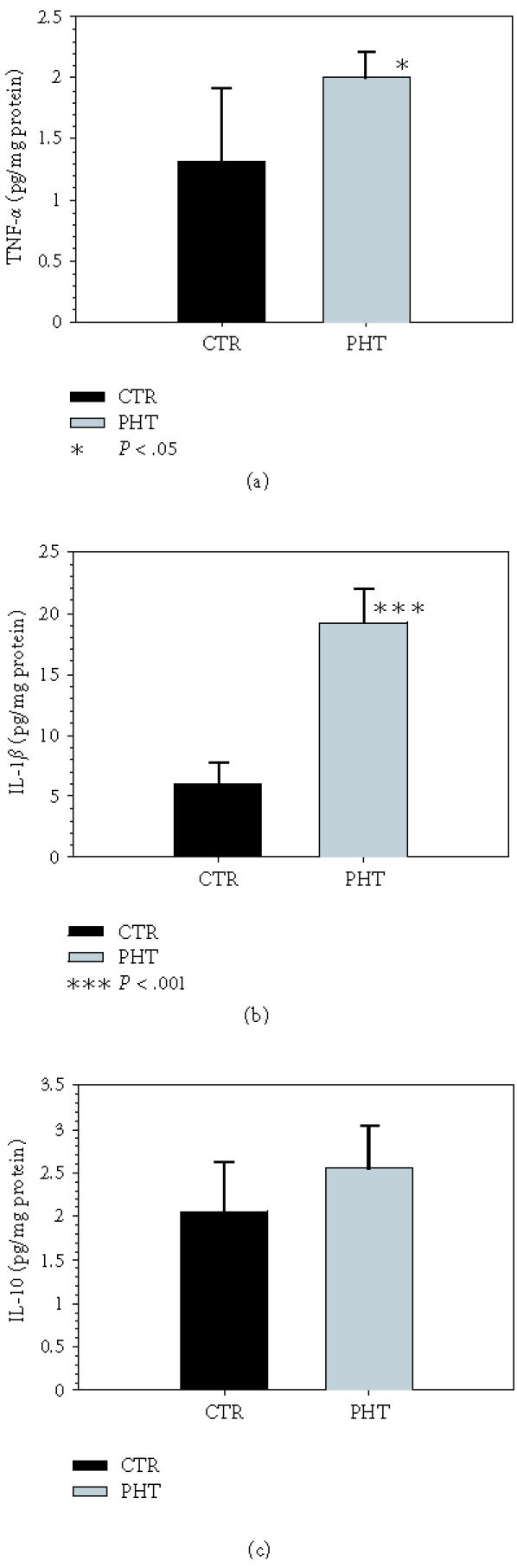
Concentrations of (a) TNF-α, (b) IL-1β, and (c) IL-10 in liver of control rats (Group I) and in rats with portal hypertension (PHT) by triple stenosing ligation of portal vein (TSLP) (Group IIb) after 28 days of evolution.
Liver and intestinal eNOS activity is similar in control and portal hypertensive-rats (Table 4). On the contrary, iNOS activity increases in liver (P < .001) and in jejunum (P < .05) in PHT rats (Table 5).
Table 4.
Liver and intestinal endothelial nitric oxide synthase (eNOS) activity in control rats (Group I) and in rats with triple stenosing ligation of portal vein (TSLP) after 28 days of evolution. The results are represented as mean ±SD.
| Group | eNOS liver | eNOS duodenum | eNOS jejunum | eNOS ileum |
| μ mol/pg protein | μ mol/pg protein | μ mol/pg protein | μ mol/pg protein | |
| I (control) | 1.1 ± 0.5 | 5.7 ± 0.95 | 5 ± 1 | 4 ± 2.8 |
| (n = 8) | ||||
| II (TSLP) | 1.45 ± 0.4 | 7 ± 0.9 | 6.5 ± 1 | 5 ± 3.2 |
| (n = 8) | ||||
Table 5.
Liver and intestinal inducible nitric oxide synthase (iNOS) activity in control rats (Group I) and in rats with triple stenosing ligation of portal vein (TSLP) (Group II) after 28 days of evolution. The results are represented as mean ±SD. ∗ denotes that P < .05 and ∗∗∗ P < .001 which is a statistically significant value in relation to the control group.
| Group | iNOS liver | iNOS duodenum | iNOS jejunum | iNOS ileum |
| μ mol/pg protein | μ mol/pg protein | μ mol/pg protein | μ mol/pg protein | |
| I (control) | 1 ± 0.4 | 6.0 ± 0.7 | 5.7 ± 0.9 | 5.7 ± 2.3 |
| (n = 8) | ||||
| II (TSLP) | 3.8 ± 0.8*** | 6.7 ± 1.4 | 7.3 ± 1.3* | 6.5 ± 1.5 |
| (n = 8) | ||||
As shown in Figures 4 and 5, in rats with PHT NO and CO of hepatic source increase (P = .005) while NO decreases in IH-IVC (P = .005).
Figure 4.
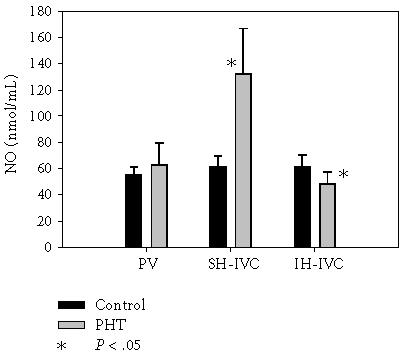
Nitric oxide (NO) in portal vein (PV), suprahepatic inferior vena cava (SH-IVC), and infrahepatic inferior vena cava (IH-IVC) in control rats (Group I) and in rats with triple stenosing ligation of portal vein (TSLP) (Group IIb) after 28 days of evolution.
Figure 5.
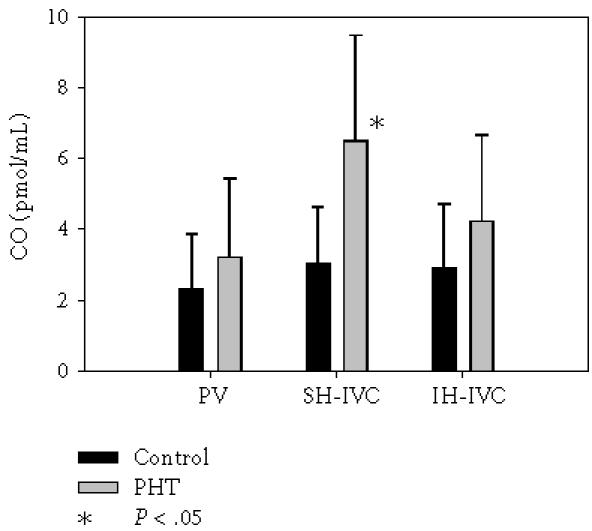
Carbon monoxide (CO) in portal vein (PV), suprahepatic inferior vena cava (SH-IVC), and infrahepatic inferior vena cava (IH-IVC) in control rats (Group I) and in rats with triple stenosing ligation of portal vein (TSLP) (Group IIb) after 28 days of evolution.
DISCUSSION
In rats with short-term prehepatic PHT, there is an ileum release of IL-10 which is associated with hepatic production of both proinflammatory mediators (TNF-α, IL-1β, and NO) and an antiinflammatory mediator (CO).
IL-10 is a pleiotropic and regulator cytokine produced principally by both T cells and macrophages, which possesses both antiinflammatory and immunosuppressive properties [24]. In PHT, IL-10 of ileum origin can be produced by T an B lymphocytes of the gut-associated-lymphatic-tissue (GALT), monocytes/macrophages, and mast cells since all these cells are well-known sources of IL-10 [25]. The production of IL-10 is triggered by several stress factors and its concentrations in the blood and in tissue compartments often reflect the magnitude of the inflammatory stress [24]. Experimental studies in rodents and primates have revealed that the primary inducers of IL-10 synthesis are in fact more proximal proinflammatory cytokines, such as TNF-α and IL-1 [24]. Moreover, glucocorticoids, endotoxin, and reactive oxygen intermediates, all raised in PHT, have been shown to induce IL-10 release [15, 24, 26, 27].
In portal hypertensive rats it has been demonstrated that in the short term (1, 14, and 45 days postoperation) the serum concentrations of TNF-α, one of the factors to which the hyperdynamic syndrome is attributed, increase [28, 29]. Bacteria colonizing the gut represent a large reservoir of microbial products, such as lipopolysacharides (LPS), endotoxins and other bacterial wall fragments capable of inducing inflammatory cytokines and lead to sustained NO production [30]. GALT has been shown to produce and release TNF in response to bacterial translocation, so the gut is a “cytokine-releasing” organ in PHT [30].
It can be hypothesized that immediately post PHT splanchnic hyperpressure could trigger an intestinal inflammatory reaction that would be highly tissue damaging and could require activation of NF-κB with ensuing generation of chemokines and cytokines in a shorter developmental period than one month p.o. Then, intestinal release of IL-10, a cytokine that controls inflammatory processes by suppressing the production of proinflammatory cytokines that are transcriptionally regulated by NF-κB, would occur. Moreover, IL-10 also inhibits the release of free oxygen radicals and NO [31]. More specifically, IL-10 is a pivotal cytokine in the control of intestinal inflammation and plays a central regulatory role in the immune responses of the intestine, limiting and ultimately terminating inflammatory responses [32]. It seems that its main function is to keep the inflammation under strict control by adjusting the intensity of the immune and inflammatory responses to the severity of the destruction produced by a pathological condition or a pathogen and, thus, minimizing damage to the host tissues caused by either the pathogen or the immune system itself [33]. In this study, the intestinal production of IL-10, possibly acting in a paracrine/autocrine fashion, would effectively inhibit intestinal release of TNF-α, IL-1β, and NO. The inhibitory effects of IL-10 on IL-1 and TNF production are crucial to its antiinflammatory activities, because these cytokines often have synergistic activities on inflammatory pathways and processes, and amplify these responses by inducing secondary mediators such as chemokines, prostaglandins, and PAF [34].
However, the increase in IL-10 intestinal production could have another meaning because IL-10, as well as inhibiting inflammation,
(1) has an important role in fetal wounds leading to reduced matrix deposition and scar-free healing [35],
(2) is expressed at elevated levels in chronic venous ulcers and may be related to the failure of these wounds to progress to final wound healing [36],
(3) has a modulatory effect on hepatic fibrogenesis, and on hepatocyte proliferation and limits liver necrosis [37].
If induction of proinflammatory cytokines in liver is not counterbalanced by antiinflammatory cytokines, especially IL-10, there are inflammatory reactions leading to massive liver damage and fibrosis with pathologic progression to cirrhosis [38, 39]. Therefore, the intestinal rise in IL-10 in PHT would prevent two severe consequences of the proinflammatory response in a tissular area as large as the intestine: an early response that would combine necrosis and edema and a later one leading to intestinal fibrosis.
It could be hypothesized that in short-term portal hypertensive rats proinflammatory gut-derived mediators enter the liver directly through the PV or by an indirect route through intestinal lymphatics and/or systemic circulation. These gut-derived mediators would be a stimulus for hepatic synthesis of TNF-α and IL-1β. Hypoxic stress has also been involved in cytokine synthesis by Kupffer cells [26] and in the liver Kupffer cells are major sources of proinflammatory cytokines that are produced in response to LPS [40]. Since PHT by PVL causes portal blood deprivation to the liver, it could be considered that this proinflammatory factor associated to LPS presence in portal circulation [41] could stimulate the Kupffer cells to produce proinflammatory compounds, including TNF-α and IL-1β.
In previous studies we have demonstrated that progressive liver steatosis is produced in rats with prehepatic PHT [42] and TNF-α and TNF-regulated cytokines are considered as effector molecules in nonalcoholic fatty liver disease (NAFLD) ranging from steatosis to cirrhosis [43]. It has been accepted that liver injury requires at least two “hits”: one that increases exposure of the hepatocytes to TNF-α and another that interferes with the fat metabolism and renders the liver more vulnerable to a second injury, such as bacteria or LPS of intestinal origin, because the hepatocytes become sensitized to TNF-mediated cell death [43, 44]. TNF-α and other cytokines inhibit mitochondrial oxidative phosphorylation, with a decrease in free fatty acids (FFA) oxidation and oxidative stress [45]. TNF-α and TNF-related cytokines can contribute to the liver steatosis which occurs in PHT both by reducing the hepatic oxidation of fatty acids and by increasing lipid synthesis because IL-1 and, in particular TNF-α, stimulate hepatic lipogenesis in the rat and increase the plasma levels of FFA and triglycerides [46].
The direct biological response of TNF-α, which is ubiquitously expressed in response to stress, also referred to as an “alarm hormone” [47], is amplified by the secondary release of other cytokines and metabolic products like IL-1 [47] and NO [48]. Our results show an increased hepatic activity of iNOS 1 month after PVL. iNOS is synthesized de novo principally in macrophages, vascular smooth muscle cells, hepatic stellate cells, and hepatocytes, only after induction by LPS and inflammatory cytokines [49], both of which are increased in this experimental model. In 1991, Vallance and Moncada proposed that the increased synthesis and release of the vascular endothelium-derived vasodilator ND, induced by endotoxin directly or indirectly by cytokines, could account for the peripheral cardiovascular abnormalities of cirrhosis [50]. However, the main enzymatic source of the NO systemic and splanchnic overproduction which occurs in PHT [51] is eNOS, the constitutive isoform of NOS. eNOS protein expression increases, eNOS activity enhances and endothelial NO release is produced in response to flow and shear stress in mesenteric and systemic vessels of PHT animals [49]. Models of PHT developed by PV stenosis are associated with a chronic increase in splanchnic shear stress, related to high blood flow, which may contribute to NO overproduction by upregulation of eNOS [52]. This could represent an adaptive mechanism of the endothelium in response to chronic increases in flow-induced shear stress [51, 53].
However, activation of iNOS in vascular smooth wall has also been described in prehepatic PHT [54, 55] leading us to deduce that possibly a small part of NO derived from iNOS, upregulated in mesentery vasculature, may also contribute to hyperdynamic circulation in PHT [56]. The contribution of each isoform of NOS to the pathogenesis of the hyperdynamic syndrome probably depends on the etiology or severity of cirrhosis in human studies and in animal studies on the PHT model [57] perhaps on the time considered after PVL and especially on the tissue in which the enzyme is measured. This could cause the increased eNOS shown in splanchnic and systemic vascular beds [50, 51, 52] while we found an increased iNOS activity in liver. Thus, 1 month after PVL our results suggest that liver proinflammatory cytokines (TNF-α and IL-1β) and endotoxin of intestinal origin could increase the hepatic synthesis of NO by iNOS upregulation.
In this early phase of PHT we have also shown the increased hepatic synthesis of CO, which besides its vasodilator and antiapoptotic effects [58], is also an antiinflammatory molecule [59]. Both isoforms of heme oxygenase (HO), the enzyme involved in the generation of CO, the inducible and the constitutive ones, can be upregulated in PHT rats. Several physical or chemical factors that may be increased during PHT, including shear stress, hypoxia, ND, glucagon, as well as the proinflammatory agents endotoxin and cytokines can induce HO-1, the inducible isoform of HO [58]. HO-1 expression is upregulated in hepatocytes and splanchnic organs from PVL rats after 7 days of the operation [59]. In relation to HO-2, the constitutive isoform of HO, the only chemical inducers identified are adrenal glucocorticoids [60]. We have previously demonstrated that there is an increase in plasma levels of corticosterone in rats with short-term PVL [13] and therefore, HO-2 could be activated by glucocorticoids in this experimental model.
The increase in serum CO levels after 1 month in portal hypertensive rats could be related to their actions. Thus, CO acts as an endogenous regulator that is required for maintaining hepatic microvascular blood flow [61]. In addition, CO acts as a potent antiinflammatory molecule that selectively inhibits expression of the proinflammatory cytokines TNF-α, IL-1β and macrophage inflammatory protein-1β (MIP-1β) [62].
In this early stage of prehepatic PHT in the rat, the hepatic hyperproduction of proinflammatory cytokines, that is, TNF-α and IL-1β, as well as of NO and CO, could favor the development of mesenteric vasculopathy with splanchnic arteriolar vasodilation and systemic hyperdynamic circulation. The dilation and tortuosity of the mesenteric vein branches, which we have called mesenteric vasculopathy and that also have been described in association with a marked dilation in microcirculation [63], are signs that can be attributed to intestinal inflammation in this PHT experimental model.
In this early state of prehepatic PHT, a gut-liver inflammatory loop could be proposed. Proinflammatory gut-derived mediators produced initially in response to sharp and sudden PHT would be a stimulus for hepatic synthesis of TNF-α and IL-1β, with iNOS activation. Liver TNF-α and IL-β, NO and CO, in turn, could leave the liver and enter the gut to complete the inflammatory loop via bile and/or the systemic recirculation.
It has been shown that in rats with portocaval anastomosis the spleen size reflects portal pressure, and preservation of portal pressure attenuates the shunt sequelae and provides metabolic and nutritional benefits [23]. However, in a group of rats with PHT, spleen weight is inversely correlated with portal pressure (Table 3). In these animals portal pressure has an inverse correlation with spleen/body weight and liver/body weight ratios. Moreover, portal pressure and spleen weight are related with a greater development of collateral circulation (Table 2). All the above mentioned findings suggest that this subgroup of animals represents one type of PHT which is characterized by hepatosplenomegaly and a great development of portosystemic collateral circulation.
PHT results from a pathological increase in either portal venous inflow (“forward hypothesis”) or resistance (“backward hypothesis”) and the maintenance of an elevated portal pressure depends, in part, on enhanced portal collateral resistance [2]. It could, therefore, be considered that in this group of animals the greater development of collateral circulation could be involved in regulating mesenteric flow and, subsequently, in controlling portal pressure.
Although PHT has been considered as a uniform experimental model, the results described previously could suggest an evolutive heterogeneity in this model. If we consider that in this subgroup of portal hypertensive rats, TNF-α and IL-1β are involved in the production of hepatomegaly and that this increased liver size is secondary to steatosis, then the hyperproduction of IL-10 by the gut would be a mechanism, not only to decrease the inflammatory response, but also to attenuate one of its pathologic consequences, steatosis. Finally, the coexistence of greater spleen weight and greater development of collateral circulation could help to formulate a physiopathological definition of an evolutive type of PHT.
In summary, a hepatointestinal relationship seems to play an essential role in regulating both the portal pressure and its complications. This is only logical because the gut-liver axis is a portal system and its functions are distributed along this axis.
ACKNOWLEDGMENT
We wish to thank Mercedes Galvez for typing the manuscript and Pedro Cuesta, from the Statistical Unit of Complutense University of Madrid, for the statistical study.
References
- 1.Bosch J, Pizcueta P, Feu F, Fernandez M, Garcia-Pagan JC. Pathophysiology of portal hypertension. Gastroenterol Clin North Am. 1992;21(1):1–14. [PubMed] [Google Scholar]
- 2.MacMathuna P, Vlavianos P, Westaby D, Williams R. Pathophysiology of portal hypertension. Dig Dis. 1992;10(suppl 1):3–15. doi: 10.1159/000171382. [DOI] [PubMed] [Google Scholar]
- 3.Bosch J, Garcia-Pagan JC. Complications of cirrhosis. I. Portal hypertension. J Hepatol. 2000;32(suppl 1):141–156. doi: 10.1016/s0168-8278(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Tsao G. Portal hypertension. Curr Opin Gastroenterol. 2002;18(3):351–359. doi: 10.1097/00001574-200205000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Lebrec D. Pharmacological treatment of portal hypertension: present and future. J Hepatol. 1998;28(5):896–907. doi: 10.1016/s0168-8278(98)80241-9. [DOI] [PubMed] [Google Scholar]
- 6.Bosch J, Abraldes JG, Groszmann RJ. Current management of portal hypertension. J Hepatol. 2003;38(suppl 1):S54–S68. doi: 10.1016/s0168-8278(02)00430-0. [DOI] [PubMed] [Google Scholar]
- 7.Iannitti DA, Henderson JM. Surgery in portal hypertension. Bailliere Clin Gastr. 1997;11(2):351–364. doi: 10.1016/s0950-3528(97)90044-0. [DOI] [PubMed] [Google Scholar]
- 8.Myking AO, Halvorsen JF. A method for graded portal vein stenosis in rats: survival related to degree of stenosis. Eur Surg Res. 1973;5(6):454–457. doi: 10.1159/000127689. [DOI] [PubMed] [Google Scholar]
- 9.Chojkier M, Groszmann RJ. Measurement of portal-systemic shunting in the rat by using gamma-labeled microspheres. Am J Physiol. 1981;240(5):G371–G375. doi: 10.1152/ajpgi.1981.240.5.G371. [DOI] [PubMed] [Google Scholar]
- 10.Sikuler E, Kravetz D, Groszmann RJ. Evolution of portal hypertension and mechanisms involved in its maintenance in a rat model. Am J Physiol. 1985;248(pt 1):G618–G625. doi: 10.1152/ajpgi.1985.248.6.G618. [DOI] [PubMed] [Google Scholar]
- 11.Vorobioff J, Bredfeldt JE, Groszmann RJ. Hyperdynamic circulation in portal-hypertensive rat model: a primary factor for maintenance of chronic portal hypertension. Am J Physiol. 1983;244(1):G52–G57. doi: 10.1152/ajpgi.1983.244.1.G52. [DOI] [PubMed] [Google Scholar]
- 12.Lin HC, Yang MC, Hou MC, et al. Effects of long-term administration of octreotide in portal vein-stenosed rats. Hepatology. 1996;23(3):537–543. doi: 10.1002/hep.510230319. [DOI] [PubMed] [Google Scholar]
- 13.Aller Reyero MA, Diéguez Fernéndez B, Nava Hidalgo MP, et al. Tipos evolutivos de hipertension portal prehepatica en la rata. An Med Interna. 2002;19(7):341–351. [PubMed] [Google Scholar]
- 14.Diez-Arias JA, Aller MA, Palma MD, et al. Increased duodenal mucosa infiltration by mast cell in rats with portal hypertension. Dig Surg. 2001;18(1):34–40. doi: 10.1159/000050094. [DOI] [PubMed] [Google Scholar]
- 15.Monterde G, Rodriguez-Fabian G, Vara E, et al. Increased plasma levels of corticosterone and prolactin and decreased T3 and T4 levels in short-term prehepatic portal hypertension in rats. Dig Dis Sci. 2000;45(9):1865–1871. doi: 10.1023/a:1005588918516. [DOI] [PubMed] [Google Scholar]
- 16.Castaneda B, Dubernardi-Venon W, Bandi JC, et al. The role of portal pressure in the severity of bleeding in portal hypertensive rats. Hepatology. 2000;31(3):581–586. doi: 10.1002/hep.510310306. [DOI] [PubMed] [Google Scholar]
- 17.Kravetz D, Sikuler E, Groszmann RJ. Splanchnic and systemic hemodynamics in portal hypertensive rats during hemorrhage and blood volume restitution. Gastroenterology. 1986;90(pt 1):1232–1240. doi: 10.1016/0016-5085(86)90390-2. [DOI] [PubMed] [Google Scholar]
- 18.Dieguez B, Aller MA, Nava MP, et al. Chronic portal hypertension in the rat by triple-portal stenosing ligation. J Inves Surg. 2002;15(6):329–336. doi: 10.1080/08941930290086146. [DOI] [PubMed] [Google Scholar]
- 19.Mester M, Carter EA, Tompkins RG, et al. Thermal injury induces very early production of interleukin-1 alpha in the rat by mechanisms other than endotoxemia. Surgery. 1994;115(5):588–596. [PubMed] [Google Scholar]
- 20.Arias-Diaz J, Vara E, Torres-Melero J, Garcia C, Hernandez J, Balibrea JL. Local production of oxygen free radicals and nitric oxide in rat diaphragm during sepsis: effects of pentoxifylline and somatostatin. Eur J Surg. 1997;163(8):619–625. [PubMed] [Google Scholar]
- 21.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnock JS, Tannenbaum SR. Analysis of nitrate, nitrite and (15N)nitrate in biological fluids. Anal Biochem. 1982;126(1):131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 22.Wolff DG. The formation of carbon monoxide during peroxidation of microsomal lipids. Biochem Biophys Res Commun. 1976;73(4):850–857. doi: 10.1016/0006-291x(76)90199-6. [DOI] [PubMed] [Google Scholar]
- 23.Dasarathy S, Mullen KD, Conjeevaram HS, Kaminsky-Russ K, Wills LA, McCullough AJ. Preservation of portal pressure improves growth and metabolic profile in the male portacaval-shunted rat. Dig Dis Sci. 2002;47(9):1936–1942. doi: 10.1023/a:1019683703951. [DOI] [PubMed] [Google Scholar]
- 24.Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: a complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med. 2002;30(suppl 1):S58–S63. [PubMed] [Google Scholar]
- 25.Del Prete G, De Carli M, Almergogna F, Giudizi MG, Biagiotti R, Romagnani S. Human IL-10 is produced by both type 1 helper (TH1) and type 2 helper (TH2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. J Immunol. 1993;150(2):353–360. [PubMed] [Google Scholar]
- 26.Yokoyama Y, Kitchens WC, Toth B, et al. Role of IL-10 in regulating proinflammatory cytokine release by Kupffer cells following trauma-hemorrhage. Am J Physiol Gastrointest Liver Physiol. 2004;286(6):G942–G946. doi: 10.1152/ajpgi.00502.2003. [DOI] [PubMed] [Google Scholar]
- 27.Swain MG, Appleyard C, Wallace J, Wong H, Le T. Endogenous glucocorticoids released during acute toxic liver injury enhance hepatic IL-10 synthesis and release. Am J Physiol Gastrointest Liver Physiol. 1999;276(pt 1):G199–G205. doi: 10.1152/ajpgi.1999.276.1.G199. [DOI] [PubMed] [Google Scholar]
- 28.Lopez-Talavera JC, Merrill WW, Groszmann RJ. Tumor necrosis factor alpha: A major contributer to the hyperdynamic circulation in prehepatic portal-hypertensive rats. Gastroenterology. 1995;108(3):761–767. doi: 10.1016/0016-5085(95)90449-2. [DOI] [PubMed] [Google Scholar]
- 29.Munoz J, Albillos A, Perez-Paramo M, Rossi I, Alvarez-Mon M. Factors mediating the hemodynamic effects of tumor necrosis factor-alpha in portal hypertensive rats. Am J Physiol. 1999;276(pt 1):G687–G693. doi: 10.1152/ajpgi.1999.276.3.G687. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Tsao G, Wiest R. Gut microflora in the pathogenesis of the complications of cirrhosis. Best Pract Res Clin Gastroenterol. 2004;18(2):353–372. doi: 10.1016/j.bpg.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Haddad JJ, Fahlman CS. Redox- and antioxidant-mediated regulation of interleukin-10: an antiinflammatory, antioxidant cytokine? Bioch Bioph Res Commun. 2002;297(2):163–176. doi: 10.1016/s0006-291x(02)02094-6. [DOI] [PubMed] [Google Scholar]
- 32.Madsen K. Combining T cells and IL-10: a new therapy for Crohn's disease? Gastroenterology. 2002;123(6):2140–2144. doi: 10.1053/gast.2002.37289. [DOI] [PubMed] [Google Scholar]
- 33.Kotenko SV. The family of IL-10-related cytokines and their receptors: related, but to what extent? Cytok Growth Factor Rev. 2002;13(3):223–240. doi: 10.1016/s1359-6101(02)00012-6. [DOI] [PubMed] [Google Scholar]
- 34.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 35.Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83(3):835–870. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 36.Lundberg JE, Roth TP, Dunn RM, Doyle JW. Comparison of IL-10 levels in chronic venous insufficiency ulcers and autologous donor tissue. Arch Dermatol Res. 1998;290(12):669–673. doi: 10.1007/s004030050371. [DOI] [PubMed] [Google Scholar]
- 37.Louis H, Le Moine O, Goldman M, Deviere J. Modulation of liver injury by interleukin-10. Acta Gastroenterol Belg. 2003;66(1):7–14. [PubMed] [Google Scholar]
- 38.Wang SC, Ohata M, Schrum L, Rippe RA, Tsukamoto H. Expression of interleukin-10 by in vitro and in vivo activated hepatic stellate cells. J Biol Chem. 1998;273(1):302–308. doi: 10.1074/jbc.273.1.302. [DOI] [PubMed] [Google Scholar]
- 39.Leifeld L, Cheng S, Ramakers J, et al. Imbalanced intrahepatic expression of interleukin-12, interferon gamma and interleukin-10 in fulminant hepatitis B. Hepatology. 2002;36(pt 1):1001–1008. doi: 10.1053/jhep.2002.35532. [DOI] [PubMed] [Google Scholar]
- 40.Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol. 2002;283(2):G256–G265. doi: 10.1152/ajpgi.00550.2001. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Tsao G, Albillos A, Barden GE, West AB. Bacterial translocation in acute and chronic portal hypertension. Hepatology. 1993;17(6):1081–1085. [PubMed] [Google Scholar]
- 42.Alonso MJ, Aller MA, Corcuera MT, et al. Progressive hepatocytic fatty infiltration in rats with prehepatic portal hypertension. Hepatogastroenterology. In press. [PubMed]
- 43.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343(20):1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 44.Diehl AM. Cytokine regulation of liver injury and repair. Immunol Rev. 2000;174:160–171. doi: 10.1034/j.1600-0528.2002.017411.x. [DOI] [PubMed] [Google Scholar]
- 45.Solga SF, Deihl AM. Non-alcoholic fatty liver disease: lumen-liver interactions and possible role for probiotics. J Hepatol. 2003;38(5):681–687. doi: 10.1016/s0168-8278(03)00097-7. [DOI] [PubMed] [Google Scholar]
- 46.Vara E, Arias-Diaz J, Torres-Melero J, Garcia C, Rodriguez JM, Balibrea JL. Effect of different sepsis related cytokines on lipid synthesis by isolated hepatocytes. Hepatology. 1994;20(pt 1):924–931. doi: 10.1002/hep.1840200422. [DOI] [PubMed] [Google Scholar]
- 47.Hallenbeck JM. The many faces of tumor necrosis factor in stroke. Nature Med. 2002;8(12):1363–1368. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- 48.Kilbourn RG, Belloni P. Endothelial cell production of nitrogen oxides in response to interferon gamma in combination with tumor necrosis factor, interleukin-1 or endotoxin. J Natl Cancer Inst. 1990;82(9):772–776. doi: 10.1093/jnci/82.9.772. [DOI] [PubMed] [Google Scholar]
- 49.Wiest R, Groszmann RJ. The paradox of nitric oxide in cirrhosis and portal hypertension: too much, not enough. Hepatology. 2002;35(2):478–491. doi: 10.1053/jhep.2002.31432. [DOI] [PubMed] [Google Scholar]
- 50.Vallance P, Moncada S. Hyperdynamic circulation in cirrhosis: a role for nitric oxide? Lancet. 1991;337(8744):776–778. doi: 10.1016/0140-6736(91)91384-7. [DOI] [PubMed] [Google Scholar]
- 51.Wiest R, Groszmann RJ. Nitric oxide and portal hypertension: its role in the regulation of intrahepatic and splanchnic vascular resistance. Semin Liver Dis. 1999;19(4):411–426. doi: 10.1055/s-2007-1007129. [DOI] [PubMed] [Google Scholar]
- 52.Pateron D, Tazi KA, Sogni P, et al. Role of aortic nitric oxide synthase 3 (eNOS) in the systemic vasodilation of portal hypertension. Gastroenterology. 2000;119(1):196–200. doi: 10.1053/gast.2000.8554. [DOI] [PubMed] [Google Scholar]
- 53.Hori N, Wiest R, Groszmann RJ. Enhanced release of nitric oxide in response to changes in flow and shear stress in the superior mesenteric arteries of portal hypertensive rats. Hepatology. 1998;28(6):1467–73. doi: 10.1002/hep.510280604. [DOI] [PubMed] [Google Scholar]
- 54.Murray BM, Paller MS. Decreased pressor reactivity to angiotensin II in cirrhotic rats. Evidence for a post-receptor defect in angiotensin action. Circ Res. 1985;57(3):424–431. doi: 10.1161/01.res.57.3.424. [DOI] [PubMed] [Google Scholar]
- 55.Michielsen PP, Boeckxstaens GE, Sys SU, Hernan AG, Pelckmans PA. The role of increased nitric oxide in the vascular hyporeactivity to noradrenaline in long-term portal vein ligated rats. J Hepatol. 1995;23(3):341–347. [PubMed] [Google Scholar]
- 56.Ai JH, Yang Z, Qiu FZ, Zhu T. Heat shock protein 90 is responsible for hyperdynamic circulation in portal hypertensive rats. World J Gastroenterol. 2003;9(11):2544–2547. doi: 10.3748/wjg.v9.i11.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cahill PA, Redmond EM, Sitzmann JV. Endothelial dysfunction in cirrhosis and portal hypertension. Pharmacol Ther. 2001;89(3):273–93. doi: 10.1016/s0163-7258(01)00128-0. [DOI] [PubMed] [Google Scholar]
- 58.Otterbein LE, Choi AMK. Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol. 2000;279(6):L1029–L1037. doi: 10.1152/ajplung.2000.279.6.L1029. [DOI] [PubMed] [Google Scholar]
- 59.Otterbein LE, Bach EH, Alam J, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated proteinkinase pathway. Nature Med. 2000;6(4):422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 60.Fernandez M, Lambrecht RW, Bonkovsky HL. Increased heme oxygenase activity in splanchnic organs from portal hypertensive rats: role in modulating mesenteric vascular reactivity. J Hepatol. 2001;34(6):812–817. doi: 10.1016/s0168-8278(01)00010-1. [DOI] [PubMed] [Google Scholar]
- 61.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 62.Suematsu M, Kashiwag S, Sano T, Goda N, Shinoda Y, Ishimura Y. Carbon monoxide as an endogenous modulator of hepatic vascular perfusion. Biochem Biophys Res Commun. 1994;205(2):1333–1337. doi: 10.1006/bbrc.1994.2811. [DOI] [PubMed] [Google Scholar]
- 63.Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: A novel target for the modulation of the inflammatory response. Nature Med. 1996;2(1):87–90. doi: 10.1038/nm0196-87. [DOI] [PubMed] [Google Scholar]
- 64.Ponce Gonzalez JF, Dominguez R, Adame Lanuza E, Martin Zurita I, Morales Mendez S. Portal hypertensive colopathy: histologic appearance of the colonic mucosa. Hepatogastroenterology. 1998;45(19):40–43. [PubMed] [Google Scholar]